Abstract
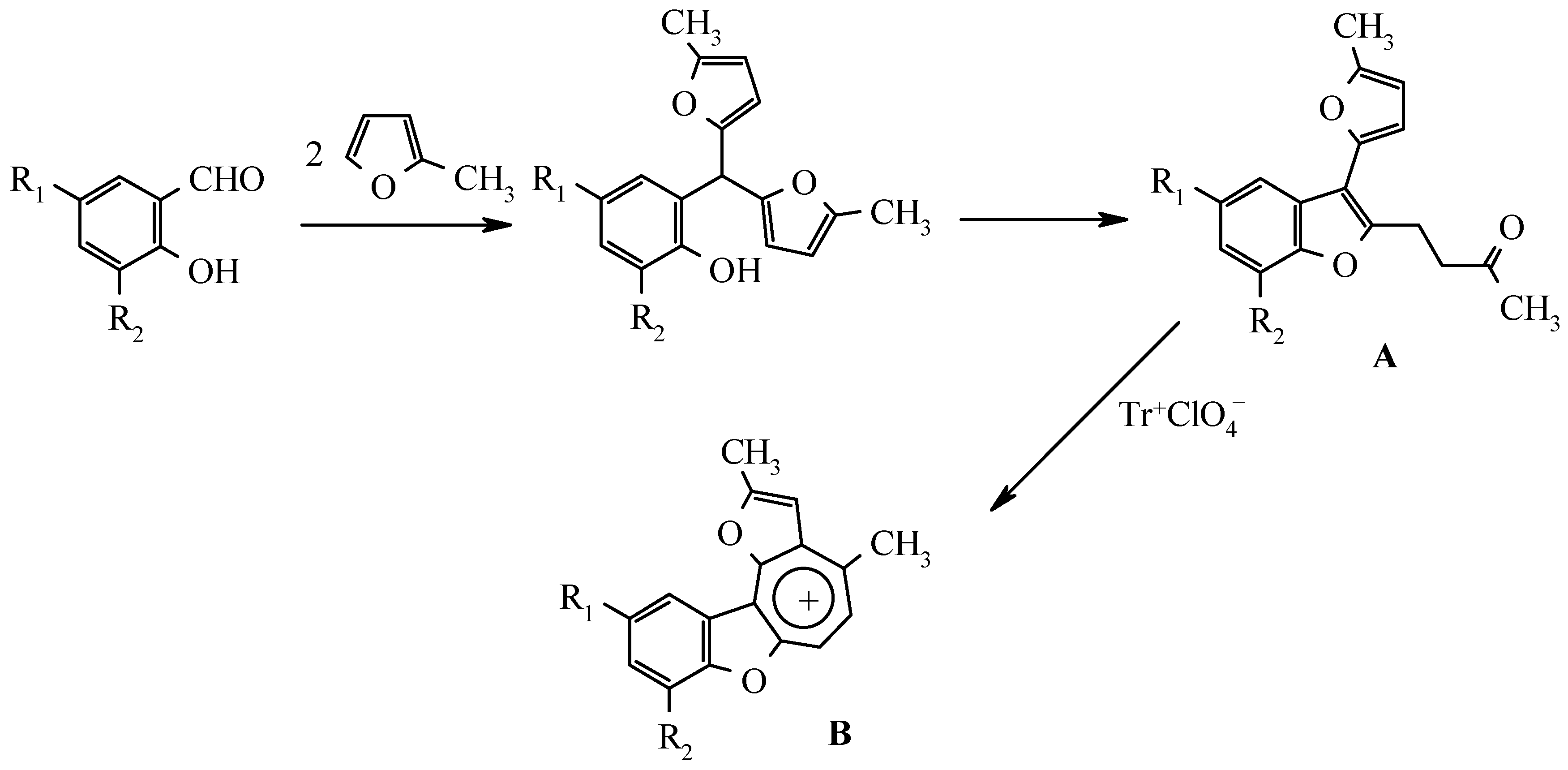
2-Hydroxyaryl(5-methylfur-2-yl)alkanes synthesized by alkylation of 2-methylfuran with various 2-hydroxybenzylic alcohols, were rearranged into corresponding 3-R-benzo[b]furan derivatives by treatment with ethanolic HCl solution. These compounds can not be transformed into dibenzoxazulenium salts.
Introduction
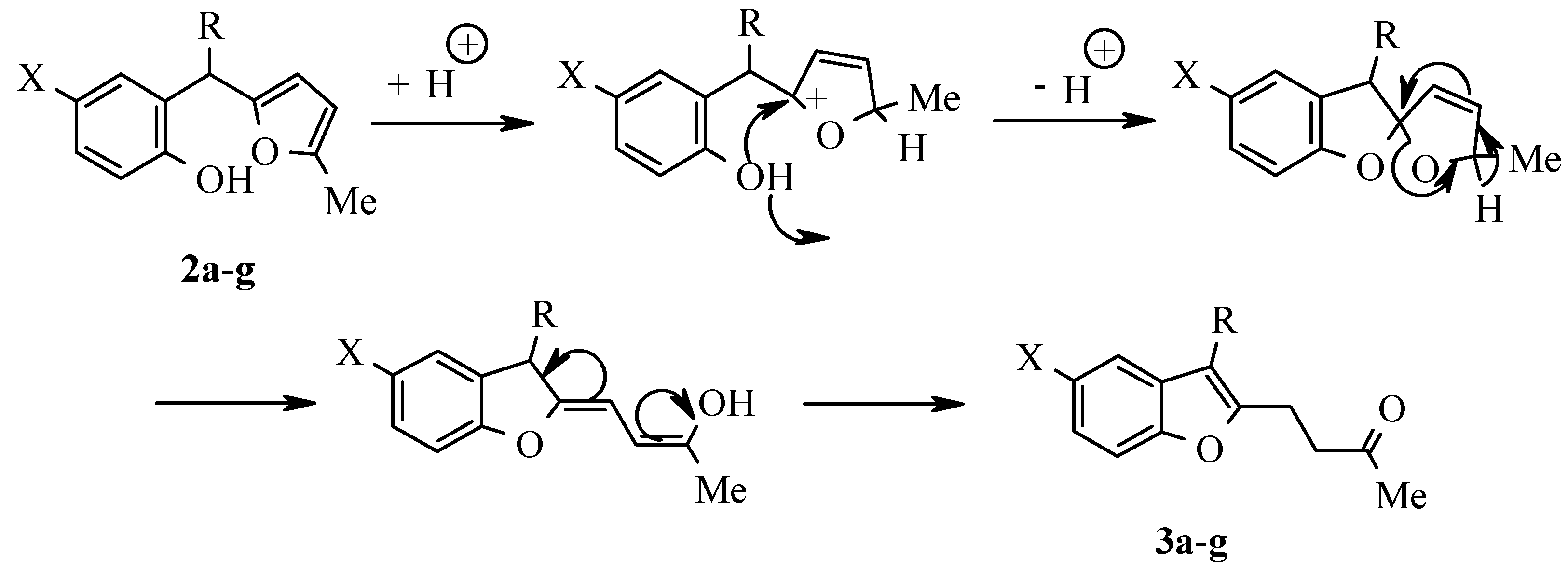
As it was shown by the authors in previous publications, 2-hydroxyarylbis(5-methylfur-2-yl)methanes, which are readily available by sylvane and salicylaldehydes condensation, catalyzed by trimethylchlorosilane or boric acid [2], undergo a rearrangement to 3-(5-methylfur-2-yl)-2-(3-oxobutyl)benzo[b]furan derivatives A by treatment with ethanolic hydrogen chloride solution (Scheme 1) [3]. The latter compounds can serve as precursors for benzo[b]furo[2,3-h]-1-oxazulenium salts B both by trityl perchlorate oxidation [4] and by disproportionation in the presence of perchloric acid [5]:
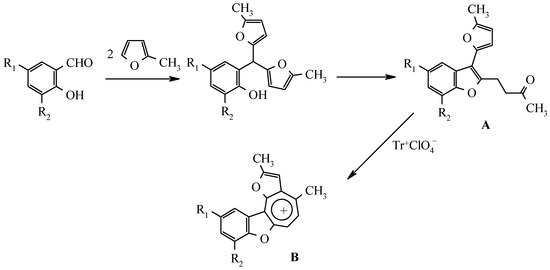
Scheme 1.
