The Importance of a Multidisciplinary Approach in the Management of a Patient with Type I Gaucher Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
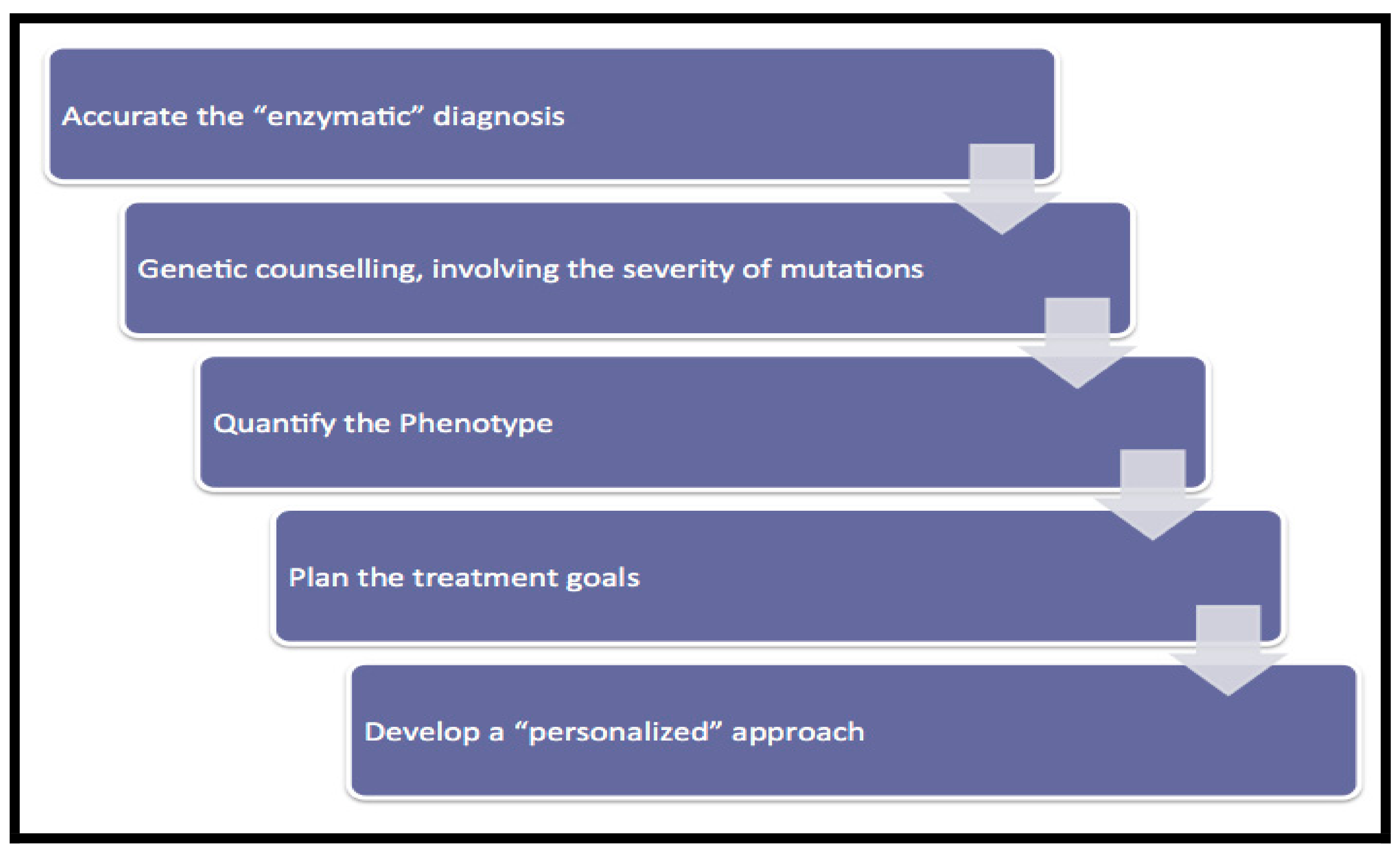
2. Optimizing Management and Follow-Up