
Neuroprotective Effect of Apolipoprotein D in Cuprizone-Induced Cell Line Models: A Potential Therapeutic Approach for Multiple Sclerosis and Demyelinating Diseases

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
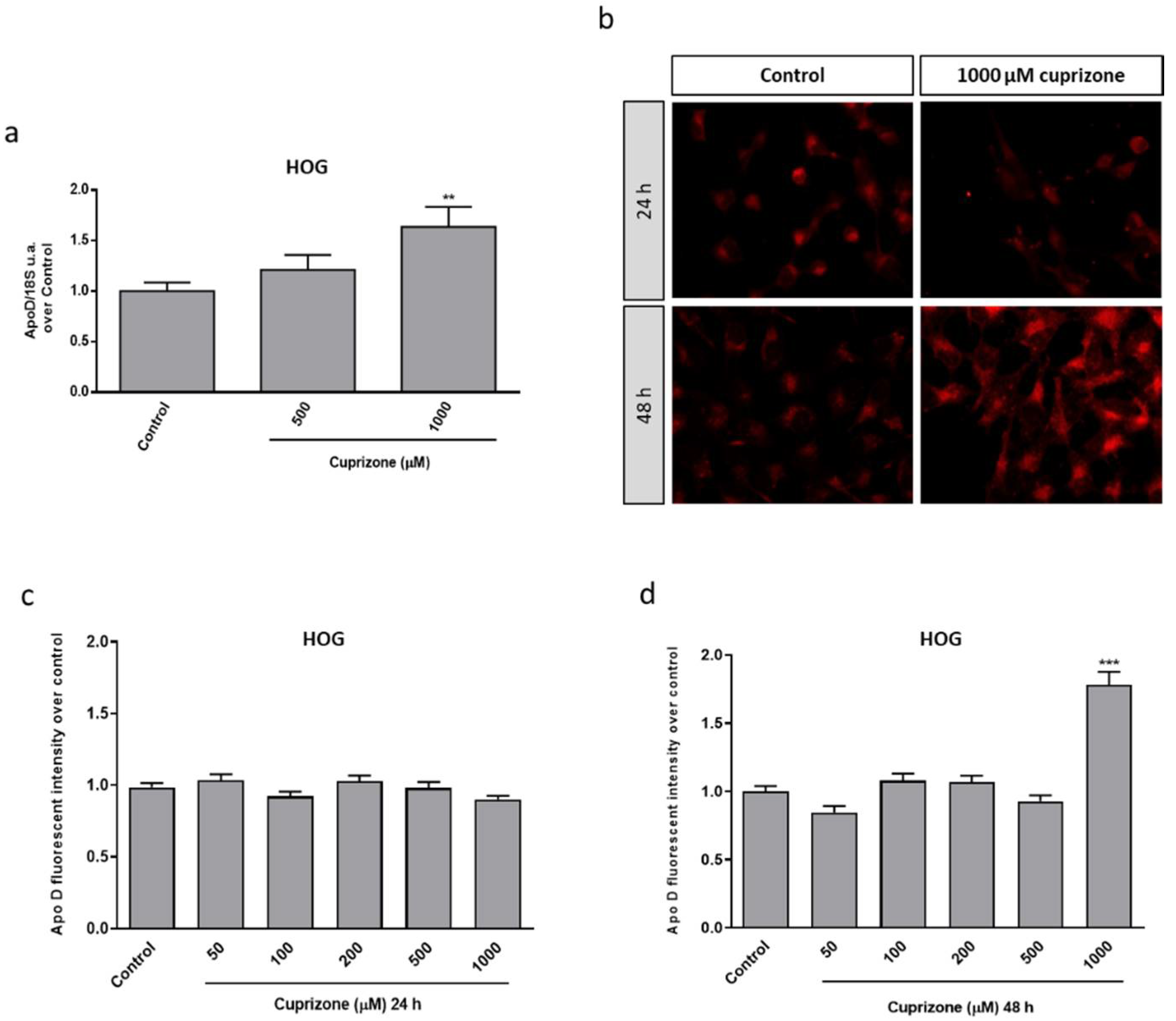
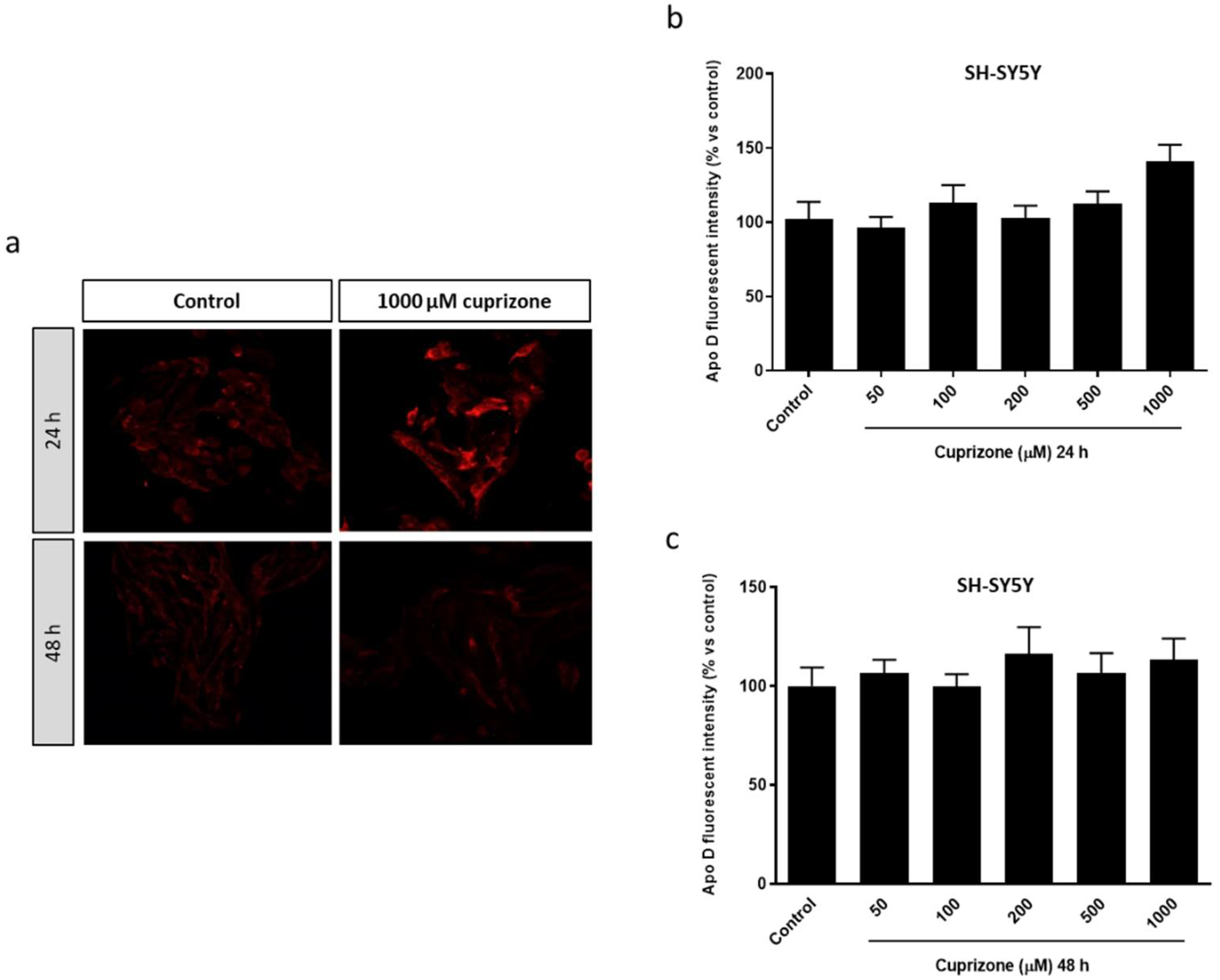
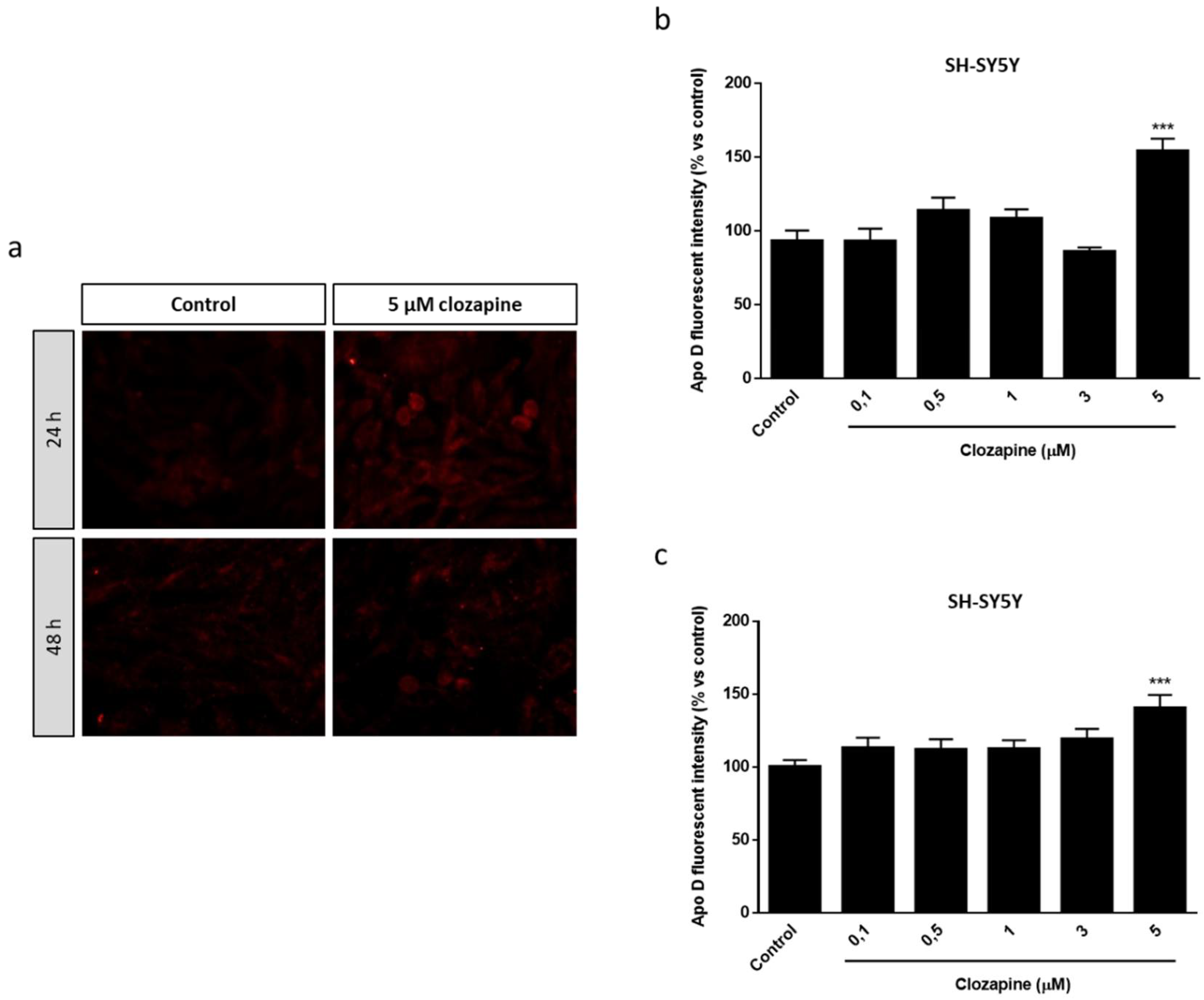
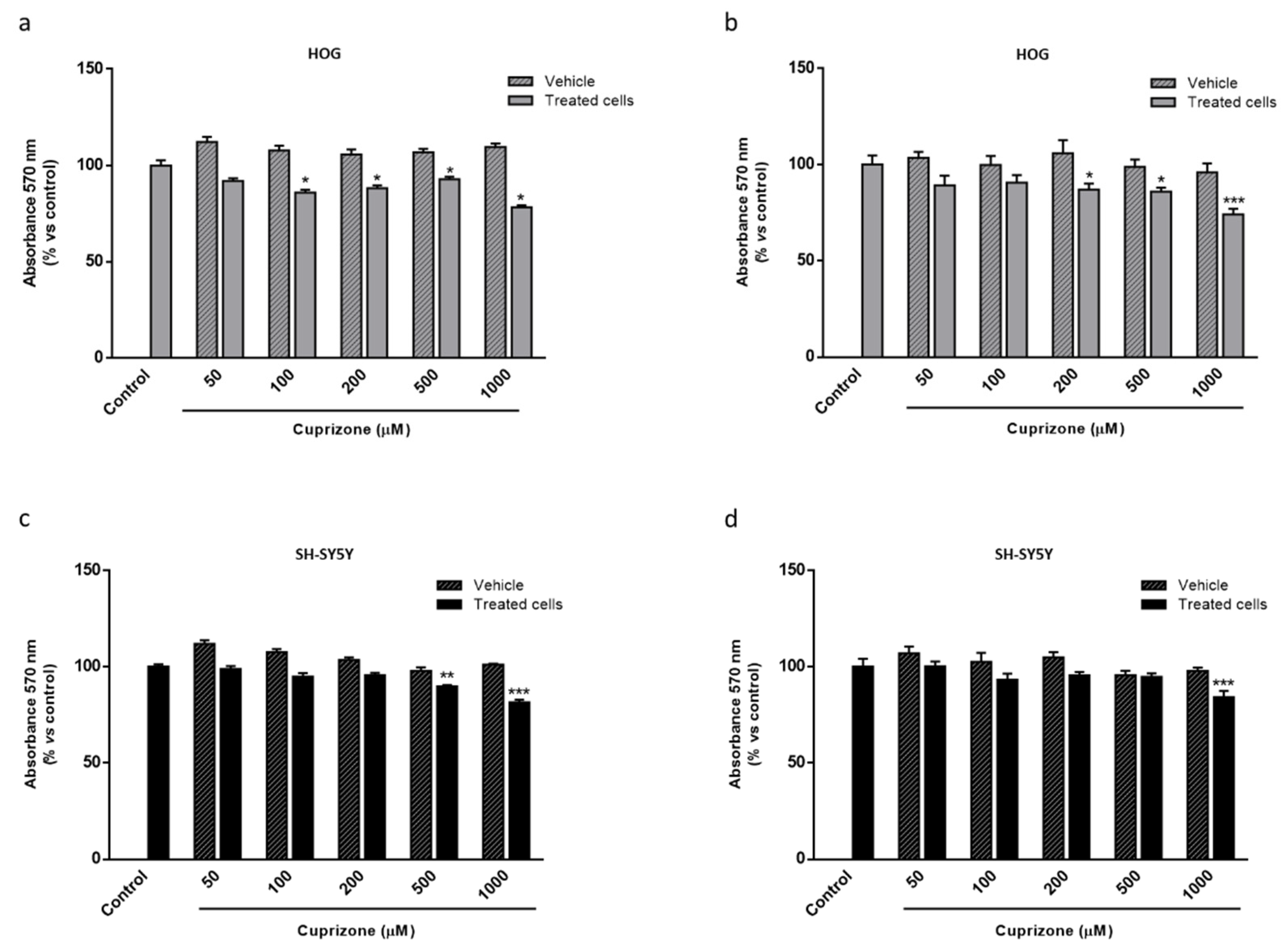
2.1. Apo D Expression in HOG and SH-SY5Y Cells in Response to Cuprizone Treatment
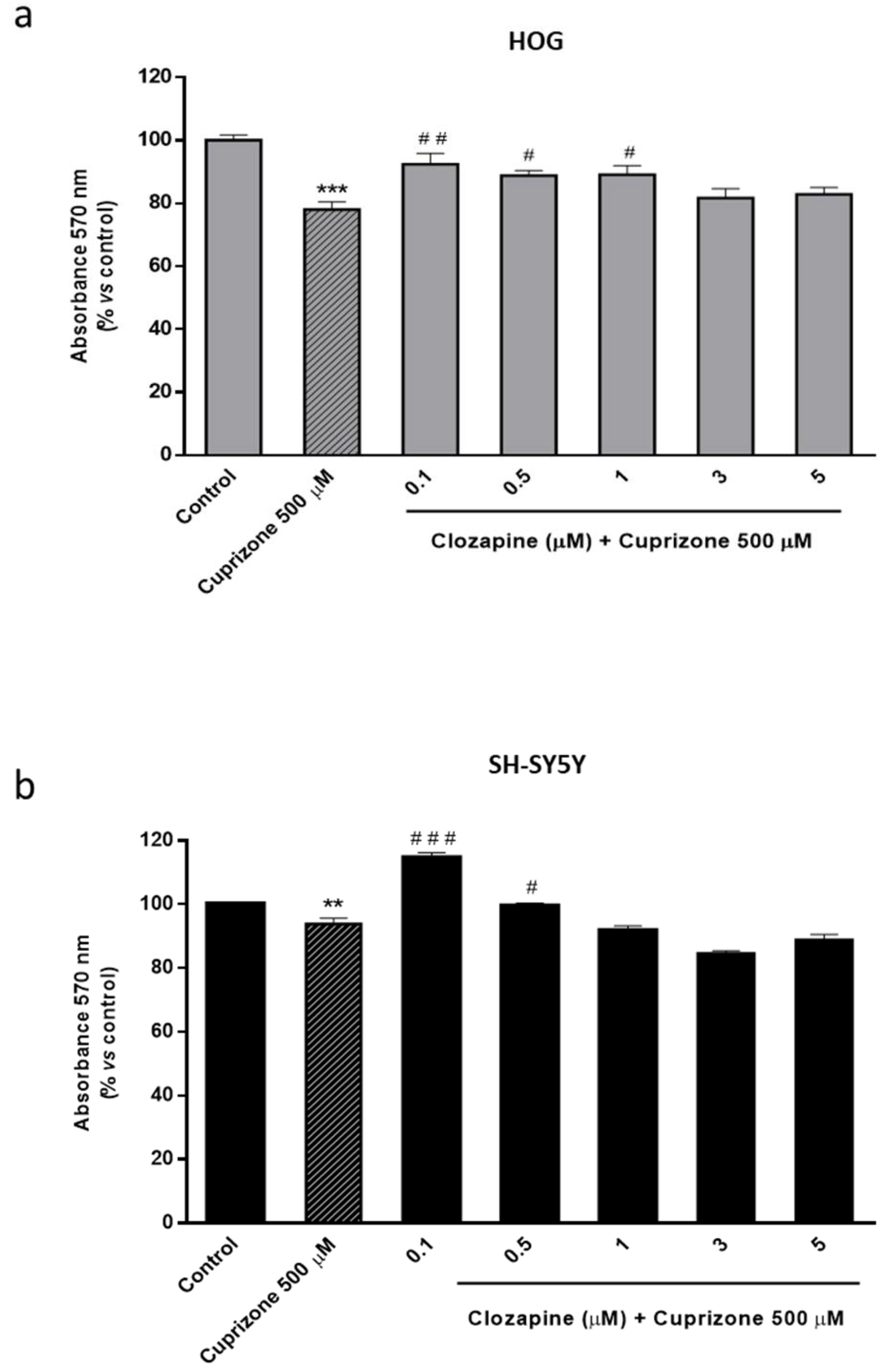
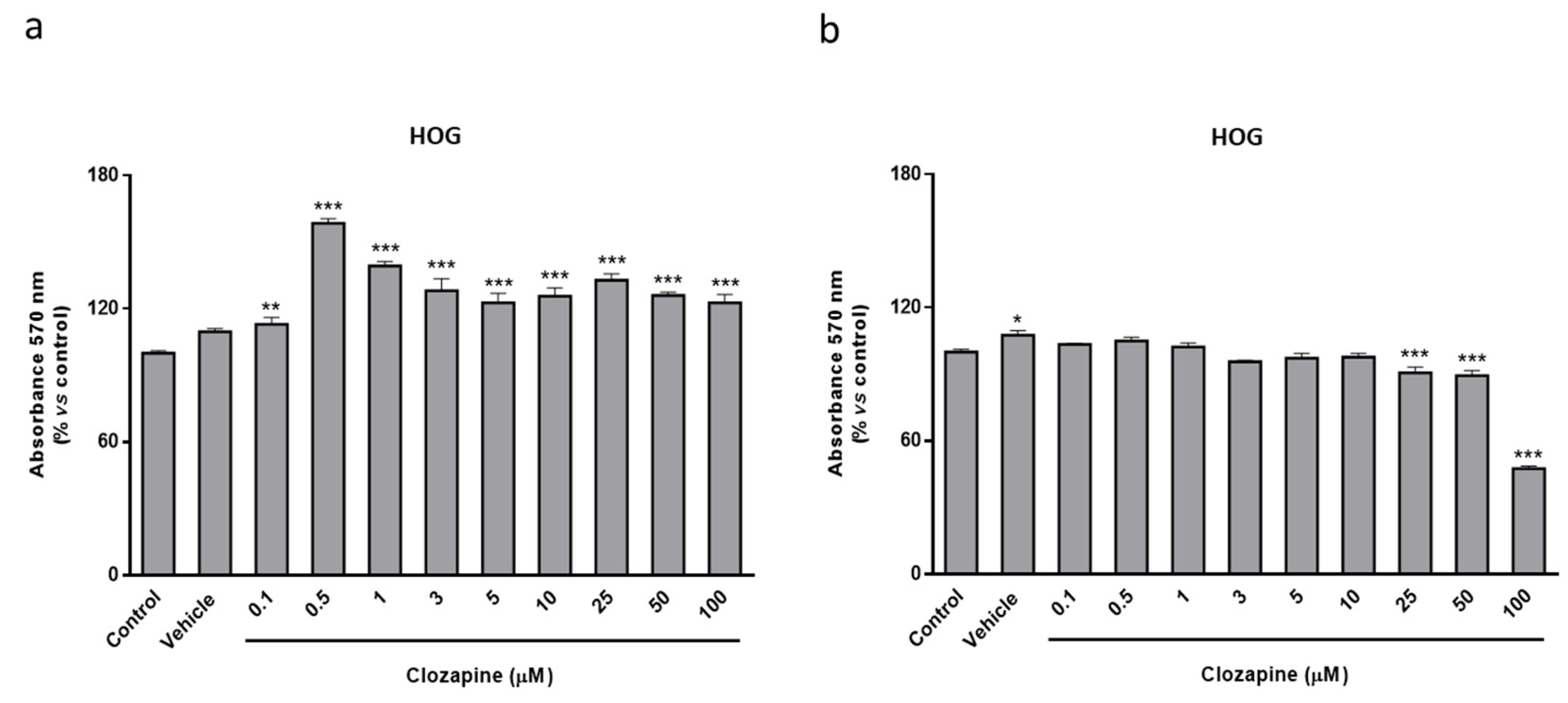
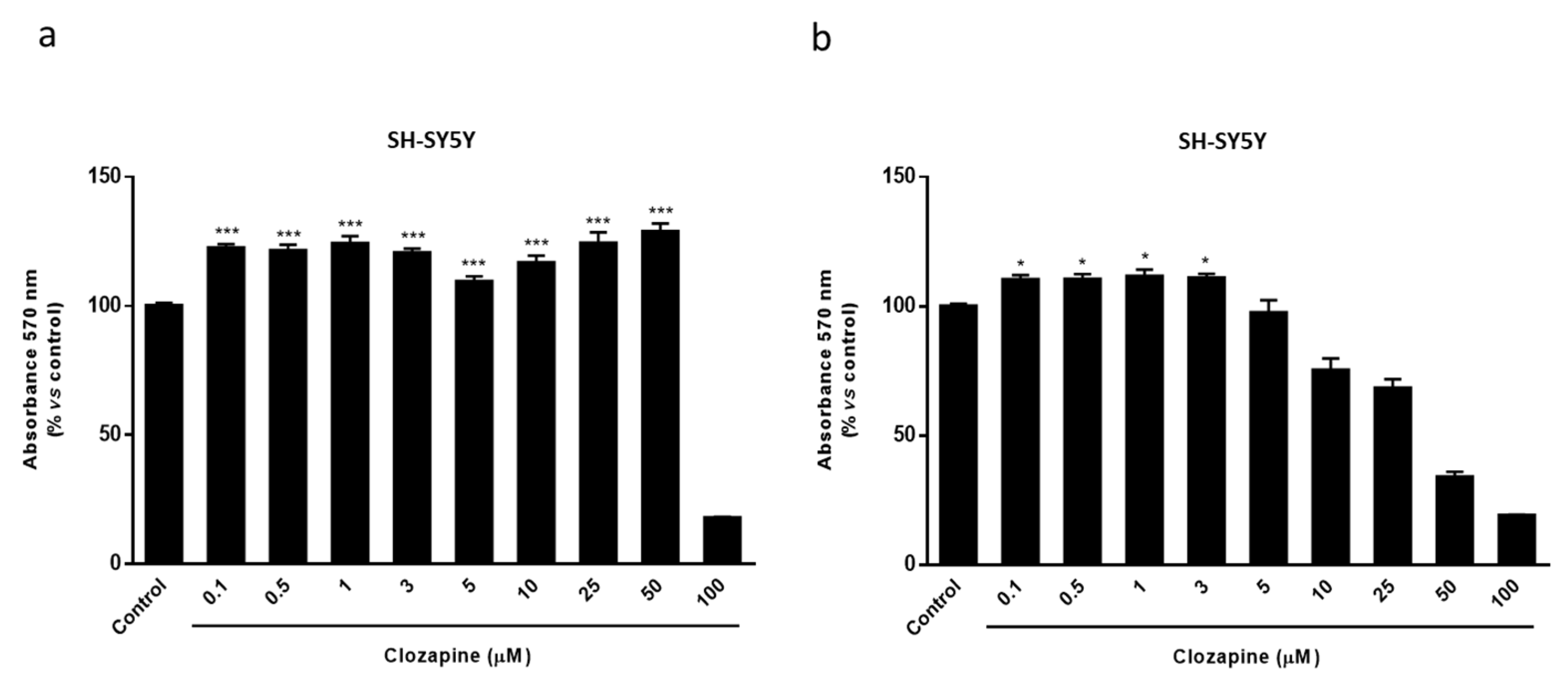
2.2. Clozapine Prevents Loss of Mitochondrial Functionality and Cell Viability in Oligodendroglial and Neuronal CPZ-Induced Models of MS
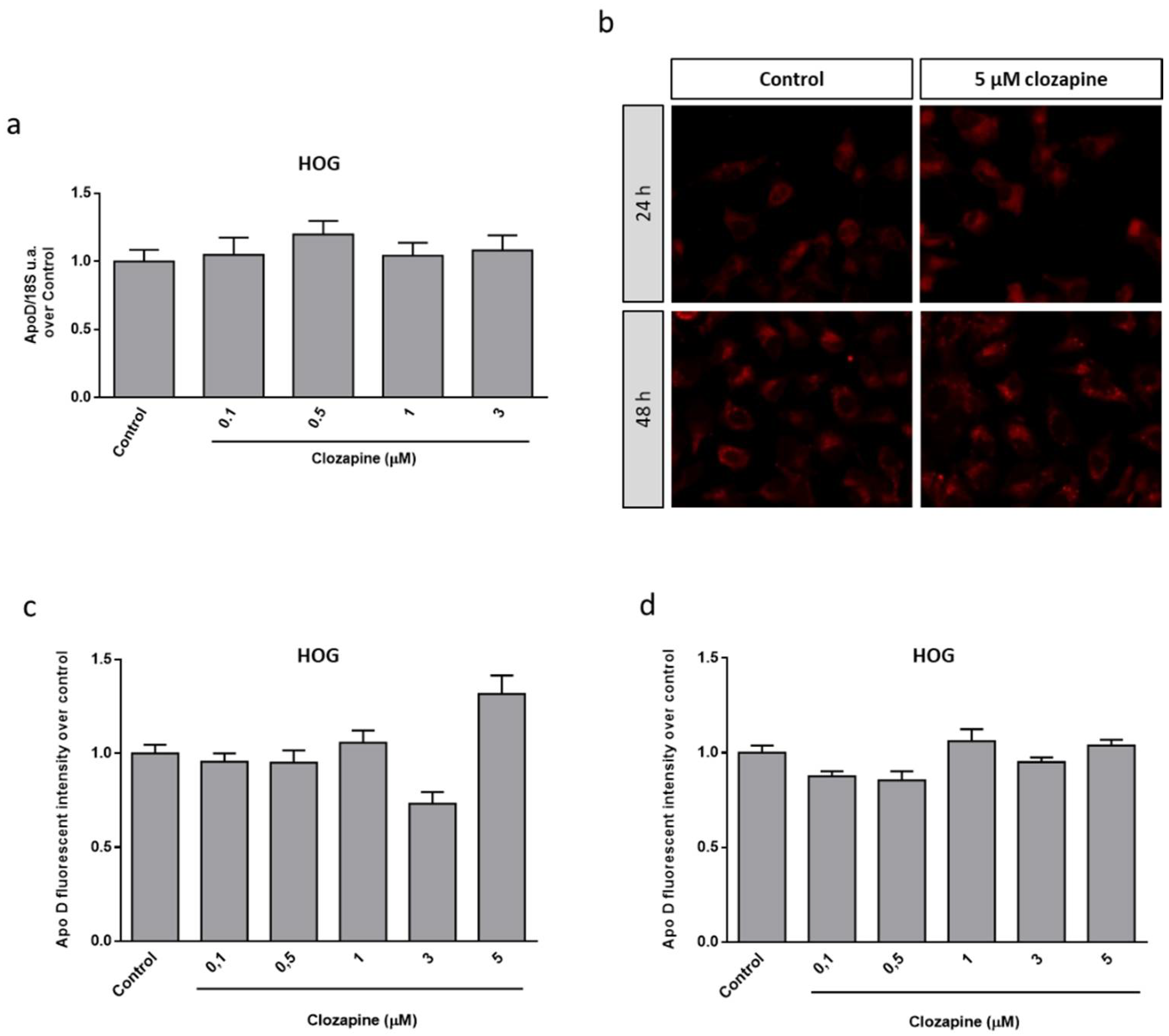
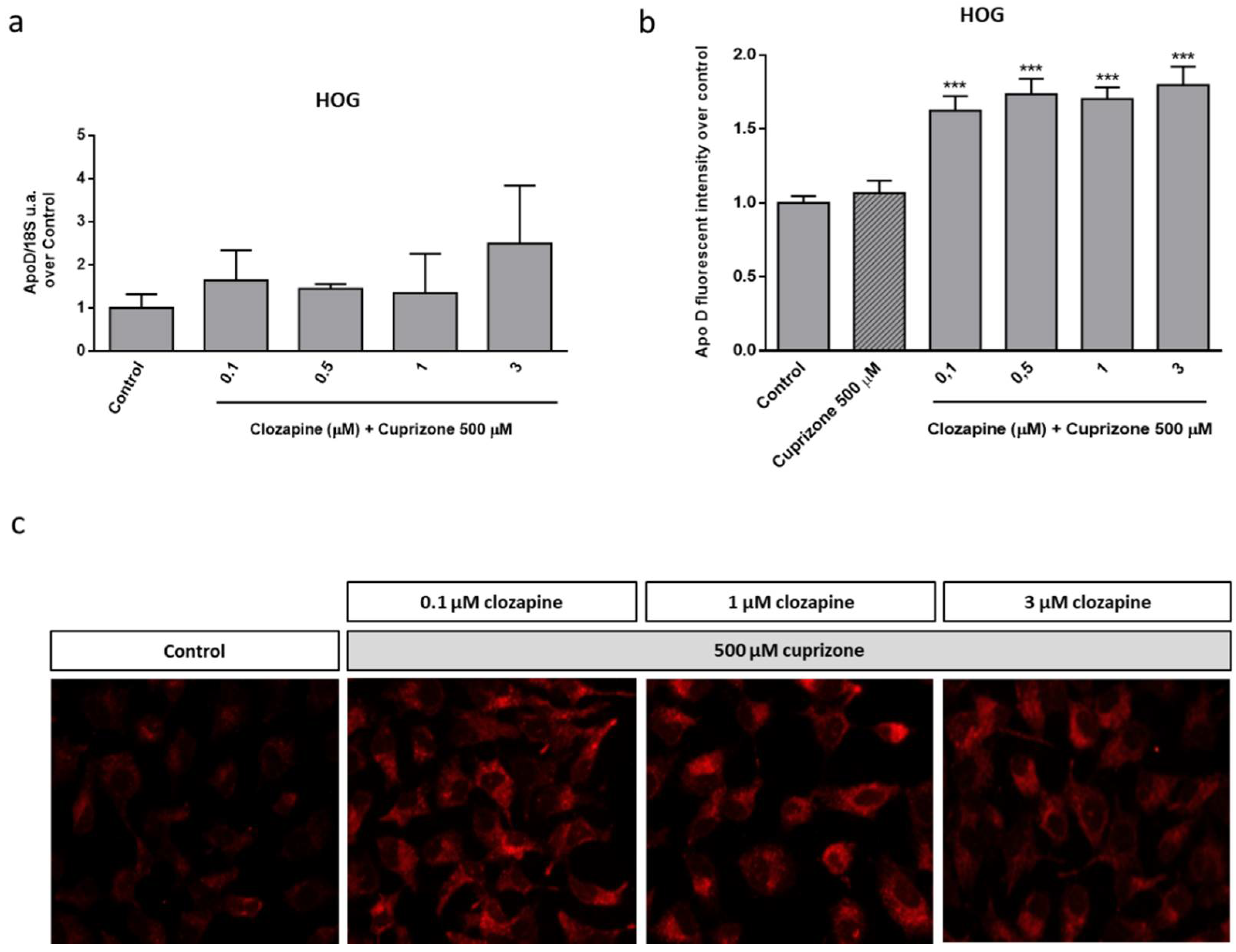
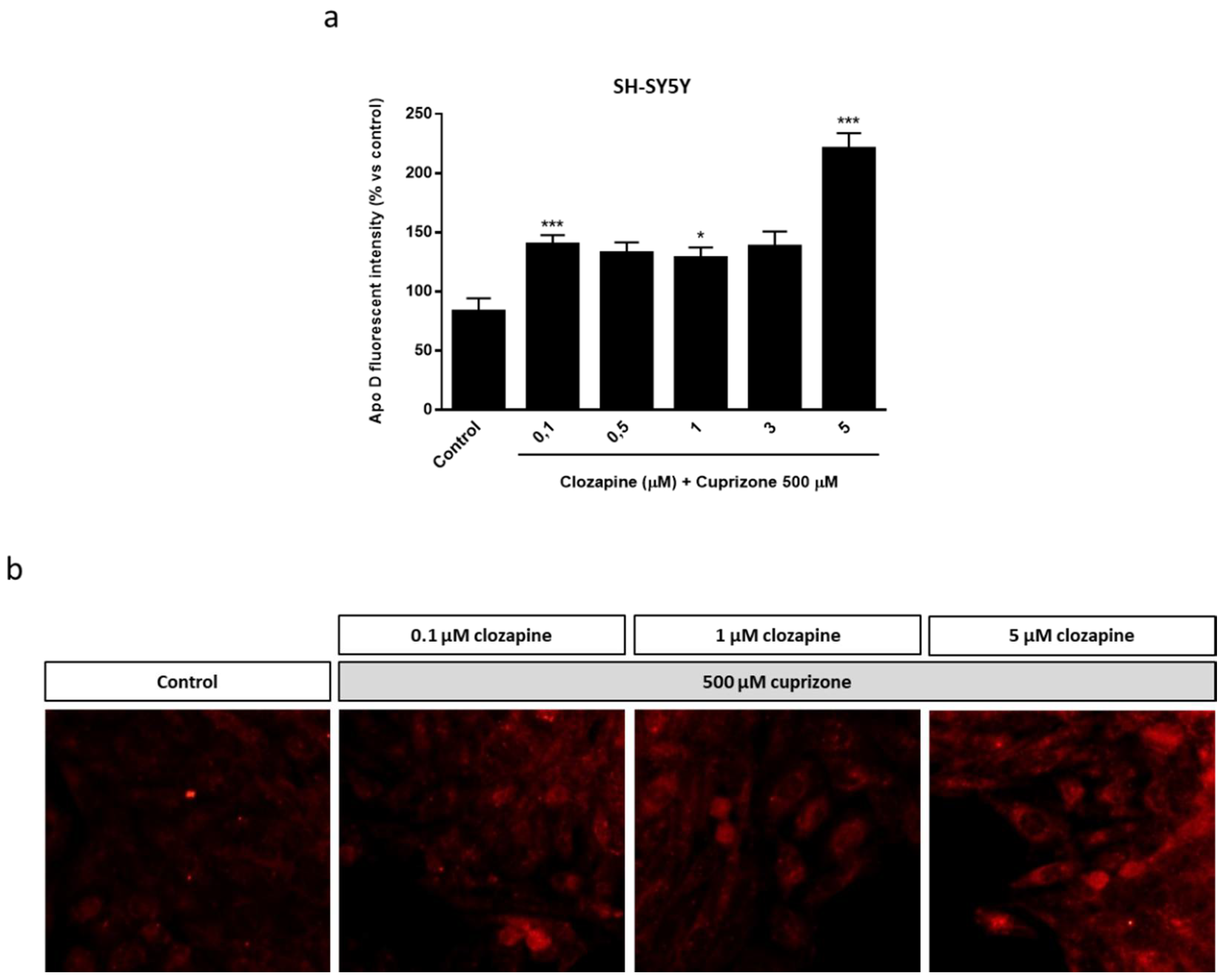
2.3. Neuroprotective Doses of Clozapine Increase Apo D Expression in the CPZ-Induced Cell Models of MS
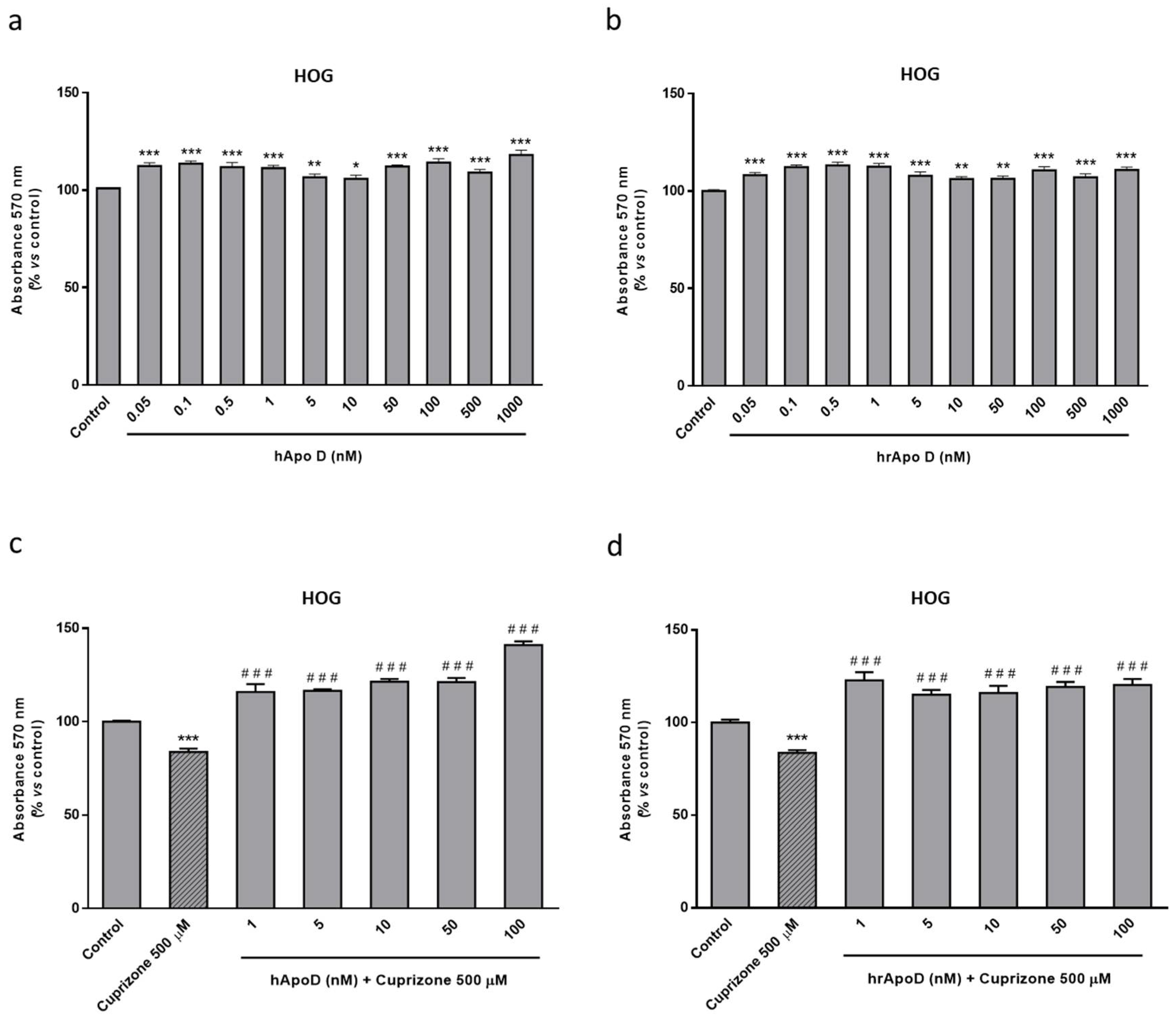
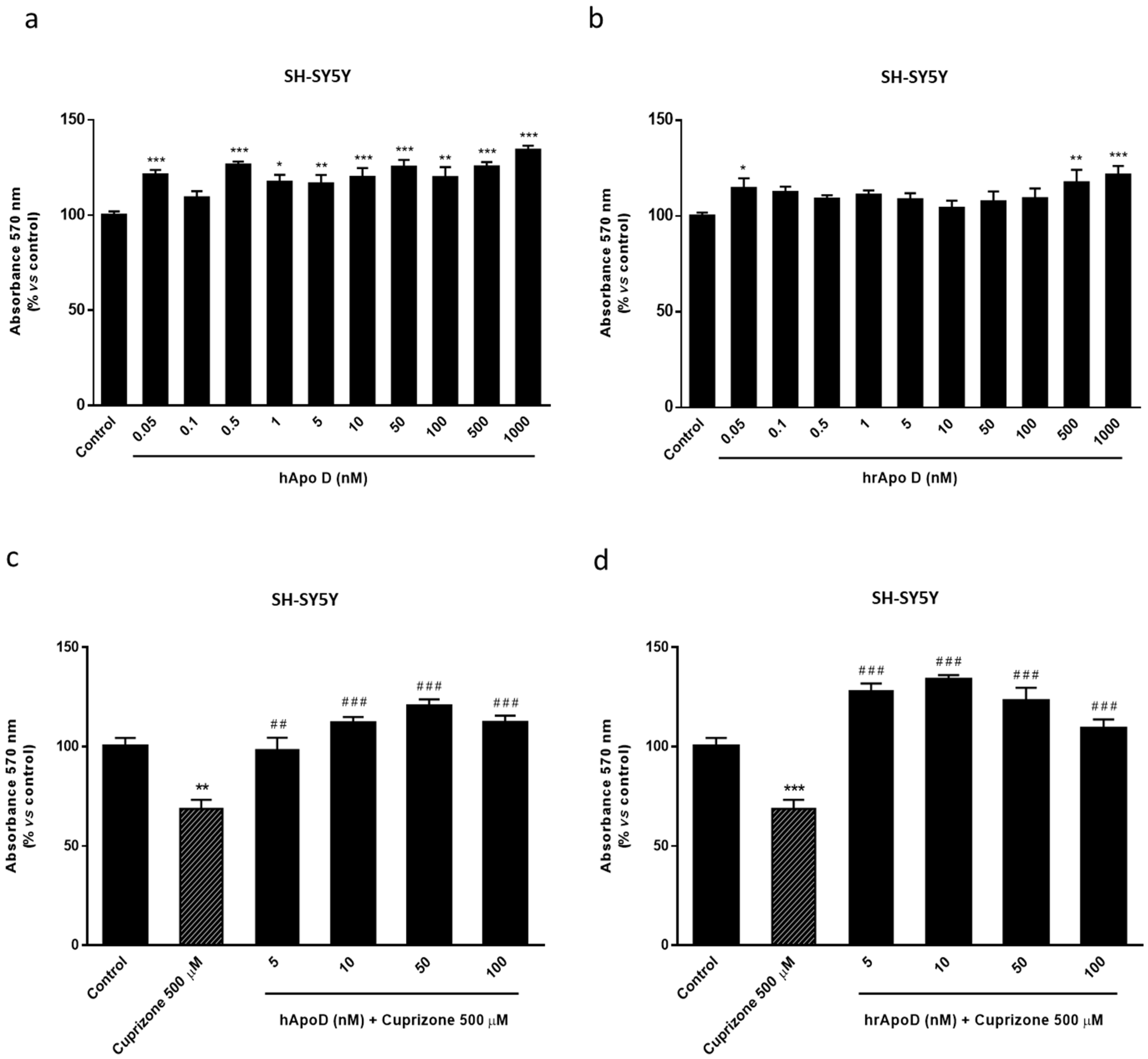
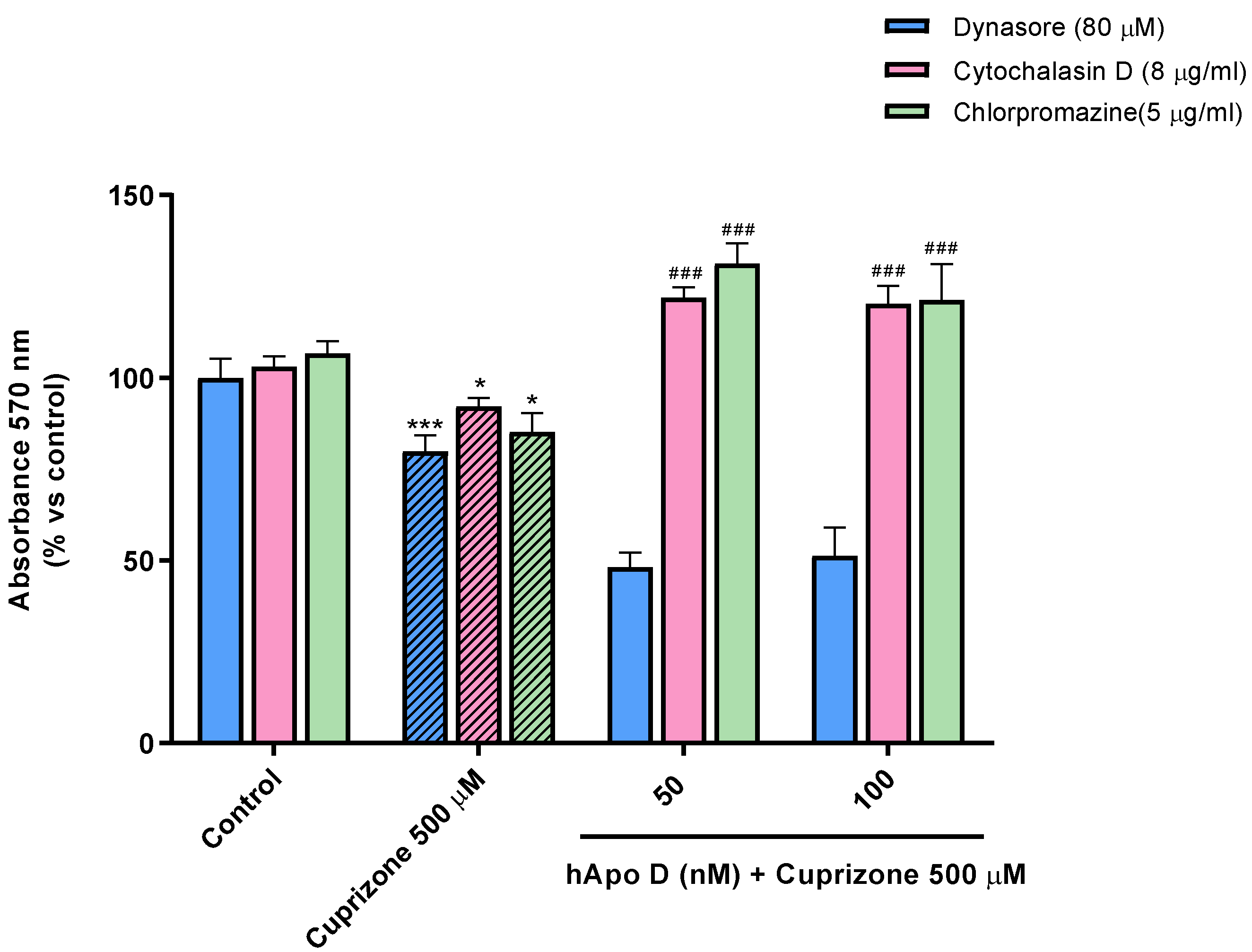
2.4. Neuroprotective Effect of the Exogenously Added hApo D in Oligodendroglial and Neuronal CPZ-Induced Models of MS
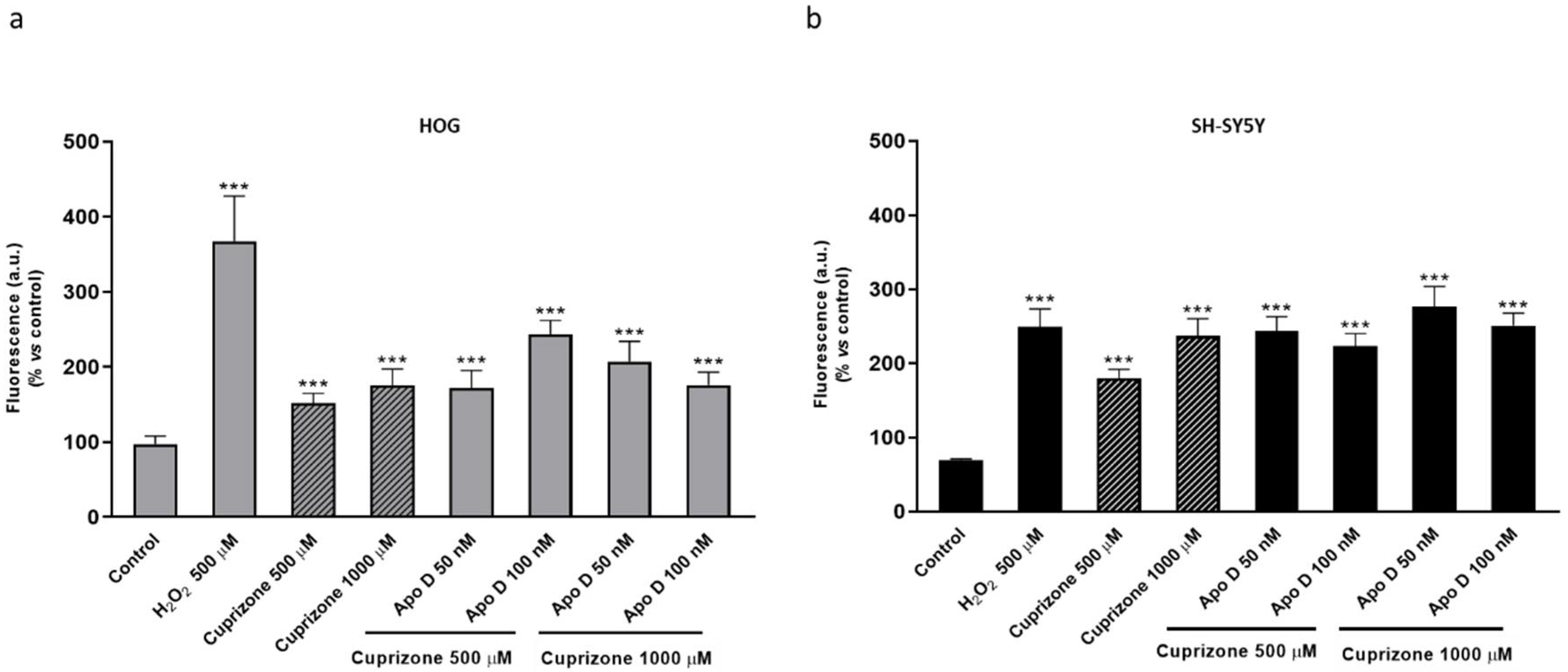
2.5. Neuroprotective Effect of hApo D is not Related to a Decrease in CPZ-Induced ROS Levels
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Human Apo D Purification
4.3. Cell Treatments
4.4. MTT Assay
4.5. Determination of ROS
4.6. Immunocytochemistry
4.7. RNA Purification
4.8. Quantitative Real-Time PCR
4.9. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Apo D | Apolipoprotein D |
| BBB | Blood–brain barrier |
| BCF | Breast cyst fluid |
| CIE | Clathrin-independent endocytosis |
| CLO | Clozapine |
| CME | Clathrin-mediated endocytosis |
| CNS | Central nervous system |
| CPZ | Cuprizone |
| CSF | Cerebrospinal fluid |
| DCF | 2′,7′-dichlorofluorescein |
| DMSO | Dimethyl sulfoxide |
| FBS | Fetal bovine serum |
| H2DCFDA | 2′7′-dichlorodihydrofluorescein diacetate |
| hApo D | Human Apo D |
| HDL | High-density plasma lipoproteins |
| hrApo D | Human recombinant Apo D |
| HUCA | Hospital Universitario Central de Asturias |
| MS | Multiple sclerosis |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| OLGs | Oligodendrocytes |
| PBS | Phosphate buffered saline |
| PD | Parkinson’s disease |
| PNS | Peripheral nervous system |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SDS | Sodium dodecyl sulfate |
| UV | Ultraviolet |
Appendix A



References
- Rassart, E.; Bedirian, A.; Do Carmo, S.; Guinard, O.; Sirois, J.; Terrisse, L.; Milne, R. Apolipoprotein D. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1482, 185–198. [Google Scholar] [CrossRef]