Previous results were summarized in review [6].
In the present work we attempt to extend the scope of such reactions.
Results and Discussion
Starting from substituted 2-hydroxybenzylic alcohols 1a-g corresponding 2-hydroxyaryl-R-(5-methylfur-2-yl)alkanes were obtained:
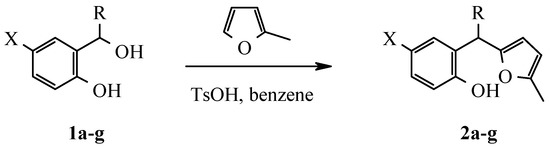
Scheme 2.
Scheme 2.
When the condensation step was conducted in the presence of a highly acid, ion-exchange resin Amberlyst-15 or boric acid in boiling benzene the reaction did not go to completion. On the other hand the corresponding methanes 2a-g were obtained quantitatively (Table 1) by refluxing the rea-gents in benzene with a catalytic amount of p-toluenesulfonic acid and with a Dean-Stark trap. The structure was proved by 1H NMR spectra (Table 2). IR-spectra of these compounds contain the char-acteristic bands of absorption of valent vibrations of the OH group.

Table 1.
Characterization of compounds 2a-g.
Table 2.
1H-NMR spectra of compounds 2a-g.
The prepared methanes were transformed smoothly into corresponding benzofuran derivatives 3a-g (Table 3) by treatment with ethanolic HCl solution (Scheme 3). The main feature of 1H NMR spectra (Table 4) of these compounds is the absence of the peaks of furan protons and the presence of two sig-nals from methylene protons α-CH2 and β-CH2. IR spectra of these compounds have an intensive band of valent vibrations of the CO group in the field of 1700 cm-1.
Table 3.
Characterization of compounds 3a-g.
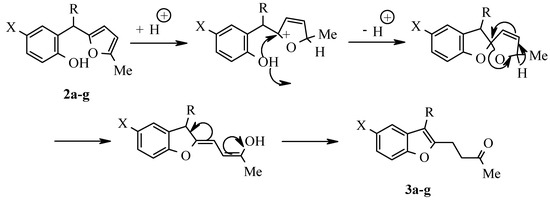
Scheme 3.
Table 4.
1H-NMR spectra of compounds 3a-g.
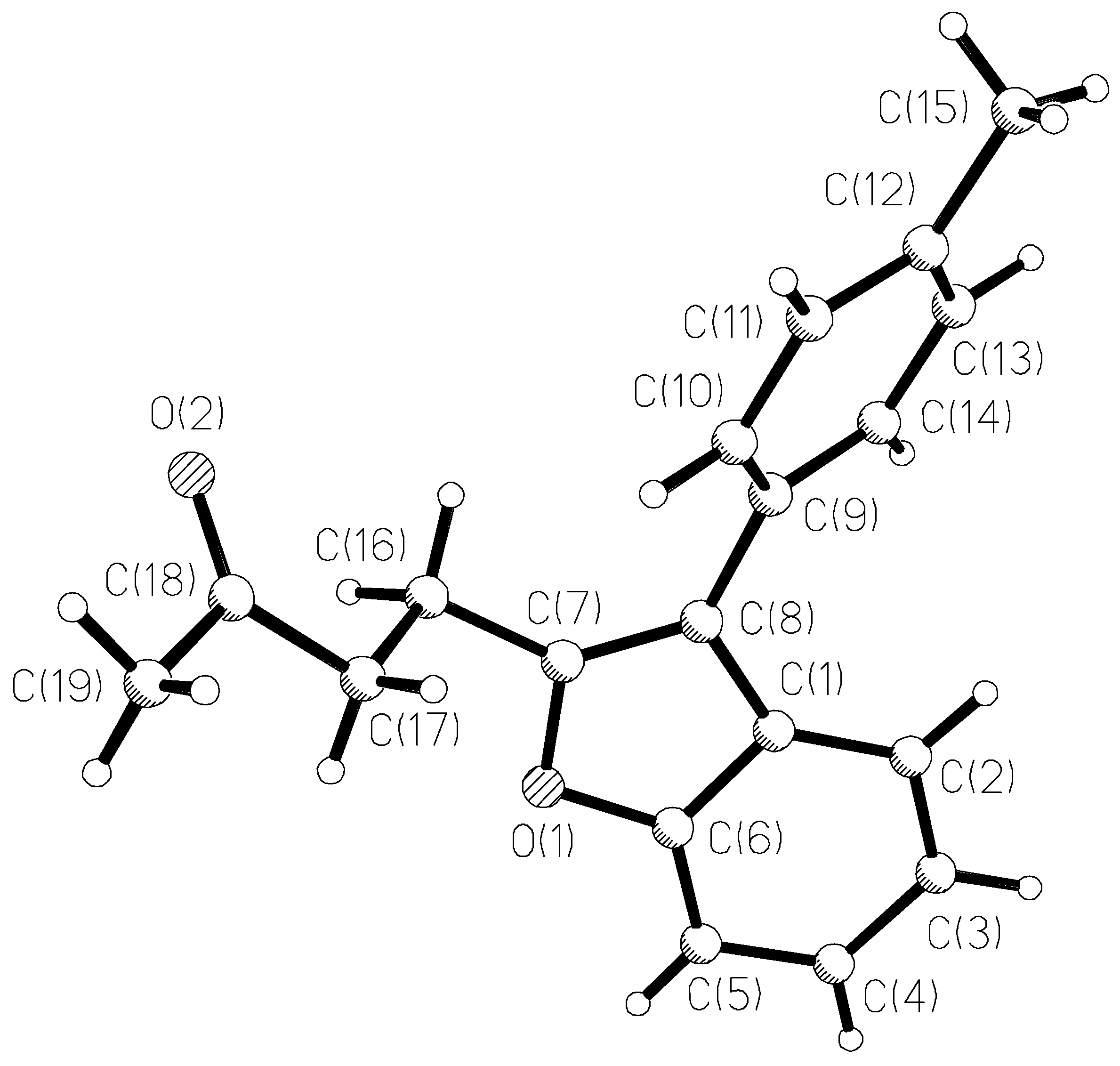
The results of X-ray study of benzofuran 3e monocrystal are given in Figure 1 and in Table 5, Table 6 and Table 7. No specific divergences from the bond lengths and valent angles in comparison with 3-furylbenzofuran A (data of X-ray study are given in [7]) are found except in the size of the dihedral angle between the benzofuran and the aromatic ring planes. In a molecule that contains a smaller furan cycle, this angle is 18°, and in a molecule 3e with a larger phenyl cycle, this angle is 40°.