of Patients with Type I Gaucher Disease
2.1. Enzymatic Analysis
2.2. Genetic Counselling
2.3. Severity of Mutations
2.4. Phenotypic Quantification
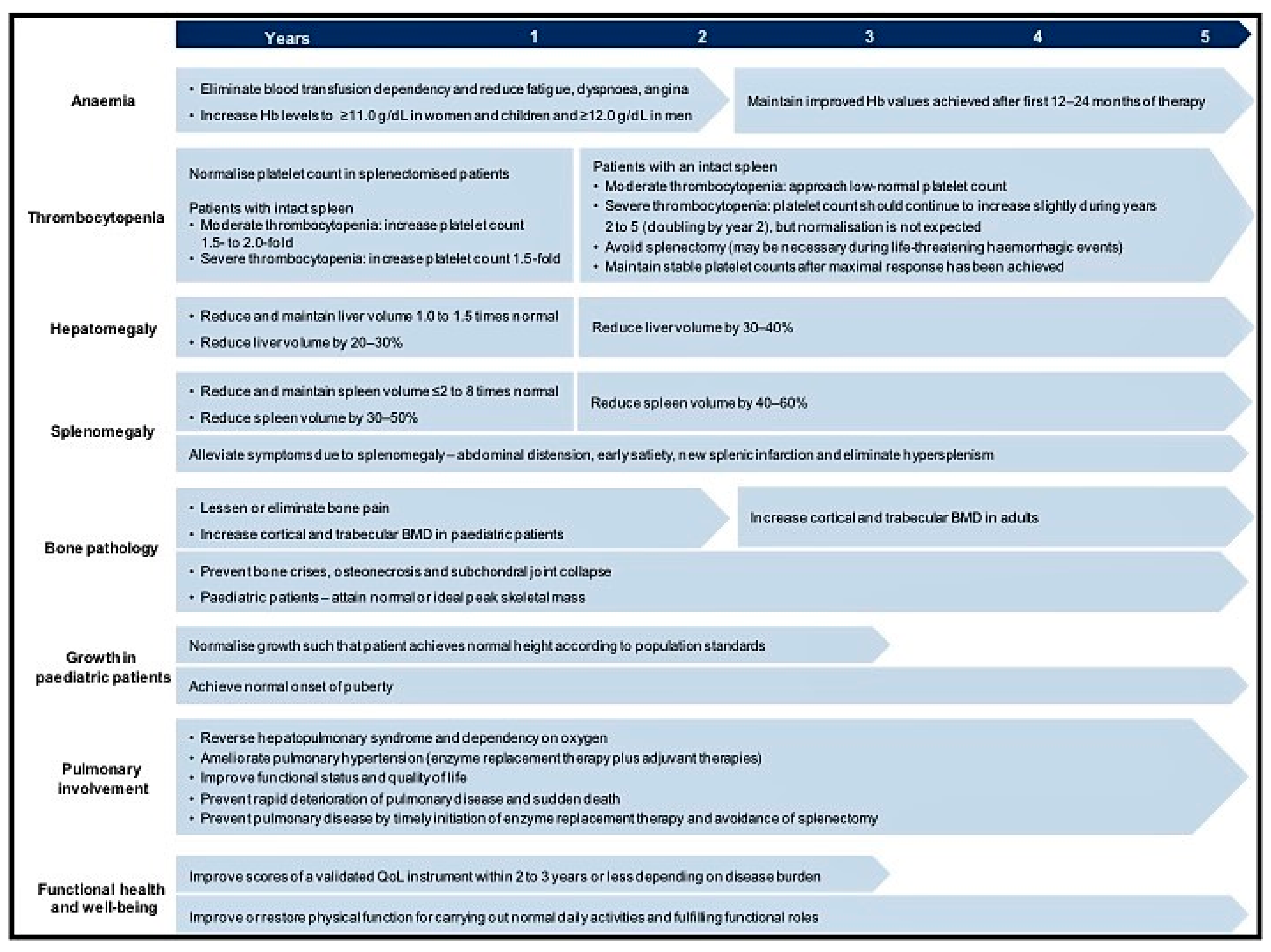
2.5. Treatment Goals
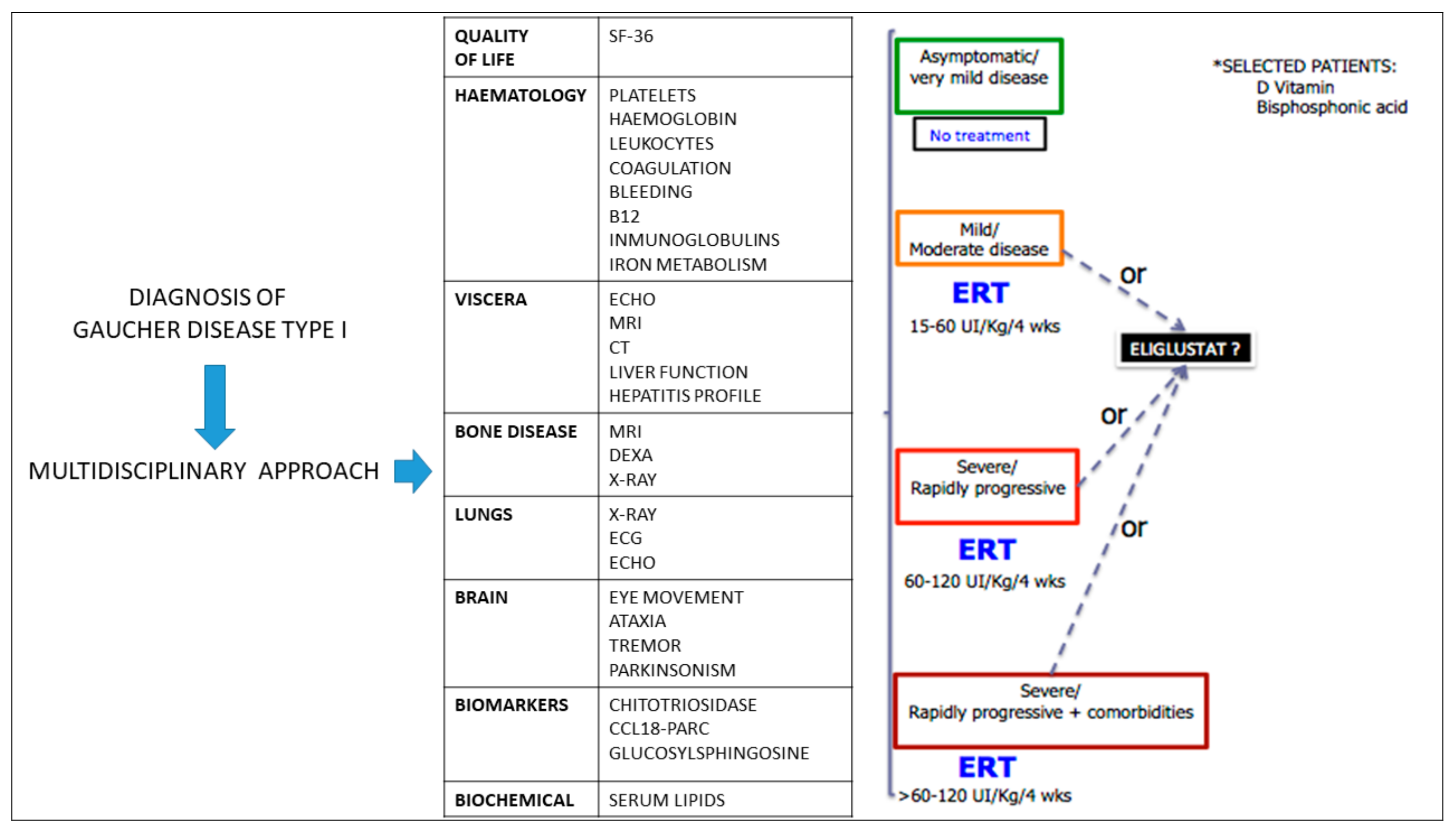
2.6. Implementing “Personalized Medicine”
- Mild/moderate disease: ERT should be started at doses of 15–60 UI/Kg/4 weeks.
- Severe/rapidly progressive: ERT may be required at doses between 60–120 UI/Kg/4 weeks.