- Rassart, E.; Desmarais, F.; Najyb, O.; Bergeron, K.F.; Mounier, C. Apolipoprotein D. Gene 2020, 756, 144874. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; Gómez-Ruiz, M.; García, C.; Hernández, M.; Ramos, J.A. Modeling Neurodegenerative Disorders for Developing Cannabinoid-Based Neuroprotective Therapies. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 593, pp. 175–198. [Google Scholar]
- Nasreen, A.; Vogt, M.; Kim, H.J.; Eichinger, A.; Skerra, A. Solubility engineering and crystallization of human apolipoprotein D. Protein Sci. 2006, 15, 190–199. [Google Scholar] [CrossRef]
- Eichinger, A.; Nasreen, A.; Kim, H.J.; Skerra, A. Structural insight into the dual ligand specificity and mode of high density lipoprotein association of apolipoprotein D. J. Biol. Chem. 2007, 282, 31068–31075. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.A.; George, R.C.; Gregor Sutcliffe, J. Apolipoprotein D modulates arachidonic acid signaling in cultured cells: Implications for psychiatric disorders. Prostaglandins Leukot. Essent. Fat. Acids 2003, 69, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Knoch, B.; Wong, J.; Kim, W.S.; Else, P.L.; Oakley, A.J.; Garner, B. Selective reduction of hydroperoxyeicosatetraenoic acids to their hydroxy derivatives by apolipoprotein D: Implications for lipid antioxidant activity and Alzheimer’s disease. Biochem. J. 2012, 442, 713–721. [Google Scholar] [CrossRef]
- Waldner, A.; Dassati, S.; Redl, B.; Smania, N.; Gandolfi, M. Apolipoprotein D Concentration in Human Plasma during Aging and in Parkinson’s Disease: A Cross-Sectional Study. Parkinsons. Dis. 2018, 2018. [Google Scholar] [CrossRef]
- Bajo-Grañeras, R.; Ganfornina, M.D.; Martín-Tejedor, E.; Sanchez, D. Apolipoprotein D mediates autocrine protection of astrocytes and controls their reactivity level, contributing to the functional maintenance of paraquat-challenged dopaminergic systems. Glia 2011, 59, 1551–1566. [Google Scholar] [CrossRef]
- Do Carmo, S.; Levros, L.C.; Rassart, E. Modulation of apolipoprotein D expression and translocation under specific stress conditions. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 954–969. [Google Scholar] [CrossRef]
- Ganfornina, M.D.; Do Carmo, S.; Lora, J.M.; Torres-Schumann, S.; Vogel, M.; Allhorn, M.; González, C.; Bastiani, M.J.; Rassart, E.; Sanchez, D. Apolipoprotein D is involved in the mechanisms regulating protection from oxidative stress. Aging Cell 2008, 7, 506–515. [Google Scholar] [CrossRef]