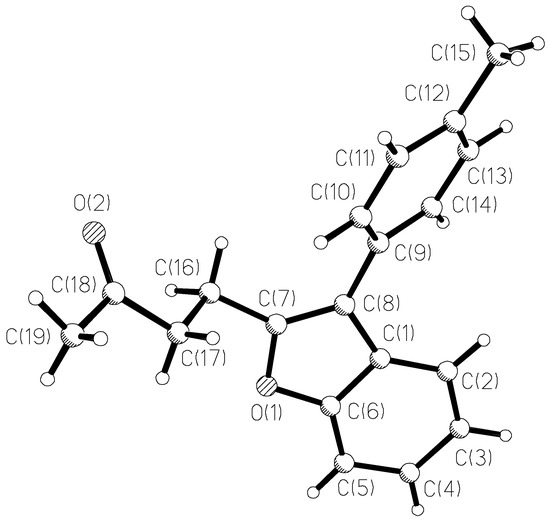
Figure 1.
The X-Ray crystal structure of 3e.
Table 5.
Crystal data and structure refinement of 3e.
Table 6.
Atom coordinates (104) and temperature factors (A2 103).
Table 7.
Bond lengths (A) and angles (deg).
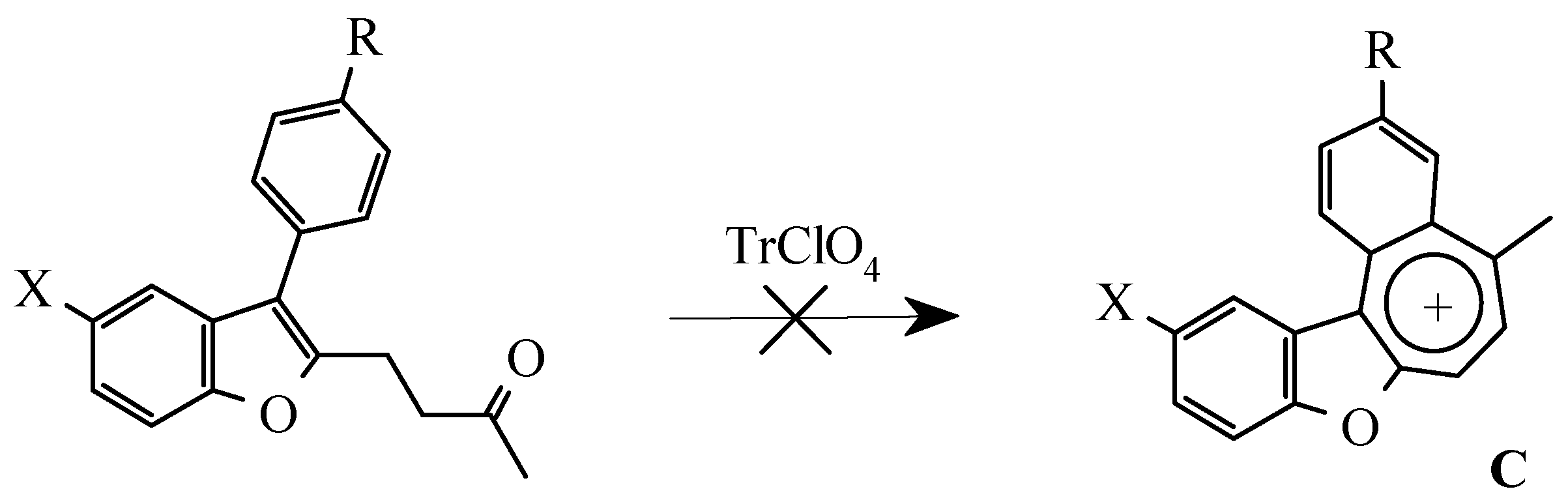
As stated above (Scheme 1) 3-furylbenzofurans A can be transformed into oxazulenium cations B in high yields by oxidation with trityl perchlorate or by treatment with perchloric acid. Attempts to obtain analogues of these salts from 3-arylbenzofurans 3a-e were unsuccessful. Neither oxidation by trityl perchlorate or chloroanil in methylene chloride nor treatment with perchloric acid in boiling di-oxane gave the desired salts.