- Severe/rapidly progressive in the presence of comorbidities: ERT should be initiated at doses higher than 60–120 UI/Kg/4 weeks.
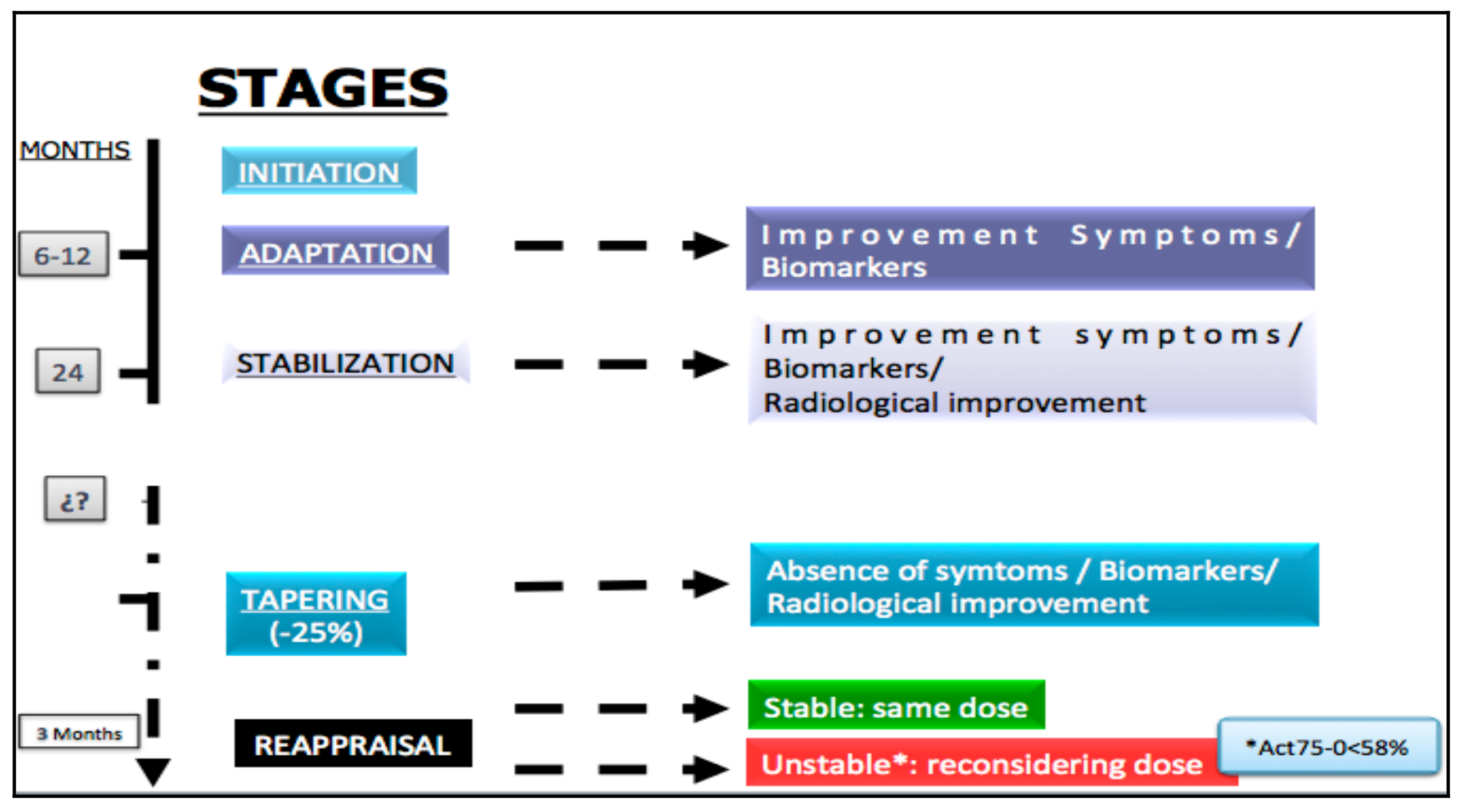
- Initiation of selected drug.
- Adaptation: In this stage a clinical improvement and the biomarkers must be demonstrated. This period of time fluctuates between 6 and 12 months.