- Sanchez, D.; Bajo-Grañeras, R.; Del Caño-Espinel, M.; Garcia-Centeno, R.; Garcia-Mateo, N.; Pascua-Maestro, R.; Ganfornina, M.D. Aging without apolipoprotein d: Molecular and cellular modifications in the hippocampus and cortex. Exp. Gerontol. 2015, 67, 19–47. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruberu, K.; Karl, T.; Garner, B. Cerebral apolipoprotein-D Is hypoglycosylated compared to peripheral tissues and is variably expressed in mouse and human brain regions. PLoS ONE 2016, 11, e0148238. [Google Scholar] [CrossRef] [PubMed]
- Scheen, L.; Brandt, L.; Bodén, R.; Tiihonen, J.; Andersen, M.; Kieler, H.; Reutfors, J. Predictors for initiation of pharmacological prophylaxis in patients with newly diagnosed bipolar disorder-A nationwide cohort study. J. Affect. Disord. 2015, 172, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Kielkopf, C.S.; Low, J.K.K.; Mok, Y.-F.; Bhatia, S.; Palasovski, T.; Oakley, A.J.; Whitten, A.E.; Garner, B.; Brown, S.H.J. Identification of a novel tetrameric structure for human apolipoprotein-D. J. Struct. Biol. 2018, 203, 205–218. [Google Scholar] [CrossRef]
- Navarro, A.; Tolivia, J.; Astudillo, A.; Del Valle, E. Pattern of apolipoprotein D immunoreactivity in human brain. Neurosci. Lett. 1998, 254, 17–20. [Google Scholar] [CrossRef]
- Navarro, A.; Del Valle, E.; Juárez, A.; Martinez, E.; Ordóñez, C.; Astudillo, A.; Tolivia, J. Apolipoprotein D synthesis progressively increases in frontal cortex during human lifespan. Age (Omaha) 2010, 32, 85–96. [Google Scholar] [CrossRef]
- Hu, C.Y.; Ong, W.Y.; Sundaram, R.K.; Chan, C.; Patel, S.C. Immunocytochemical localization of apolipoprotein D in oligodendrocyte precursor-like cells, perivascular cells, and pericytes in the human cerebral cortex. J. Neurocytol. 2001, 30, 209–218. [Google Scholar] [CrossRef]
- García-Mateo, N.; Pascua-Maestro, R.; Pérez-Castellanos, A.; Lillo, C.; Sanchez, D.; Ganfornina, M.D. Myelin extracellular leaflet compaction requires apolipoprotein D membrane management to optimize lysosomal-dependent recycling and glycocalyx removal. Glia 2018, 66, 670–687. [Google Scholar] [CrossRef]
- Bhatia, S.; Kim, W.S.; Shepherd, C.E.; Halliday, G.M. Apolipoprotein D Upregulation in Alzheimer’s Disease but Not Frontotemporal Dementia. J. Mol. Neurosci. 2018, 67, 125. [Google Scholar] [CrossRef]
- Muffat, J.; Walker, D.W. Apolipoprotein D: An overview of its role in aging and age-related diseases. Cell Cycle 2010, 9, 269–273. [Google Scholar] [CrossRef]
- Dassati, S.; Waldner, A.; Schweigreiter, R. Apolipoprotein D takes center stage in the stress response of the aging and degenerative brain. Neurobiol. Aging 2014, 35, 1632–1642. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019. [Google Scholar] [CrossRef]
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, J.M. Multiple sclerosis: Diagnosis, differential diagnosis, and clinical presentation. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 122, pp. 269–290. [Google Scholar]
