
Structure-Guided Identification of JAK2 Inhibitors: From Similarity to Stability and Specificity


 ,
,  ,
,  ,
,
Abstract
1. Introduction
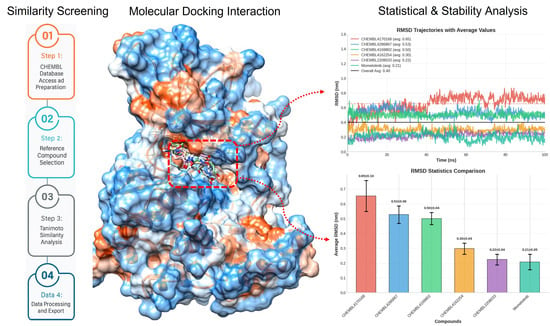
2. Methodology
2.1. Chembl Library Accession and Screening
2.2. Receptor 3D Model Accession and Pharmacophore Modeling
2.3. Validation of Pharmacophore Model
- A is the total number of active compounds in the dataset.
- D is the total number of decoy compounds in the dataset.
- Ht is the number of true positives (correctly identified active compounds).
- Hf is the number of false positives (decoys incorrectly identified as active).
- N is the total number of compounds in the dataset.
2.4. Molecular Docking
2.5. Molecular Dynamics Simulations
2.6. Free Energy Calculation
3. Results
3.1. Chembl Library Accession
3.2. Receptor 3D Model Accession
3.3. Pharmacophore Model Generation
3.4. Pharmacophore Model Validation and Ligand Screening
3.5. Molecular Docking Analysis
3.6. Molecular Docking Interactions
3.7. Molecular Dynamics Simulation
3.7.1. Root Mean Square Deviation Analysis
3.7.2. Hydrogen Bonds Plot Analysis
3.7.3. MD Interaction Energy
3.8. gmxMMPBSA Free Energy Calculation
3.9. Additional MD Duplicates Run
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hubbard, S.R. Mechanistic insights into regulation of JAK2 tyrosine kinase. Front. Endocrinol. 2018, 8, 361. [Google Scholar] [CrossRef]
- Ferrao, R.; Lupardus, P.J. The Janus kinase (JAK) FERM and SH2 domains: Bringing specificity to JAK–receptor interactions. Front. Endocrinol. 2017, 8, 71. [Google Scholar] [CrossRef]
- Virtanen, A.T.; Haikarainen, T.; Sampathkumar, P.; Palmroth, M.; Liukkonen, S.; Liu, J.; Nekhotiaeva, N.; Hubbard, S.R.; Silvennoinen, O. Identification of novel small molecule ligands for JAK2 pseudokinase domain. Pharmaceuticals 2023, 16, 75. [Google Scholar] [CrossRef]
- Purohit, M.; Gupta, G.; Afzal, O.; Altamimi, A.S.A.; Alzarea, S.I.; Kazmi, I.; Almalki, W.H.; Gulati, M.; Kaur, I.P.; Singh, S.K. Janus kinase/signal transducers and activator of transcription (JAK/STAT) and its role in Lung inflammatory disease. Chem. Biol. Interact. 2023, 371, 110334. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal. Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Kiu, H.; Nicholson, S.E. Biology and significance of the JAK/STAT signalling pathways. Growth Factors 2012, 30, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Choe, J.; Hassan, M.; Kloczkowski, A.; Chun, W. Recent advances and future perspectives in small molecule JAK2 inhibitors. Future Med. Chem. 2025, 17, 1175–1191. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.G.; Paes, J.; da Costa, A.G.; Malheiro, A.; Silva, G.V.; Mourão, L.P.d.S.; Tarragô, A.M. JAK2 variant signaling: Genetic, hematologic and immune implication in chronic myeloproliferative neoplasms. Biomolecules 2022, 12, 291. [Google Scholar] [CrossRef]
- Bertsias, G. Therapeutic targeting of JAKs: From hematology to rheumatology and from the first to the second generation of JAK inhibitors. Mediterr. J. Rheumatol. 2020, 31, 105–111. [Google Scholar] [CrossRef]
- Behrens, K.; Alexander, W.S. Cytokine control of megakaryopoiesis. Growth Factors 2018, 36, 89–103. [Google Scholar] [CrossRef]