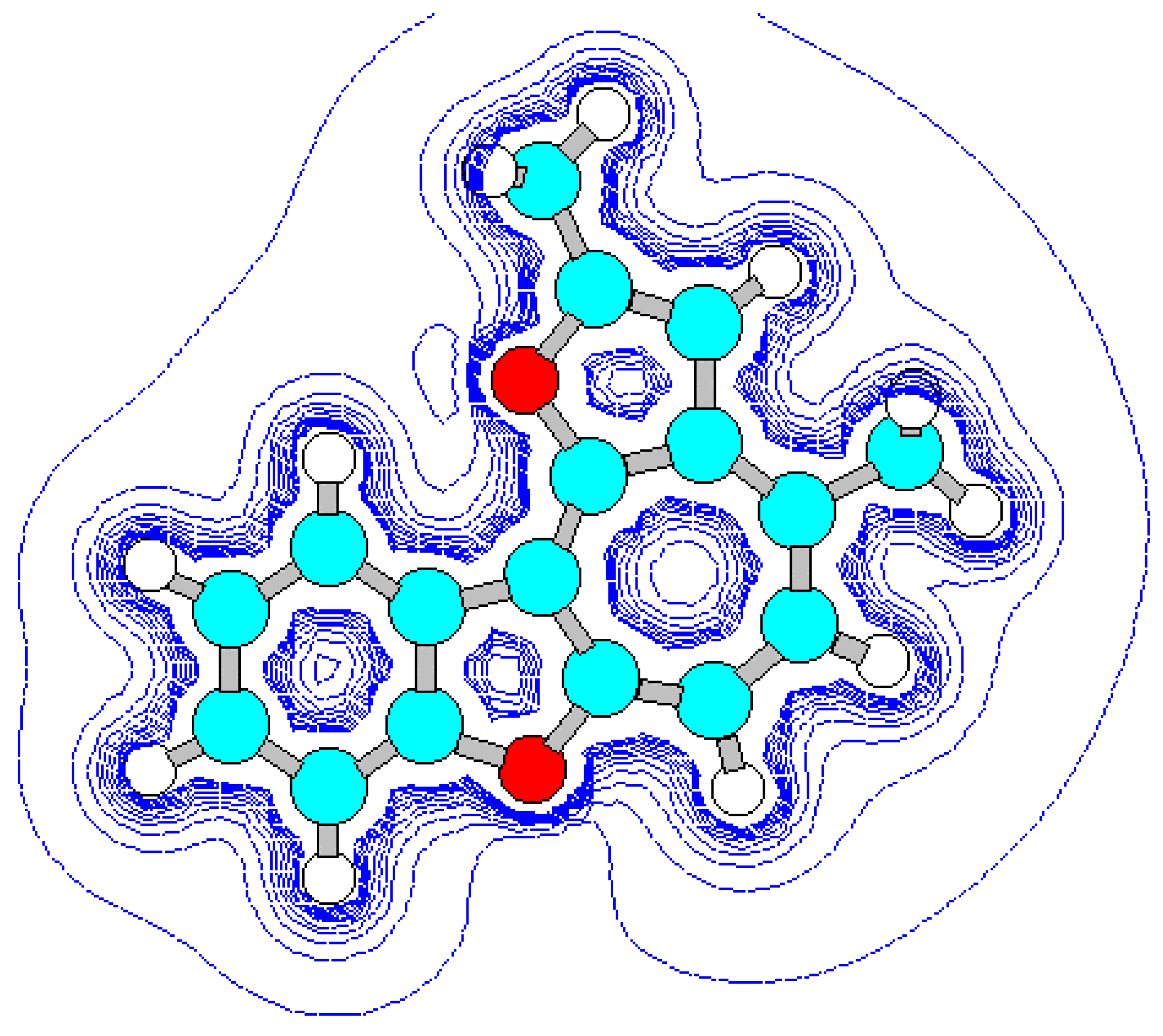
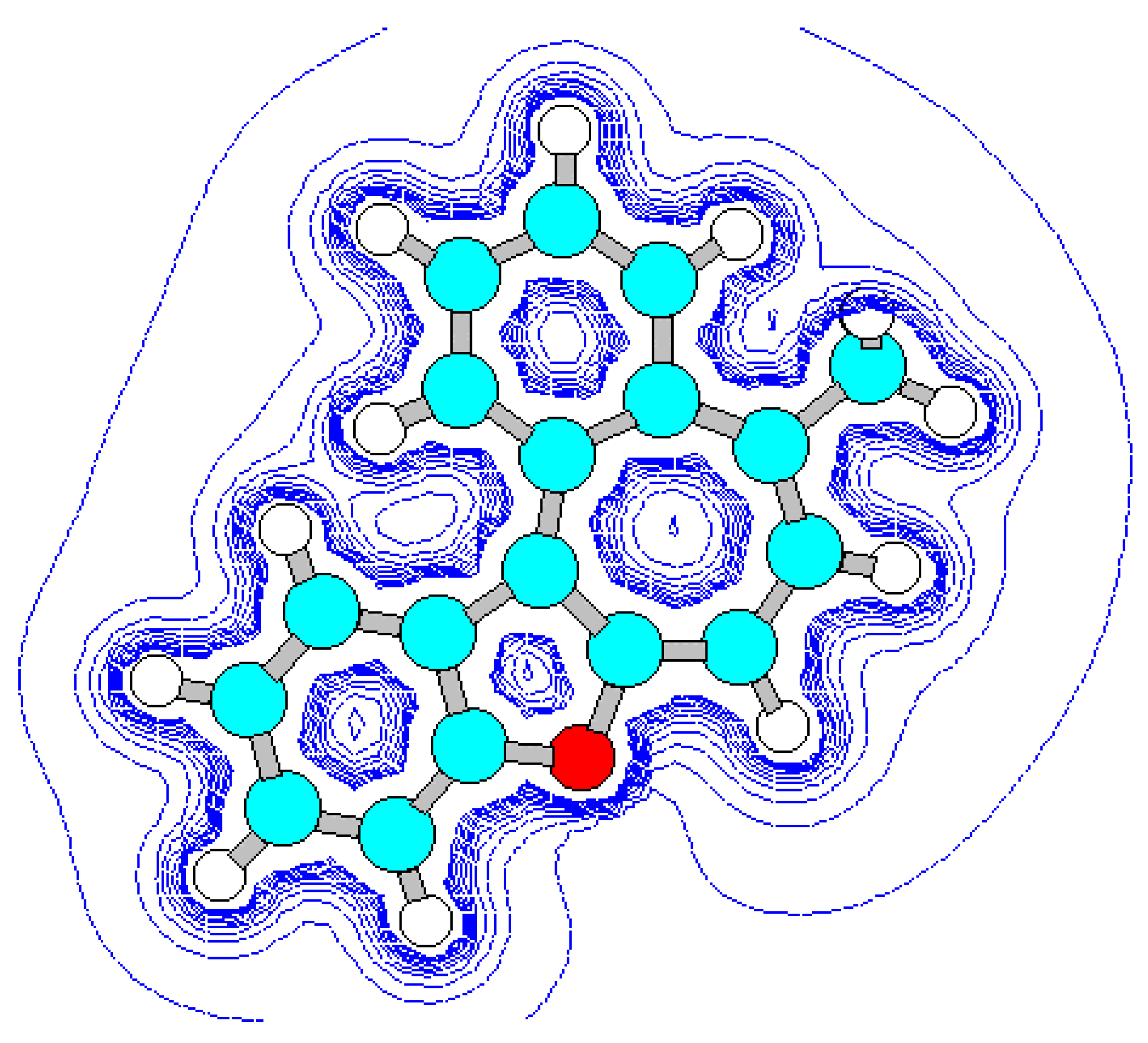
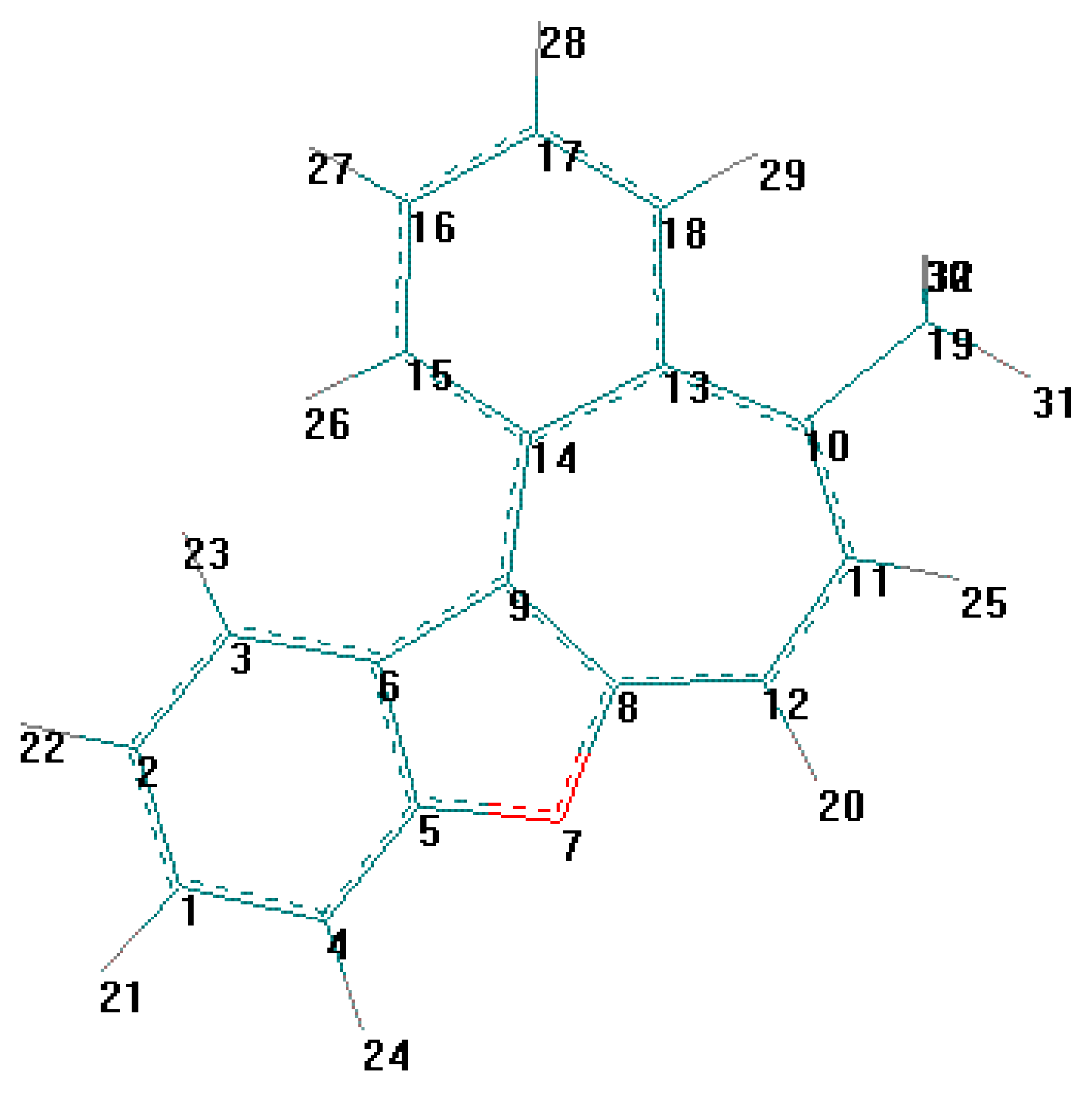
For the detection of the existing cation B features and the hypothetical cation C constitution (Scheme 4) their quantum-chemical calculation was conducted (preliminary optimization of geometry by a method MM+ and finally by a semiempirical method AM1). The outcomes of the cation B ge-ometry calculation is shown in Figure 2, Figure 4 and in Table 8, Table 9, in which