- Correale, J.; Gaitán, M.I.; Ysrraelit, M.C.; Fiol, M.P. Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain 2017, 140, 527–546. [Google Scholar] [CrossRef] [PubMed]
- Haines, J.D.; Inglese, M.; Casaccia, P. Axonal damage in multiple sclerosis. Mt. Sinai J. Med. 2011, 78, 231–243. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, J.E. Practical Review of Neuropathology.: Gregory N, Fuller J, Goodman C. 2001, Lippincott Williams & Wilkins, $69.95. ISBN 0 7817 2778 2. J. Clin. Pathol. 2011, 55, 158. [Google Scholar] [CrossRef]
- Bö, L.; Geurts, J.J.G.; Mörk, S.J.; Van Der Valk, P. Grey matter pathology in multiple sclerosis. In Acta Neurologica Scandinavica; Wiley-Blackwell: Hoboken, NJ, USA, 2006; Volume 113, pp. 48–50. [Google Scholar]
- Zindler, E.; Zipp, F. Neuronal injury in chronic CNS inflammation. Best Pract. Res. Clin. Anaesthesiol. 2010, 24, 551–562. [Google Scholar] [CrossRef]
- McQualter, J.L.; Bernard, C.C.A. Multiple sclerosis: A battle between destruction and repair. J. Neurochem. 2007, 100, 295–306. [Google Scholar] [CrossRef]
- McGuire, C.; Beyaert, R.; van Loo, G. Death receptor signalling in central nervous system inflammation and demyelination. Trends Neurosci. 2011, 34, 619–628. [Google Scholar] [CrossRef]
- Disanto, G.; Morahan, J.M.; Barnett, M.H.; Giovannoni, G.; Ramagopalan, S.V. The evidence for a role of B cells in multiple sclerosis. Neurology 2012, 78, 823–832. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Stys, P.K.; Zamponi, G.W.; Van Minnen, J.; Geurts, J.J.G. Will the real multiple sclerosis please stand up? Nat. Rev. Neurosci. 2012, 13, 507–514. [Google Scholar] [CrossRef]
- Caprariello, A.V.; Rogers, J.A.; Morgan, M.L.; Hoghooghi, V.; Plemel, J.R.; Koebel, A.; Tsutsui, S.; Dunn, J.F.; Kotra, L.P.; Ousman, S.S.; et al. Biochemically altered myelin triggers autoimmune demyelination. Proc. Natl. Acad. Sci. USA 2018, 115, 5528–5533. [Google Scholar] [CrossRef] [PubMed]
- Titus, H.E.; Chen, Y.; Podojil, J.R.; Robinson, A.P.; Balabanov, R.; Popko, B.; Miller, S.D. Pre-clinical and Clinical Implications of “Inside-Out” vs. “Outside-In” Paradigms in Multiple Sclerosis Etiopathogenesis. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Ontaneda, D.; Hyland, M.; Cohen, J.A. Multiple Sclerosis: New Insights in Pathogenesis and Novel Therapeutics. Annu. Rev. Med. 2012, 63, 389–404. [Google Scholar] [CrossRef]
- Leibowitz, S.M.; Yan, J. NF-κB pathways in the pathogenesis of multiple sclerosis and the therapeutic implications. Front. Mol. Neurosci. 2016, 9, 84. [Google Scholar] [CrossRef]