- Stabilization: For most patients it occurs after the first 24 months following the beginning of treatment. Besides the improvement of the clinical aspects and the biomarkers, radiological enhancement must be exhibited.
- Tapering: After stabilization of the patient, which is defined by the absence of symptoms, normalization of biomarkers and demonstration of a radiological improvement, the tapering stage can be initiated, with a dose reduction of 25% in the case of ERT.
- Maintenance: 3 Months after modification of the doses, patients must be carefully re-evaluated, according to the “evaluation and monitoring recommendations”. If the patient remains stable the same dosage should be continued, but in an unstable patient the multidisciplinary team should reconsider the appropriate dose. Recently, the usefulness of a new biomarker (Act 75–0 <58%) has been published, which has proved extremely practical for discriminating whether the dose administered is sufficient in the patient with ERT [34].
3. Summary
- (1)
- Social: Enhancing access to services, improving the quality of care and lowering overall health care expenditure. Moreover, it allows more efficient use of interventions by setting individualized objectives, varying dosing schedules, and types of therapy.
- (2)
- Medical: This model provides a framework that enriches the clinical interpretation and the applicability of clinical assessment tools which represents the best model for the translation of results from research to clinical practice.
Author Contributions
Funding
Conflicts of Interest
References
- Brady, R.O.; Kanfer, J.N.; Bradley, R.M.; Shapiro, D. Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher’s disease. J. Clin. Investig. 1966, 45, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Grabowski, G.A. Gaucher disease. In The Metabolic Basis of Inherited Disease, 7th ed.; Scriver, C.R., Beudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 1995; pp. 2641–2670. [Google Scholar]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Sainsbury, R.; Haward, B.; Rider, L.; Johnston, C.; Round, C. Influence of clinician workload and patterns of treatment on survival from breast cancer. Lancet 1995, 345, 1265–1270. [Google Scholar] [CrossRef]
- Chang, J.H.; Vines, E.; Bertsch, H.; Fraker, D.L.; Czerniecki, B.J.; Rosato, E.F.; Lawton, T.; Conant, E.F.; Orel, S.G.; Schuchter, L.; et al. The impact of a multidisciplinary breast cancer center on recommendations for patient management: The University of Pennsylvania experience. Cancer 2001, 91, 1231–1237. [Google Scholar] [CrossRef]
- Zimran, A. How I treat Gaucher Disease. Blood 2011, 118, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Kuhl, W. The diagnosis of the adult type of Gaucher’s disease and its carrier state by demonstration of deficiency of beta-glucosidase activity in peripheral blood leukocytes. J. Lab. Clin. Med. 1970, 76, 747–755. [Google Scholar] [PubMed]
- Beutler, E.; Saven, A. Misuse of marrow examination in the diagnosis of Gaucher disease. Blood 1990, 76, 646–648. [Google Scholar] [PubMed]
- Torralba, M.A.; Alfonso, P.; Pérez-Calvo, J.I.; Cenarro, A.; Pastores, G.M.; Giraldo, P.; Civeira, F.; Pocovi, M. High prevalence of the 55-bp deletion (c.1263del55) in Exon 9 of the glucocerebrosidase gene causing misdiagnosis (for homozygous N370S (c.1226A>G) mutation) in Spanish Gaucher Disease patients. Blood Cells Mol. Dis. 2002, 29, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Mensink, K.A.; Jennifer, L.; Hand, J.L. Autosomal recessive inheritance: Un updated review. Pediatr. Dermatol. 2006, 23, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Grace, M.E.; Graves, P.N.; Smith, F.I.; Grabowski, G.A. Analyses of catalytic activity and inhibitor binding of human acid beta-glucosidase by site-directed mutagenesis. Identification of residues critical to catalysis and evidence for causality of two Ashkenazi Jewish Gaucher disease type 1 mutations. J. Biol. Chem. 1990, 265, 6827–6835. [Google Scholar] [PubMed]
