



Biomolecules 2026, 16(2), 252; https://doi.org/10.3390/biom16020252 - 4 Feb 2026
Viewed by 893
Abstract
►
Show Figures
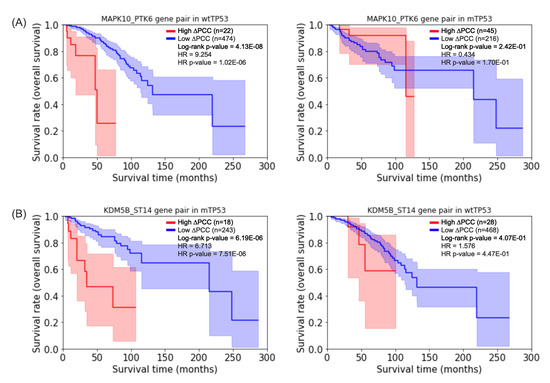
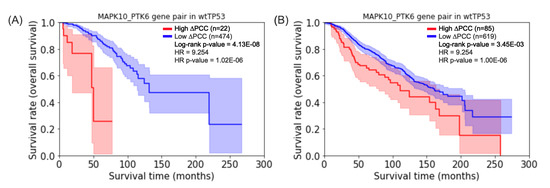
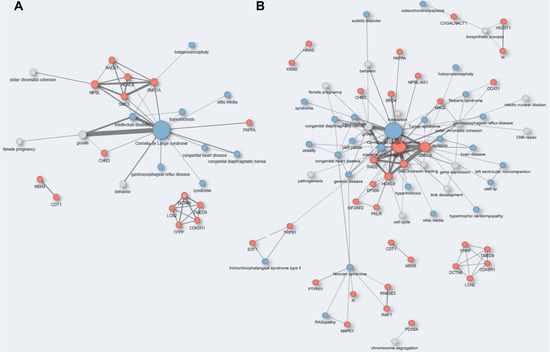
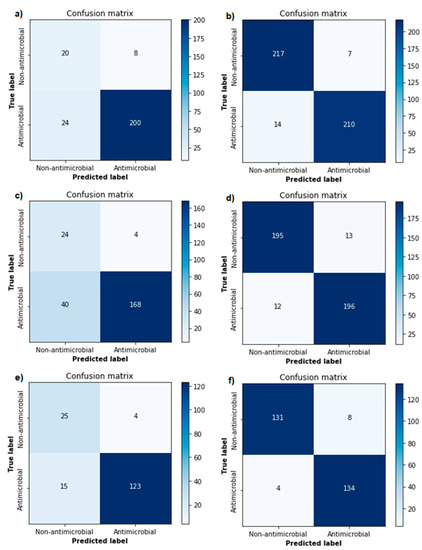
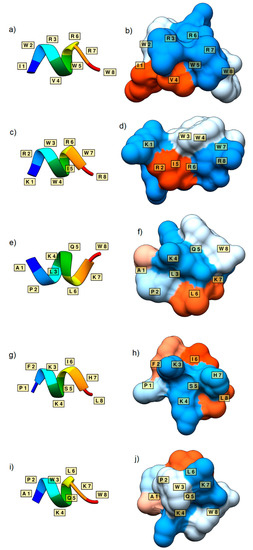
Cancer immunoprevention leverages the immune system’s surveillance mechanisms to mitigate tumor development. Vaccines that constitute a tumor antigen and an immune adjuvant are perceived as immunoprevention modalities. However, relevant tumor antigens are unknown for non-viral cancers, which constitute most human cancers. Our group
[...] Read more.
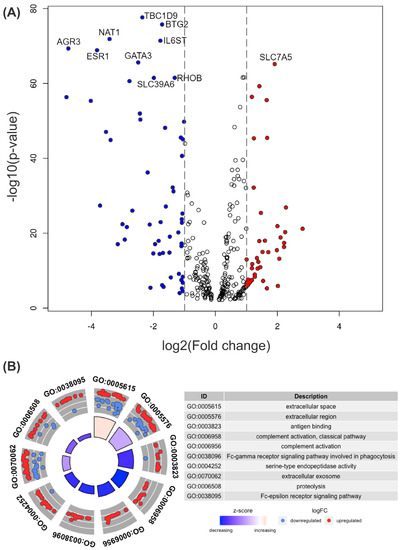
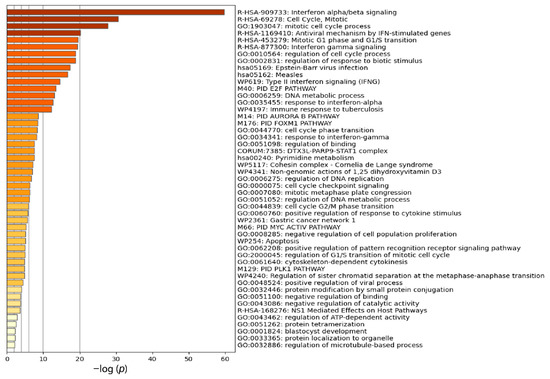
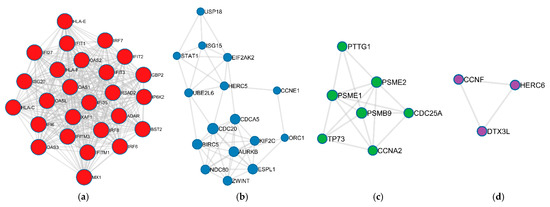
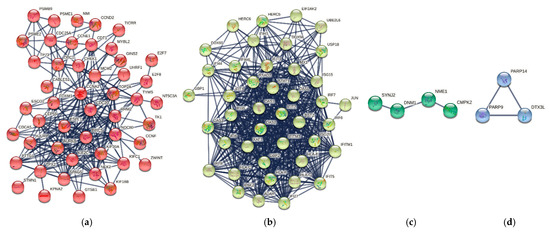
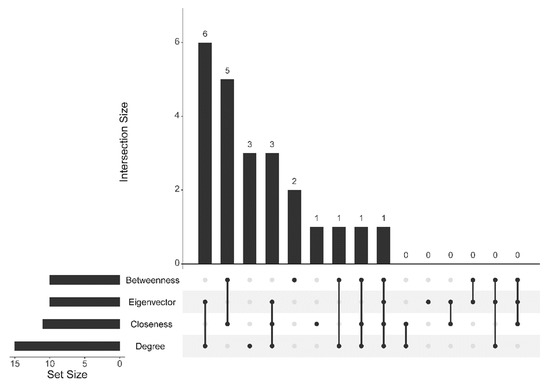
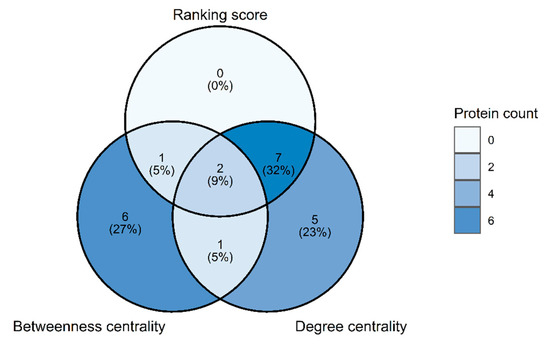
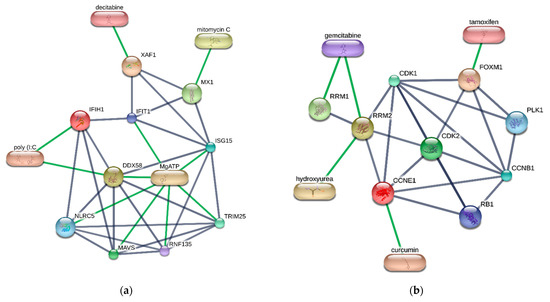
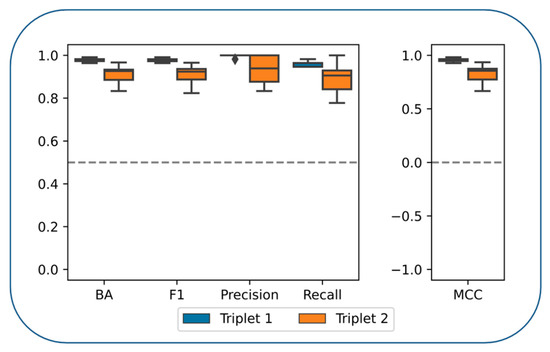
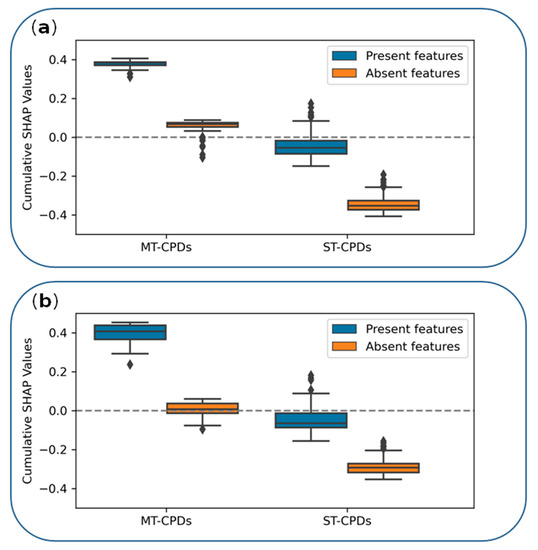
Cancer immunoprevention leverages the immune system’s surveillance mechanisms to mitigate tumor development. Vaccines that constitute a tumor antigen and an immune adjuvant are perceived as immunoprevention modalities. However, relevant tumor antigens are unknown for non-viral cancers, which constitute most human cancers. Our group has recently shown that SA–4–1BBL, a novel agonist of CD137 receptor, but not antibodies, shows immunoprevention efficacy against