- Vandenbark, A.A.; Culbertson, N.E.; Bartholomew, R.M.; Huan, J.; Agotsch, M.; LaTocha, D.; Yadav, V.; Mass, M.; Whitham, R.; Lovera, J.; et al. Therapeutic vaccination with a trivalent T-cell receptor (TCR) peptide vaccine restores deficient FoxP3 expression and TCR recognition in subjects with multiple sclerosis. Immunology 2008, 123, 66–78. [Google Scholar] [CrossRef]
- Goldberg, P.; Fleming, M.C.; Picard, E.H. Multiple sclerosis: Decreased relapse rate through dietary supplementation with calcium, magnesium and vitamin D. Med. Hypotheses 1986, 21, 193–200. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Denic, A.; Johnson, A.J.; Bieber, A.J.; Warrington, A.E.; Rodriguez, M.; Pirko, I. The relevance of animal models in multiple sclerosis research. Pathophysiology 2011, 18, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Vega-Riquer, J.M.; Mendez-Victoriano, G.; Morales-Luckie, R.A.; Gonzalez-Perez, O. Five Decades of Cuprizone, an Updated Model to Replicate Demyelinating Diseases. Curr. Neuropharmacol. 2019, 17, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Nyamoya, S.; Schweiger, F.; Kipp, M.; Hochstrasser, T. Cuprizone as a model of myelin and axonal damage. Drug Discov. Today Dis. Model. 2017, 25–26, 63–68. [Google Scholar] [CrossRef]
- Procaccini, C.; De Rosa, V.; Pucino, V.; Formisano, L.; Matarese, G. Animal models of Multiple Sclerosis. Eur. J. Pharmacol. 2015, 759, 182–191. [Google Scholar] [CrossRef]
- Stoop, M.P.; Singh, V.; Dekker, L.J.; Titulaer, M.K.; Stingl, C.; Burgers, P.C.; Sillevis Smitt, P.A.E.; Hintzen, R.Q.; Luider, T.M. Proteomics comparison of cerebrospinal fluid of relapsing remitting and primary progressive multiple sclerosis. PLoS ONE 2010, 5, e12442. [Google Scholar] [CrossRef]
- Reindl, M.; Knipping, G.; Wicher, I.; Dilitz, E.; Egg, R.; Deisenhammer, F.; Berger, T. Increased intrathecal production of apolipoprotein D in multiple sclerosis. J. Neuroimmunol. 2001, 119, 327–332. [Google Scholar] [CrossRef]
- Navarro, A.; Rioseras, B.; Del Valle, E.; Martínez-Pinilla, E.; Astudillo, A.; Tolivia, J. Expression Pattern of Myelin-Related Apolipoprotein D in Human Multiple Sclerosis Lesions. Front. Aging Neurosci. 2018, 10, 254. [Google Scholar] [CrossRef]
- Pascua-Maestro, R.; González, E.; Lillo, C.; Ganfornina, M.D.; Falcón-Pérez, J.M.; Sanchez, D. Extracellular vesicles secreted by astroglial cells transport apolipoprotein D to neurons and mediate neuronal survival upon oxidative stress. Front. Cell. Neurosci. 2019, 12, 526. [Google Scholar] [CrossRef]
- Thomas, E.A.; Danielson, P.E.; Austin Nelson, P.; Pribyl, T.M.; Hilbush, B.S.; Hasel, K.W.; Gregor Sutcliffe, J. Clozapine increases apolipoprotein D expression in rodent brain: Towards a mechanism for neuroleptic pharmacotherapy. J. Neurochem. 2001, 76, 789–796. [Google Scholar] [CrossRef]