- Torralba, M.A.; Olivera, S.; Bureo, J.C.; Dalmau, J.; Nuñez, R.; León, P.; Villarrubia, J. Residual enzymatic activity as a prognostic factor in patients with Gaucher disease type 1: Correlation with Zimran and GAUSS-I index and the severity of bone disease. QJM Int. J. Med. 2016, 109, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Goker-Alpan, O.; Hruska, K.S.; Orvisky, E.; Kishnani, P.S.; Stubblefield, B.K.; Schiffmann, R.; Sidransky, E. Divergent phenotypes in Gaucher disease implicate the role of modifiers. J. Med. Genet. 2005, 42, e37. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Kay, A.; Gelbart, T.; Garver, P.; Thurston, D.; Saven, A.; Beutler, E. Gaucher disease. Clinical, laboratory, radiologic, and genetic features of 53 patients. Medicine 1992, 71, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Di Rocco, M.; Giona, F.; Carubbi, F.; Linari, S.; Minichilli, F.; Brady, R.O.; Mariani, G.; Cappellini, M.D. A new severity score index for phenotypic classification and evaluation of responses to treatment in type I Gaucher disease. Haematologica 2008, 93, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, N.J.; Cappellini, M.D.; Cox, T.M.; Giannini, E.H.; Grabowski, G.A.; Hwu, W.L.; Mankin, H.; Martins, A.M.; Sawyer, C.; Vom Dahl, S.; et al. A validated disease severity scoring system for adults with type 1 Gaucher disease. Genet Med. 2010, 12, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Hollack, C.E.; van Weely, S.; van Oers, M.H.; Aerts, J.M. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J. Clin. Investig. 1994, 93, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Boot, R.G.; Verhoek, M.; De Fost, M.; Hollack, C.E.; Maas, M.; Bleijlevens, B.; van Breemen, M.J.; van Meurs, M.; Boven, L.A.; Laman, J.D.; et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: A novel surrogate marker for assessing therapeutic intervention. Blood 2003, 103, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, V.; Chuang, W.L.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Pastores, G.M.; Weinreb, N.J.; Aerts, H.; Andria, G.; Cox, T.M.; Giralt, M.; Grabowski, G.A.; Mistry, P.K.; Tylki-Szymańska, A. Therapeutic goals in the treatment of Gaucher disease. Semin. Hematol. 2004, 41, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Biegstraaten, M.; Cox, T.M.; Belmatoug, N.; Berger, M.G.; Collin-Histed, T.; Vom Dahl, S.; Di Rocco, M.; Fraga, C.; Giona, F.; Giraldo, P.; et al. Management goals for type 1 Gaucher disease: An expert consensus document from the European working group on Gaucher disease. Blood Cells Mol. Dis. 2018, 68, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Charrow, J.; Andersson, H.C.; Kaplan, P.; Kolodny, E.H.; Mistry, P.; Pastores, G.; Rosenbloom, B.E.; Scott, C.R.; Wappner, R.S.; Weinreb, N.J. The Gaucher registry: Demographics and disease characteristics of 1698 patients with Gaucher disease. Arch. Intern. Med. 2000, 160, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Lau, H.; Belmatoug, N.; Deegan, P.; Goker-Alpan, O.; Schwartz, I.V.D.; Shankar, S.P.; Panahloo, Z.; Zimran, A. Reported outcomes of 453 pregnancies in patients with Gaucher disease: An analysis from the Gaucher outcome survey. Blood Cells Mol. Dis. 2018, 68, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, N.J.; Aggio, M.C.; Andersson, H.C.; Andria, G.; Charrow, J.; Clarke, J.T.; Erikson, A.; Giraldo, P.; Goldblatt, J.; Hollak, C.; et al. Gaucher disease type 1: Revised recommendations on evaluations and monitoring for adult patients. Semin. Hematol. 2004, 41, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Baldellou, A.; Andria, G.; Campbell, P.E.; Charrow, J.; Cohen, I.J.; Grabowski, G.A.; Harris, C.M.; Kaplan, P.; McHugh, K.; Mengel, E.; et al. Paediatric non-neuronopathic Gaucher disease: Recommendations for treatment and monitoring. Eur. J. Pediatr. 2004, 163, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Gardner, D. Ten lessons in collaboration. Online J. Issues Nurs. 2005, 10, 2. [Google Scholar] [PubMed]