the result of the X-ray study of its monocrystal is also shown [4]. As it can be seen, values of interatomic spacing intervals and va-lent angles obtained by computational are in good agreement with experimental X-Ray data, except for the lengths of furan C-O bonds for which the calculation gives higher values. There are no in-tramolecular contacts causing sterical strain in the cation B.
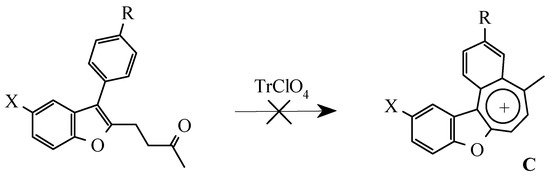
Scheme 4.
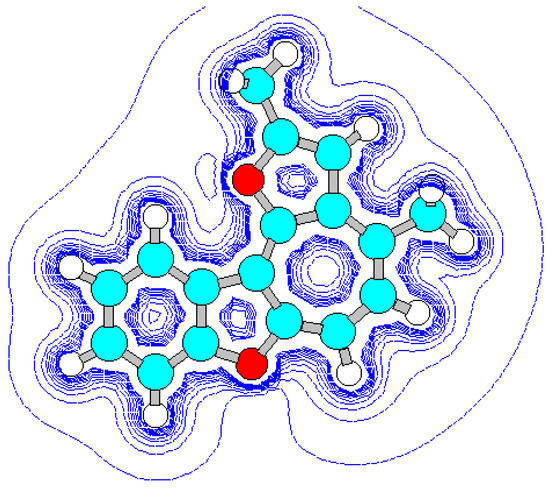
Figure 2.