- Mahadik, S.P.; Khan, M.M.; Evans, D.R.; Parikh, V.V. Elevated plasma level of apolipoprotein D in schizophrenia and its treatment and outcome. Schizophr. Res. 2002, 58, 55–62. [Google Scholar] [CrossRef]
- Do Carmo, S.; Séguin, D.; Milne, R.; Rassart, E. Modulation of apolipoprotein D and apolipoprotein E mRNA expression by growth arrest and identification of key elements in the promoter. J. Biol. Chem. 2002, 277, 5514–5523. [Google Scholar] [CrossRef] [PubMed]
- Sarjeant, J.M.; Lawrie, A.; Kinnear, C.; Yablonsky, S.; Leung, W.; Massaeli, H.; Prichett, W.; Veinot, J.P.; Rassart, E.; Rabinovitch, M. Apolipoprotein D Inhibits Platelet-Derived Growth Factor-BB–Induced Vascular Smooth Muscle Cell Proliferated by Preventing Translocation of Phosphorylated Extracellular Signal Regulated Kinase 1/2 to the Nucleus. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2172–2177. [Google Scholar] [CrossRef] [PubMed]
- Martínez, E.; Navarro, A.; Ordóez, C.; Del Valle, E.; Tolivia, J. Amyloid-β25-35 induces apolipoprotein D synthesis and growth arrest in HT22 hippocampal cells. J. Alzheimer’s Dis. 2012, 30, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Martínez, E.; Navarro, A.; Ordóñez, C.; Del Valle, E.; Tolivia, J. Oxidative stress induces apolipoprotein d overexpression in hippocampus during aging and alzheimer’s disease. J. Alzheimer’s Dis. 2013, 36, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, L.A.; Calatayud, C.A.; Bertone Uña, A.L.; Millet, V.; Pasquini, J.M.; Soto, E.F. The neurotoxic effect of cuprizone on oligodendrocytes depends on the presence of pro-inflammatory cytokines secreted by microglia. Neurochem. Res. 2007, 32, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Benetti, F.; Ventura, M.; Salmini, B.; Ceola, S.; Carbonera, D.; Mammi, S.; Zitolo, A.; D’Angelo, P.; Urso, E.; Maffia, M.; et al. Cuprizone neurotoxicity, copper deficiency and neurodegeneration. Neurotoxicology 2010, 31, 509–517. [Google Scholar] [CrossRef]
- Yao, J.K.; Thomas, E.A.; Reddy, R.D.; Keshavan, M.S. Association of plasma apolipoproteins D with RBC membrane arachidonic acid levels in schizophrenia. Schizophr. Res. 2005, 72, 259–266. [Google Scholar] [CrossRef]
- Thomas, E.A.; Yao, J.K. Clozapine specifically alters the arachidonic acid pathway in mice lacking apolipoprotein D. Schizophr. Res. 2007, 89, 147–153. [Google Scholar] [CrossRef]
- Dutta, D.; Donaldson, J.G. Search for inhibitors of endocytosis. Cell. Logist. 2012, 2, 203–208. [Google Scholar] [CrossRef]
- Patel, S.C.; Asotra, K.; Patel, Y.C.; McConathy, W.J.; Patel, R.C.; Suresh, S. Astrocytes synthesize and secrete the lipophilic ligand carrier apolipoprotein D. Neuroreport 1995, 6, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.Y.; Lau, C.P.; Leong, S.K.; Kumar, U.; Suresh, S.; Patel, S.C. Apolipoprotein d gene expression in the rat brain and light and electron microscopic immunocytochemistry of apolipoprotein D expression in the cerebellum of neonatal, immature and adult rats. Neuroscience 1999, 90, 913–922. [Google Scholar] [CrossRef]