- Pasquier, F.; Cabagnols, X.; Secardin, L.; Plo, I.; Vainchenker, W. Myeloproliferative neoplasms: JAK2 signaling pathway as a central target for therapy. Clin. Lymphoma. Myeloma. Leuk. 2014, 14, S23–S35. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Yao, Q.; Gu, X.; Shi, Q.; Yuan, X.; Chu, Q.; Bao, Z.; Lu, J.; Li, L. Evolving cognition of the JAK-STAT signaling pathway: Autoimmune disorders and cancer. Signal. Transduct. Target. Ther. 2023, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Singh, M.K.; Shyam, H.; Mishra, A.; Kumar, S.; Kumar, A.; Kushwaha, J. Role of JAK/STAT in the neuroinflammation and its association with neurological disorders. Ann. Neurosci. 2021, 28, 191–200. [Google Scholar] [CrossRef]
- Gou, P.; Zhang, W.; Giraudier, S. Insights into the potential mechanisms of JAK2V617F somatic mutation contributing distinct phenotypes in myeloproliferative neoplasms. Int. J. Mol. Sci. 2022, 23, 1013. [Google Scholar] [CrossRef]
- Oh, S.T.; Gotlib, J. JAK2 V617F and beyond: Role of genetics and aberrant signaling in the pathogenesis of myeloproliferative neoplasms. Expert. Rev. Hematol. 2010, 3, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, G.; McMullin, M.F.; Mills, K. Molecular pathogenesis of the myeloproliferative neoplasms. J. Hematol. Oncol. 2021, 14, 103. [Google Scholar] [CrossRef]
- Sonbol, M.B.; Firwana, B.; Zarzour, A.; Morad, M.; Rana, V.; Tiu, R.V. Comprehensive review of JAK inhibitors in myeloproliferative neoplasms. Ther. Adv. Hematol. 2013, 4, 15–35. [Google Scholar] [CrossRef]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in aging, inflammation, hematopoiesis and malignant transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Li, S.; Ma, J.; Dai, X.; Lu, J. Deciphering JAK/STAT signaling pathway: A multifaceted approach to tumorigenesis, progression and therapeutic interventions. Int. Immunopharmacol. 2024, 131, 111846. [Google Scholar] [CrossRef]
- Obeagu, E.I. JAK2 in pediatric leukemia: Mechanisms of pathogenesis and drug development–a narrative review. Ann. Med. Surg. 2025, 87, 3410–3423. [Google Scholar] [CrossRef] [PubMed]
- Masarova, L.; Bose, P.; Verstovsek, S. The rationale for immunotherapy in myeloproliferative neoplasms. Curr. Hematol. Malig. Rep. 2019, 14, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, B.; Liu, Y.; Yu, Y.; Wan, Y.; Wu, J.; Wang, Y. JAK2 inhibitors for the treatment of Philadelphia-negative myeloproliferative neoplasms: Current status and future directions. Mol. Divers. 2023, 28, 3445–3456. [Google Scholar] [CrossRef]
- Spinelli, F.R.; Meylan, F.; O’Shea, J.J.; Gadina, M. JAK inhibitors: Ten years after. Eur. J. Immunol. 2021, 51, 1615–1627. [Google Scholar] [CrossRef]
- Shawky, A.M.; Almalki, F.A.; Abdalla, A.N.; Abdelazeem, A.H.; Gouda, A.M. A Comprehensive Overview of Globally Approved JAK Inhibitors. Pharmaceutics 2022, 14, 1001. [Google Scholar] [CrossRef]
- Tokareva, K.; Reid, P.; Yang, V.; Liew, D.; Peterson, A.C.; Baraff, A.; Giles, J.; Singh, N. JAK inhibitors and black box warnings: What is the future for JAK inhibitors? Expert. Rev. Clin. Immunol. 2023, 19, 1385–1397. [Google Scholar] [CrossRef]
- Sardana, K.; Bathula, S.; Khurana, A. Which is the Ideal JAK Inhibitor for Alopecia Areata—Baricitinib, Tofacitinib, Ritlecitinib or Ifidancitinib—Revisiting the Immunomechanisms of the JAK Pathway. Indian Dermatol. Online J. 2023, 14, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Ritlecitinib: First Approval. Drugs 2023, 83, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Deucravacitinib: First Approval. Drugs 2022, 82, 1671–1679. [Google Scholar] [CrossRef]