Energy minimized structure (AM1) of cation B generated by HyperChem 5.0 program (with 2Dcontour map of electrostatic potential).
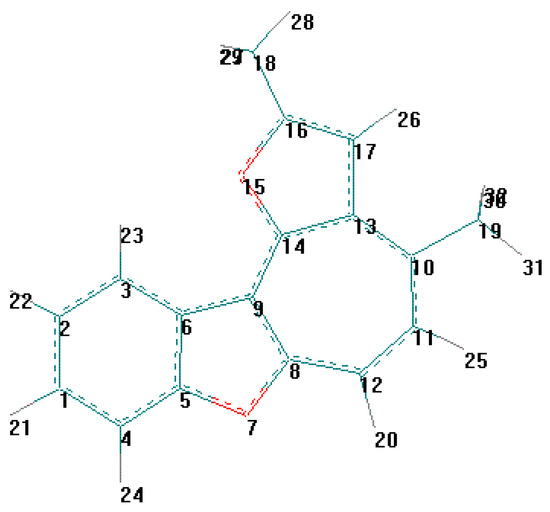
Figure 4.
Table 8.
Bond lengths (A) for compound B from X-ray and AM1 data.*
Table 9.
Bond angles (deg) for compound B from X-ray and AM1 data.*
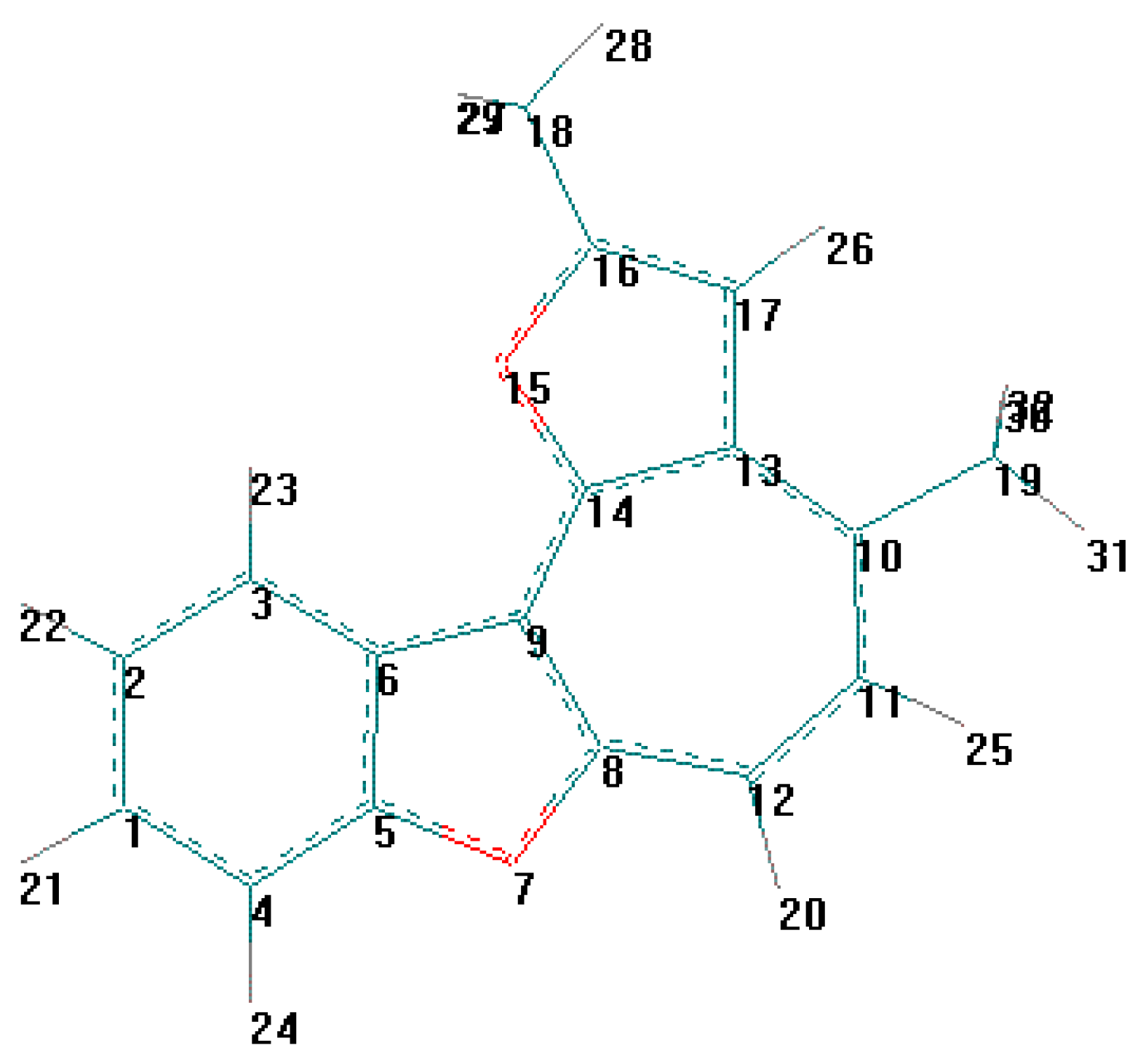
Vice-versa, flat geometry of cation C (Figure 3, Figure 5, Table 10, Table 11) should be accompanied by strong intramolecular strain leading to a noticeable distortion of some valent angles and bond lengths as compared with standard values and with the corresponding values in cation B.
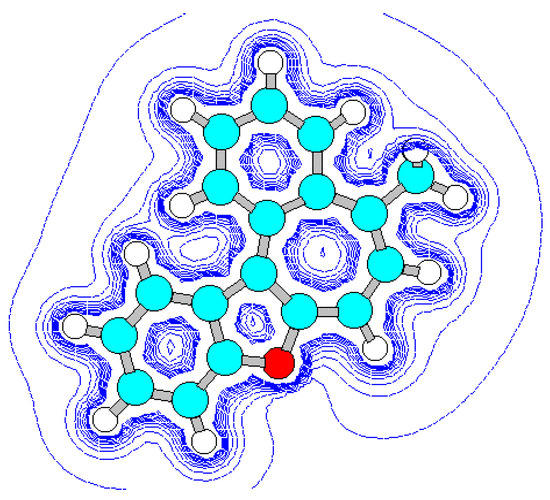
Figure 3.
Energy minimized structure (AM1) of hypotetic cation C generated by HyperChem 5.0 pro-gram (with 2Dcontour map of electrostatic potential).