- Pascua-Maestro, R.; Diez-Hermano, S.; Lillo, C.; Ganfornina, M.D.; Sanchez, D. Protecting cells by protecting their vulnerable lysosomes: Identification of a new mechanism for preserving lysosomal functional integrity upon oxidative stress. PLoS Genet. 2017, 13, e1006603. [Google Scholar] [CrossRef] [PubMed]
- Najyb, O.; Brissette, L.; Rassart, E. Apolipoprotein D internalization is a basigin-dependent mechanism. J. Biol. Chem. 2015, 290, 16077–16087. [Google Scholar] [CrossRef] [PubMed]
- Najyb, O.; Do Carmo, S.; Alikashani, A.; Rassart, E. Apolipoprotein D Overexpression Protects Against Kainate-Induced Neurotoxicity in Mice. Mol. Neurobiol. 2017, 54, 3948–3963. [Google Scholar] [CrossRef]
- He, X.; Jittiwat, J.; Kim, J.H.; Jenner, A.M.; Farooqui, A.A.; Patel, S.C.; Ong, W.Y. Apolipoprotein D modulates F2-isoprostane and 7-ketocholesterol formation and has a neuroprotective effect on organotypic hippocampal cultures after kainate-induced excitotoxic injury. Neurosci. Lett. 2009, 455, 183–186. [Google Scholar] [CrossRef]
- Zhang, Y.; Cong, Y.; Wang, S.; Zhang, S. Antioxidant activities of recombinant amphioxus (Branchiostoma belcheri) apolipoprotein D. Mol. Biol. Rep. 2011, 38, 1847–1851. [Google Scholar] [CrossRef]
- Oakley, A.J.; Bhatia, S.; Ecroyd, H.; Garner, B. Molecular dynamics analysis of apolipoprotein-D - lipid hydroperoxide interactions: Mechanism for selective oxidation of Met-93. PLoS ONE 2012, 7, e34057. [Google Scholar] [CrossRef]
- Åkerstrom, B.; Flower, D.R.; Salier, J.P. Lipocalins: Unity in diversity. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1482, 1–8. [Google Scholar] [CrossRef]
- Post, G.R.; Dawson, G. Characterization of a cell line derived from a human oligodendroglioma. Mol. Chem. Neuropathol. 1992, 16, 303–317. [Google Scholar] [CrossRef]
- Bello-Morales, R.; Crespillo, A.J.; García, B.; Dorado, L.Á.; Martín, B.; Tabarés, E.; Krummenacher, C.; De Castro, F.; López-Guerrero, J.A. The effect of cellular differentiation on HSV-1 infection of oligodendrocytic cells. PLoS ONE 2014, 9, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Cammer, W. The neurotoxicant, cuprizone, retards the differentiation of oligodendrocytes in vitro. J. Neurol. Sci. 1999, 168, 116–120. [Google Scholar] [CrossRef]