- Cai, W.; Tong, R.; Sun, Y.; Yao, Y.; Zhang, J. Comparative efficacy of five approved Janus kinase inhibitors as monotherapy and combination therapy in patients with moderate-to-severe active rheumatoid arthritis: A systematic review and network meta-analysis of randomized controlled trials. Front. Pharmacol. 2024, 15, 1387585. [Google Scholar] [CrossRef]
- Bose, P.; Verstovsek, S. JAK2 inhibitors for myeloproliferative neoplasms: What is next? Am. Society. Hematol. 2017, 130, 115–125. [Google Scholar] [CrossRef]
- Pardanani, A.; Tefferi, A. The Journal of the American Society of Hematology, How I treat myelofibrosis after failure of JAK inhibitors. Blood 2018, 132, 492–500. [Google Scholar] [CrossRef]
- Yasir, M.; Park, J.; Han, E.T.; Park, W.S.; Han, J.H.; Chun, W. Drug Repositioning via Graph Neural Networks: Identifying Novel JAK2 Inhibitors from FDA-Approved Drugs through Molecular Docking and Biological Validation. Molecules 2024, 29, 1363. [Google Scholar] [CrossRef]
- Yasir, M.; Park, J.; Han, E.T.; Park, W.S.; Han, J.H.; Kwon, Y.S.; Lee, H.J.; Chun, W. Machine Learning-Based Drug Repositioning of Novel Janus Kinase 2 Inhibitors Utilizing Molecular Docking and Molecular Dynamic Simulation. J. Chem. Inf. Model. 2023, 63, 6487–6500. [Google Scholar] [CrossRef]
- Schaller, D.; Šribar, D.; Noonan, T.; Deng, L.; Nguyen, T.N.; Pach, S.; Machalz, D.; Bermudez, M.; Wolber, G. Next generation 3D pharmacophore modeling. WIREs Comp. Mol. Sci. 2020, 10, e1468. [Google Scholar] [CrossRef]
- Naqvi, A.A.; Mohammad, T.; Hasan, G.M.; Hassan, M.I. Advancements in docking and molecular dynamics simulations towards ligand-receptor interactions and structure-function relationships. Curr. Top. Med. Chem. 2018, 18, 1755–1768. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhong, A.; Wang, Q.; Zheng, T. Structure-based pharmacophore modeling, virtual screening, molecular docking, ADMET, and molecular dynamics (MD) simulation of potential inhibitors of PD-L1 from the library of marine natural products. Mar. Drugs 2021, 20, 29. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory. Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Rácz, A.; Bajusz, D.; Héberger, K. Life beyond the Tanimoto coefficient: Similarity measures for interaction fingerprints. J. Cheminform. 2018, 10, 48. [Google Scholar] [CrossRef]
- Horvath, D. Pharmacophore-based virtual screening. Methods Mol. Biol. 2011, 672, 261–298. [Google Scholar] [PubMed]
- Qing, X.; Yin Lee, X.; De Raeymaeker, J.; RH Tame, J.; YJ Zhang, K.; De Maeyer, M.; RD Voet, A. Pharmacophore modeling: Advances, limitations, and current utility in drug discovery. J. Rec. Lig. Chann. Res. 2014, 2014, 81–92. [Google Scholar]
- Miao, Y.; Virtanen, A.; Zmajkovic, J.; Hilpert, M.; Skoda, R.C.; Silvennoinen, O.; Haikarainen, T. Functional and Structural Characterization of Clinical-Stage Janus Kinase 2 Inhibitors Identifies Determinants for Drug Selectivity. J. Med. Chem. 2024, 67, 10012–10024. [Google Scholar] [CrossRef]
- Banat, R.; Daoud, S.; Taha, M.O. Ligand-based pharmacophore modeling and machine learning for the discovery of potent aurora A kinase inhibitory leads of novel chemotypes. Mol. Divers. 2024, 28, 4241–4257. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Ho, Y.; Liu, H.-L. Structure-Based Pharmacophore Modeling to Discover Novel CCR5 Inhibitors for HIV-1/Cancers Therapy. J. Biom. Sci. Eng. 2019, 12, 10–30. [Google Scholar] [CrossRef]
- Kumar, S.P. Receptor pharmacophore ensemble (REPHARMBLE): A probabilistic pharmacophore modeling approach using multiple protein-ligand complexes. J. Mol. Model. 2018, 24, 282. [Google Scholar] [CrossRef]