various tumors. Advanced bioinformatics analyses of bulk RNA-seq data were conducted to elucidate mechanisms underlying cancer immunoprevention. Mice received subcutaneous injections of SA–4–1BBL or agonistic 3H3 antibody, and the injection-site tissue (IS) and draining lymph nodes (LN) were analyzed for differential gene expression. SA–4–1BBL induced a compartmentalized and temporally dynamic immune program characterized by early effector activation at IS and sustained immune regulation in draining LN. K-means clustering of 4564 DEGs identified eight functionally distinct clusters. IS-enriched clusters contained activation genes for CD4+ T and NK cells, including Cd28, Klra1, Cd4, Cd40, and Cd40l, while LN clusters were enriched for regulatory genes (Tnfaip3, Irf5, Col1a2) that ensure immune priming and homeostatic restraint for a balanced response. SA–4–1BBL generated a more selective and durable activation of adaptive immunity, TCR signaling, Th1/Th2 differentiation, and NK cytotoxicity. 3H3 activated broader innate inflammatory programs, including Toll-like receptor and neurodegeneration-linked pathways. IMPRes analysis showed that SA–4–1BBL activates sequential immune-regulatory circuits centered on Stat1, Cd247, and Ifng and modulates the CD151–TGF-β axis. These findings demonstrate that SA–4–1BBL elicits a balanced immune response, ensuring both safety and efficacy in preventing cancer development.
Full article

Figure 1




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}