- Bénardais, K.; Kotsiari, A.; Škuljec, J.; Koutsoudaki, P.N.; Gudi, V.; Singh, V.; Vulinović, F.; Skripuletz, T.; Stangel, M. Cuprizone [bis(cyclohexylidenehydrazide)] is selectively toxic for mature oligodendrocytes. Neurotox. Res. 2013, 24, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Díez-Itza, I.; Vizoso, F.; Merino, A.M.; Sánchez, L.M.; Tolivia, J.; Fernández, J.; Ruibal, A.; López-Otín, C. Expression and prognostic significance of apolipoprotein D in breast cancer. Am. J. Pathol. 1994, 144, 310–320. [Google Scholar]
- Navarro, A.; Del Valle, E.; Tolivia, J. Differential expression of apolipoprotein D in human astroglial and oligodendroglial cells. J. Histochem. Cytochem. 2004, 52, 1031–1036. [Google Scholar] [CrossRef]
- Tolivia, J.; Navarro, A.; Del Valle, E.; Perez, C.; Ordoñez, C.; Martínez, E. Application of photoshop and scion image analysis to quantification of signals in histochemistry, immunocytochemistry and hybridocytochemistry. Anal. Quant. Cytol. Histol. 2006, 28, 43–53. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide | Sequence | |
|---|---|---|
| R2-ApoD-F | -------- | TGCATCCAGGCCAACTACTC |
| R2-ApoD-Rev | -------- | GGGTGGCTTCACCTTCGATT |
| 18S-Fw | -------- | ATGCTCTTAGCTGAGTGTCCCG |
| 18S-Rev | -------- | ATTCCTAGCTGCGGTATCCAGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Pinilla, E.; Rubio-Sardón, N.; Peláez, R.; García-Álvarez, E.; del Valle, E.; Tolivia, J.; Larráyoz, I.M.; Navarro, A. Neuroprotective Effect of Apolipoprotein D in Cuprizone-Induced Cell Line Models: A Potential Therapeutic Approach for Multiple Sclerosis and Demyelinating Diseases. Int. J. Mol. Sci. 2021, 22, 1260. https://doi.org/10.3390/ijms22031260
Martínez-Pinilla E, Rubio-Sardón N, Peláez R, García-Álvarez E, del Valle E, Tolivia J, Larráyoz IM, Navarro A. Neuroprotective Effect of Apolipoprotein D in Cuprizone-Induced Cell Line Models: A Potential Therapeutic Approach for Multiple Sclerosis and Demyelinating Diseases. International Journal of Molecular Sciences. 2021; 22(3):1260. https://doi.org/10.3390/ijms22031260
Chicago/Turabian StyleMartínez-Pinilla, Eva, Núria Rubio-Sardón, Rafael Peláez, Enrique García-Álvarez, Eva del Valle, Jorge Tolivia, Ignacio M. Larráyoz, and Ana Navarro. 2021. "Neuroprotective Effect of Apolipoprotein D in Cuprizone-Induced Cell Line Models: A Potential Therapeutic Approach for Multiple Sclerosis and Demyelinating Diseases" International Journal of Molecular Sciences 22, no. 3: 1260. https://doi.org/10.3390/ijms22031260
APA StyleMartínez-Pinilla, E., Rubio-Sardón, N., Peláez, R., García-Álvarez, E., del Valle, E., Tolivia, J., Larráyoz, I. M., & Navarro, A. (2021). Neuroprotective Effect of Apolipoprotein D in Cuprizone-Induced Cell Line Models: A Potential Therapeutic Approach for Multiple Sclerosis and Demyelinating Diseases. International Journal of Molecular Sciences, 22(3), 1260. https://doi.org/10.3390/ijms22031260