- Gupta, N.; Sitwala, N.; Patel, K. Pharmacophore modelling, validation, 3D virtual screening, docking, design and in silico ADMET simulation study of histone deacetylase class-1 inhibitors. Med. Chem. Res. 2014, 23, 4853–4864. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An overview of scoring functions used for protein–ligand interactions in molecular docking. Interdiscip. Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Biovia, D.S. Discovery Studio Modeling Environment, Release 2017; DassaultSystèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Wu, G.; Robertson, D.H.; Brooks, C.L., III; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Jo, S.; MacKerell, A.D.; Klauda, J.B.; Im, W. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. Biophys. J. 2016, 110, 641a. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Willard, L.; Ranjan, A.; Zhang, H.; Monzavi, H.; Boyko, R.F.; Sykes, B.D.; Wishart, D.S. VADAR: A web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 2003, 31, 3316–3319. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery: Miniperspective. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Dantas, R.F.; Evangelista, T.C.S.; Neves, B.J.; Senger, M.R.; Andrade, C.H.; Ferreira, S.B.; Silva-Junior, F.P. Dealing with frequent hitters in drug discovery: A multidisciplinary view on the issue of filtering compounds on biological screenings. Expert Opin. Drug Discov. 2019, 14, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Kaserer, T.; Beck, K.R.; Akram, M.; Odermatt, A.; Schuster, D. Pharmacophore models and pharmacophore-based virtual screening: Concepts and applications exemplified on hydroxysteroid dehydrogenases. Molecules 2015, 20, 22799–22832. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Park, J.; Han, E.-T.; Han, J.-H.; Park, W.S.; Chun, W. Identification of Malaria-Selective Proteasome β5 Inhibitors Through Pharmacophore Modeling, Molecular Docking, and Molecular Dynamics Simulation. Int. J. Mol. Sci. 2024, 25, 11881. [Google Scholar] [CrossRef]
- Mukherjee, T.; Kumar, N.; Chawla, M.; Philpott, D.J.; Basak, S. The NF-κB signaling system in the immunopathogenesis of inflammatory bowel disease. Sci. Signal. 2024, 17, eadh1641. [Google Scholar] [CrossRef]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef]
- Lin, F.-Y.; MacKerell, A.D., Jr. Do halogen–hydrogen bond donor interactions dominate the favorable contribution of halogens to ligand–protein binding? J. Phys. Chem. B 2017, 121, 6813–6821. [Google Scholar] [CrossRef]
- Rafiq, H.; Hu, J.; Hakami, M.A.; Hazazi, A.; Alamri, M.A.; Alkhatabi, H.A.; Mahmood, A.; Alotaibi, B.S.; Wadood, A.; Huang, X. Identification of novel STAT3 inhibitors for liver fibrosis, using pharmacophore-based virtual screening, molecular docking, and biomolecular dynamics simulations. Sci. Rep. 2023, 13, 20147. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Di, B.; Niu, M.-M. Structure-based pharmacophore design and virtual screening for novel tubulin inhibitors with potential anticancer activity. Molecules 2019, 24, 3181. [Google Scholar] [CrossRef]
- Yasir, M.; Park, J.; Han, E.-T.; Han, J.-H.; Park, W.S.; Choe, J.; Chun, W. Integration of Deep Learning with Molecular Docking and Molecular Dynamics Simulation for Novel TNF-α-Converting Enzyme Inhibitors. Future Pharmacol. 2025, 5, 55. [Google Scholar] [CrossRef]
- Yasir, M.; Park, J.; Chun, W. Discovery of Novel Aldose Reductase Inhibitors via the Integration of Ligand-Based and Structure-Based Virtual Screening with Experimental Validation. ACS Omega 2024, 9, 20338–20349. [Google Scholar] [CrossRef]