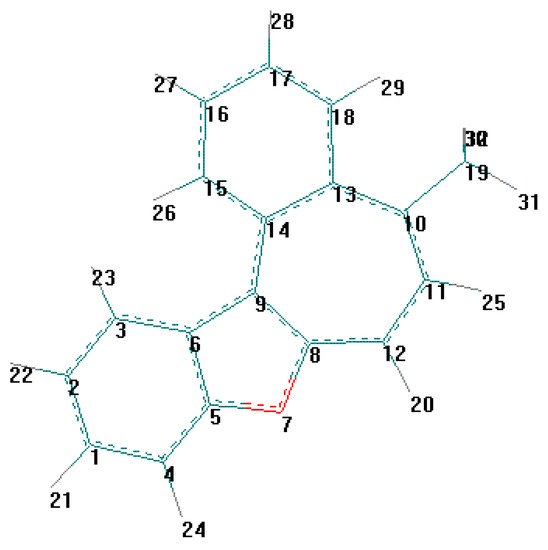
Figure 5.
Table 10.
Bond lengths (A) for C from AM1 data.
Table 11.
Bond angles (deg). For C from AM1 calculation data.
The main cause of sterical strain is intramolecular interaction H (23) ⃛ H (26) (Figure 5). Intera-tomic spacing interval H (23) ⃛ H (26) according to calculation is 1.628 Å, which is much less then the sum of their Van-der-Vaals radii. As a result, the bond lengths and values of the valence angles in exocycle H(23)-C(3)-C(6)-C(9)-C(14)-C(15)-H(26) change.
Similar type of intramolecular interaction, leading to torsion of a molecule into a spiral with preser-vation of standard values of interatomic spacing intervals and valence angles, can be seen in the ben-zophenantrene molecule (Figure 6) [8]. Probably in cation C the energy of conjugating would be so great, that it does not allow the structure to bend in a spiral to decrease sterical tension. Obviously the inability of the molecule to undergo removal of intramolecular strain is the main reason why it can not be synthesized.

Figure 6.
This example is not the only one known. It is of interest that the dibenzo[a,c]tropylium cation was not obtained by the route shown in Scheme 5 due to steric hindrance caused by two overlapping hy-drogens which makes complanarity of the aromatic cation impossible (Scheme 5) [9]:
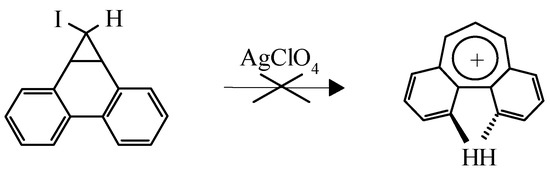
Scheme 5.
Instead a green polymer was isolated despite the high stability of the cation predicted by MO LCAO. However dibenzo[a,d]tropylium salts are stable compounds [10].
Experimental
General
1H NMR spectra were registered on Tesla BS-587 (80 MHz) in CDCl3, with hexamethyldisiloxane as internal standard. IR spectra were obtained on Specord M80 in vaseline. The course of a reaction was controled by TLC on Sorbfil plates (bromine and dinitrophenylhydrazine solution as a developer).
General method of synthesis of 2-hydroxybenzyl alcohols 1
To a Grignard reagent prepared from 0.25 mol of the corresponding halide and 6.8 g (0.28 mol) of Mg in 250 ml of dry ether 13.6 g (0.1 mol) of substituted salicylaldehyde was added dropwise. The mixture was stirred for 5 minutes and then decomposed by slow addition of water until magnesium salts coagulated. The ether layer was separated and the solid residue in the flask repeatedly extracted with ether, combined extracts were dried over Na2SO4 and evaporated. The oily residue was dissolved in a hot benzene-hexane mixture, filtered through a pad of Al2O3 and left to crystallize overnight.
1a Yield 73%. M. p. 102-103°C. Analysis: Found C, 78.31; H 6.25; C14H14O2, Mr 214.26 requires C, 78.48; H 6.58. 1H NMR δ 2.13 (s, 3H, CH3), 2.91 (b. s, 1H, CHOH), 5.87 (b. s, 1H, CH), 6.61-6.86 (m, 3H, HAr), 7,29 (s, 5H, HAr), and 7.60 (b. s, 1H, OH). IR 3190 (b. s, OH) and 3530 cm-1(s, OH).
1b Yield 72%. M. p. 106-107°C. Analysis: Found C, 57.01; H 4.79; C14H13BrO2, Mr 293.16 requires C, 57.36; H 4.47. 1H NMR δ 2.14 (s, 3H, CH3), 3.17 (b. s, 1H, CHOH), 5.78 (s, 1H, CH), 6.61-6.86 (m, 3H, HAr), 7,17 (d, J = 8.2 Hz, 2H, HAr), 7.30 (d, J = 8.2 Hz, 2H, HAr) and 7.62 (s, 1H, OH). IR 3210 (b. s, OH) and 3520 cm-1 (s, OH).
1c Yield 81%. M. p. 97-98°C. Analysis: Found C, 79.11; H 7.25; C15H16O2, Mr 228.29 requires C, 78.92; H 7.06. 1H NMR δ 2.12 (s, 3H, CH3), 2.26 (s, 3H, CH3), 3.02 (b. s, 1H, CHOH), 5.79 (b. s, 1H, CH), 6.58-6.82 (m, 3H, HAr), 7,13 (s, 4H, HAr) and 7.73 (s, 1H, OH). IR 3310 cm-1 (b. s, OH).
1d Yield 78%. M. p. 87-88°C. Analysis: Found C, 78.16; H 5.89; C13H12O2, Mr 200.23 requires C, 77.98; H 6.04. 1H NMR 2.99 (b. s, 1H, CHOH), 5.92 (b. s, 1H, CH), 6.69-7.40 (m, 9H, HAr) and 7.83 (b. s, 1H, OH). IR 3215 (b. s, OH) and 3520 cm-1 (s, OH).
1e Yield 73%. M. p. 104-105°C. Analysis: Found C, 78.24; H 6.89; C14H14O2, Mr 214.26 requires C, 78.48; H 6.58. 1H NMR δ 2.25 (s, 3H, CH3), 3.07 (b. s, 1H, CHOH), 5.84 (s, 1H, CH), 6.64-7.30 (m, 8H, HAr) and 7.95 (b. s, 1H, OH). IR 3205 (b. s, OH) and 3530 cm-1 (s, OH).