- Headrick, L.A.; Wilcock, P.M.; Batalden, P.B. Interprofessional working and continuing medical education. BMJ 1998, 316, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Belmatoug, N.; Di Rocco, M.; Fraga, C.; Giraldo, P.; Hughes, D.; Lukina, E.; Maison-Blanche, P.; Merkel, M.; Niederau, C.; Plöckinger, U.; et al. Management and monitoring recommendations for the use of eliglustat in adults with type 1 Gaucher disease in Europe. Eur. J. Intern. Med. 2017, 37, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Cappellini, M.D.; Lukina, E.; Ozsan, H.; Mach Pascual, S.; Rosenbaum, H.; Helena Solano, M.; Spigelman, Z.; Villarrubia, J.; Watman, N.P.; et al. A reappraisal of Gaucher disease-diagnosis and disease management algorithms. Am. J. Hematol. 2011, 86, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.M.; Aerts, J.M.; Andria, G.; Beck, M.; Belmatoug, N.; Bembi, B.; Chertkoff, R.; Vom Dahl, S.; Elstein, D.; Erikson, A.; et al. The role of the iminosugar N-butyldeoxynojirimycin (miglustat) in the management of type I (non-neuronopathic) Gaucher disease: A position statement. J. Inherit. Metab. Dis. 2003, 26, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J.; Mehta, A.; Hollak, C.; Giraldo, P.; Hughes, D.; Belmatoug, N.; Brand, M.; Muller, A.; Schaaf, B.; Giorgino, R.; et al. Miglustat therapy in type 1 Gaucher disease: Clinical and safety outcomes in a multicenter retrospective cohort study. Blood Cells Mol. Dis. 2013, 51, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Burrow, T.A.; Charrow, J.; Goker-Alpan, O.; Kaplan, P.; Kishnani, P.S.; Mistry, P.; Ruskin, J.; Weinreb, N. Recommendations for the use of eliglustat in the treatment of adults with Gaucher disease type 1 in the United States. Mol. Genet. Metab. 2016, 117, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Poll, L.W.; vom Dahl, S. Therapy of adult Gaucher disease. Haematologica 2007, 92, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Gras-Colomer, E.; Martínez-Gómez, M.A.; Climente-Martí, M.; Fernandez-Zarzoso, M.; Almela-Tejedo, M.; Giner-Galvañ, V.; Marcos-Rodríguez, J.A.; Rodríguez-Fernández, A.; Torralba-Cabeza, M.Á.; Merino-Sanjuan, M. Relationship between glucocerebrosidase activity and clinical response to enzyme replacement therapy in patients with Gaucher Disease type I. Basic Clin. Pharmacol. Toxicol. 2018, 123, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Gorman, P. Managing Multi-Disciplinary Teams in the NHS; McGraw-Hill Education: London, UK, 1998; ISBN 0749427876. [Google Scholar]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torralba-Cabeza, M.-Á.; Olivera-González, S.; Sierra-Monzón, J.-L. The Importance of a Multidisciplinary Approach in the Management of a Patient with Type I Gaucher Disease. Diseases 2018, 6, 69. https://doi.org/10.3390/diseases6030069
Torralba-Cabeza M-Á, Olivera-González S, Sierra-Monzón J-L. The Importance of a Multidisciplinary Approach in the Management of a Patient with Type I Gaucher Disease. Diseases. 2018; 6(3):69. https://doi.org/10.3390/diseases6030069
Chicago/Turabian StyleTorralba-Cabeza, Miguel-Ángel, Susana Olivera-González, and José-Luis Sierra-Monzón. 2018. "The Importance of a Multidisciplinary Approach in the Management of a Patient with Type I Gaucher Disease" Diseases 6, no. 3: 69. https://doi.org/10.3390/diseases6030069
APA StyleTorralba-Cabeza, M.-Á., Olivera-González, S., & Sierra-Monzón, J.-L. (2018). The Importance of a Multidisciplinary Approach in the Management of a Patient with Type I Gaucher Disease. Diseases, 6(3), 69. https://doi.org/10.3390/diseases6030069