1f Yield 69%. M. p. 82-83°C. Analysis: Found C, 78.59; H 6.72; C15H16O2, Mr 228.29 requires C, 78.92; H 7.06. 1H NMR δ 2.16 (s, 3H, CH3), 2.57 (b. s, 1H, CHOH), 3,01 (d, J = 7.0 Hz, 2H, CH2), 4.87 (t, J = 7.0 Hz, 1H, CH), 6.61-6.82 (m, 3H, HAr), 7,20 (s, 5H, HAr), and 7.70 (b. s, 1H, OH). IR 3170 (b. s, OH) and 3480 cm-1 (s, OH).
1g Yield 53%. M. p. 142-143°C. Analysis: Found C, 71.89; H 4.78; C10H14O2, Mr 166.22 requires C, 72.26; H 4.49. 1H NMR δ 0.72 (t, J = 7.2 Hz, 3H, CH2CH3), 1.84 (k. d, 2.16 J = 7.2 Hz, J = 7.1 Hz, CH2CH3) 2.17 (s, 3H, CH3), 4.23 (t, J = 7.1 Hz, 1H, CH), 6.55-6.87 (m, 3H, HAr), and 7.08 (s, 1H, OH). IR 3190 (b. s, OH) and 3370 cm-1 (s, OH).
General method of synthesis of 2-hydroxyaryl-R - (5-methylfur-2-yl)methanes 2
The mixture of 0.02 mol of alcohol 1, 0.022 mol of 2-methylfuran and 50 mg of p-toluenesulphonic acid in 20 ml of benzene was refluxed with Dean-Stark trap for 5 minutes. Cooled solution was fil-tered through a pad of Al2O3 and evaporated to dryness, leaving a product as a colourless oil.
General method of synthesis of 3-R-2 - (3-oxobutyl)benzo[b]furans 3
To a boiling solution of 0.01 mol of compound 2 in 5 ml of ethanol, 5 ml of saturated ethanolic HCl was added all at once. The reaction mixture was refluxed for 5 minutes, then cooled, diluted with water. The thick oil was washed with water and extracted with hot hexane. The warm extract was fil-tered through a pad of Al2O3, the mother liquor concentrated and left to crystallize at -5°C to give product as white crystals.
References and Notes
- Butin, A. V.; Stroganova, T. A.; Abaev, V. T.; Kul'nevich, V. G. Khim. Geterotsikl. Soedin. 1998, 1250.
- Gutnov, A. V.; Abaev, V. T.; Butin, A. V.; Zavodnik, V. E.; Kul'nevich, V. G. Khim. Geterotsikl. Soedin. 1996, 162.
- Abaev, V. T.; Gutnov, A. V.; Butin, A. V. Khim. Geterotsikl. Soedin. 1998, 603.
- Butin, A. V.; Abaev, V. T.; Zavodnik, V. E.; Kul'nevich, V. G. Khim. Geterotsikl. Soedin. 1993, 627.
- Butin, A. V.; Gutnov, A. V.; T. A. Abaev, V. T.; Krapivin, G. D. Khim. Geterotsikl. Soedin. 1998, 883.
- Butin, A. V.; Gutnov, A. V.; T. A. Abaev, V. T.; Krapivin, G. D. Molecules 1999, 52.
- Butin, A. V.; Krapivin, G. D.; Zavodnik, V. E.; Kul'nevich, V. G. Khim. Geterotsikl. Soedin. 1993, 616.
- Herbstein, F.N.; Schmidt, G.M.J. J. Chem. Soc. 1954, 3302.
- Dewar, M. J. S.; Ganellin, C.R. J.Chem. Soc. 1959, 3139.
- Berti, G.; Da Settimo, A. Ann. Chimica 1962, 995.
- Samples Availability: Available from MDPI.
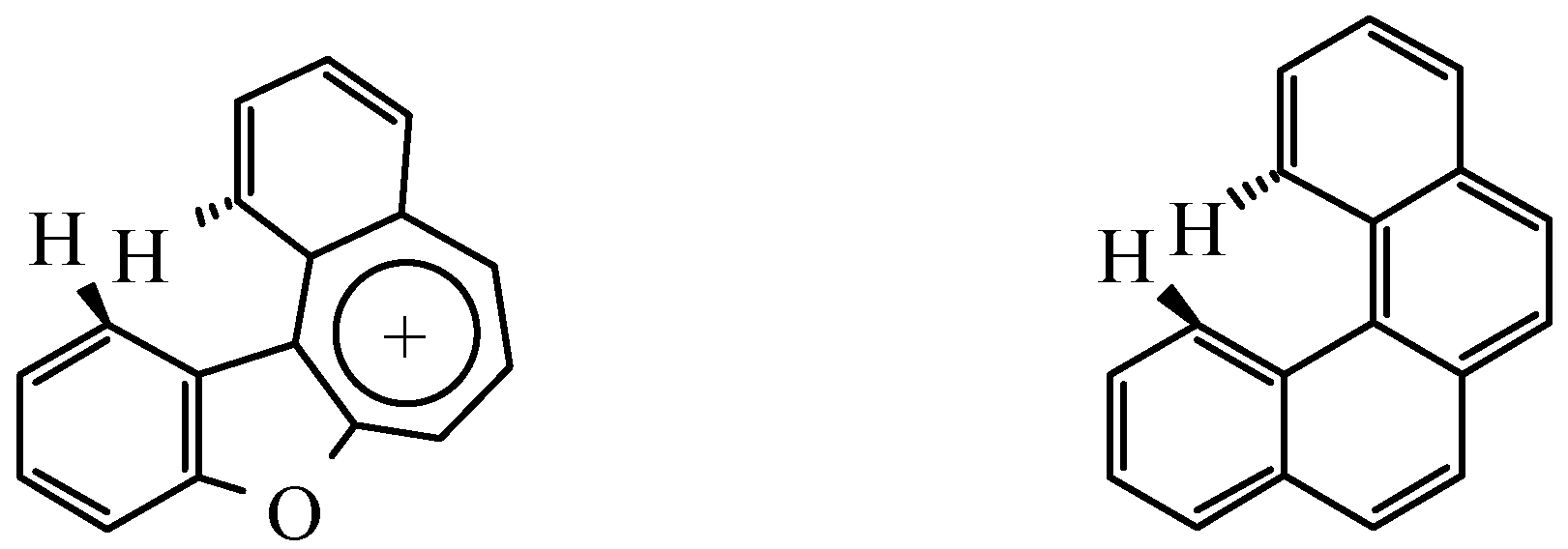
© 1999 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/3.0/).