- Yasir, M.; Park, J.; Han, E.-T.; Park, W.S.; Han, J.-H.; Chun, W. Identification of Potential Tryptase Inhibitors from FDA-Approved Drugs Using Machine Learning, Molecular Docking, and Experimental Validation. ACS Omega 2024, 9, 38820–38831. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.E.; HuangFu, W.-C.; Chao, M.-W.; Sung, T.-Y.; Chang, C.-D.; Chen, Y.-Y.; Hsieh, J.-H.; Tu, H.-J.; Huang, H.-L.; Pan, S.-L. A novel selective JAK2 inhibitor identified using pharmacological interactions. Front. Pharmacol. 2018, 9, 1379. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Jiménez, L.K.; Rivera, G.; Juárez-Saldivar, A.; Ortega-Balleza, J.L.; Ortiz-Pérez, E.; Jaime-Sánchez, E.; Paz-González, A.; Lara-Ramírez, E.E. Biological Evaluations and Computer-Aided Approaches of Janus Kinases 2 and 3 Inhibitors for Cancer Treatment: A Review. Pharmaceutics 2024, 16, 1165. [Google Scholar] [CrossRef]
- Oliveira, M.P.; Hünenberger, P.H. Influence of the Lennard-Jones Combination Rules on the Simulated Properties of Organic Liquids at Optimal Force-Field Parametrization. J. Chem. Theory Comput. 2023, 19, 2048–2063. [Google Scholar] [CrossRef] [PubMed]
- Galano-Frutos, J.J.; Sancho, J. Energy, water, and protein folding: A molecular dynamics-based quantitative inventory of molecular interactions and forces that make proteins stable. Protein Sci. 2024, 33, e4905. [Google Scholar] [CrossRef]
- Singh, A.; Mishra, A. Molecular modelling study to discover novel JAK2 signaling pathway inhibitor. J. Biomol. Struct. Dyn. 2023, 41, 5827–5838. [Google Scholar] [CrossRef]
- Baskin, R.; Majumder, A.; Sayeski, P.P. The recent medicinal chemistry development of Jak2 tyrosine kinase small molecule inhibitors. Curr. Med. Chem. 2010, 17, 4551–4558. [Google Scholar] [CrossRef]
- Gorantla, S.P.; Prince, G.; Osius, J.; Dinesh, D.C.; Boddu, V.; Duyster, J.; von Bubnoff, N. Type II mode of JAK2 inhibition and destabilization are potential therapeutic approaches against the ruxolitinib resistance driven myeloproliferative neoplasms. Front. Oncol. 2024, 14, 1430833. [Google Scholar] [CrossRef]
- Davis, R.R.; Li, B.; Yun, S.Y.; Chan, A.; Nareddy, P.; Gunawan, S.; Ayaz, M.; Lawrence, H.R.; Reuther, G.W.; Lawrence, N.J. Structural insights into JAK2 inhibition by ruxolitinib, fedratinib, and derivatives thereof. J. Med. Chem. 2021, 64, 2228–2241. [Google Scholar] [CrossRef]
- Zhao, C.; Khadka, D.B.; Cho, W.-J. Insights into the structural features essential for JAK2 inhibition and selectivity. Curr. Med. Chem. 2016, 23, 1331–1355. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.C.; Piehler, J.; Tvorogov, D.; Ross, D.M.; Lopez, A.F.; Gotlib, J.; Thomas, D. Next-generation JAK2 inhibitors for the treatment of myeloproliferative neoplasms: Lessons from structure-based drug discovery approaches. Blood Cancer Discov. 2023, 4, 352–364. [Google Scholar] [CrossRef]
- Pippis, E.J.; Yacyshyn, B.R. Clinical and mechanistic characteristics of current JAK inhibitors in IBD. Inflamm. Bowel Dis. 2021, 27, 1674–1683. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef]
- Lv, Y.; Mi, P.; Babon, J.J.; Fan, G.; Qi, J.; Cao, L.; Lang, J.; Zhang, J.; Wang, F.; Kobe, B. Small molecule drug discovery targeting the JAK-STAT pathway. Pharmacol. Res. 2024, 204, 107217. [Google Scholar] [CrossRef]
- Kavanagh, M.E.; Horning, B.D.; Khattri, R.; Roy, N.; Lu, J.P.; Whitby, L.R.; Ye, E.; Brannon, J.C.; Parker, A.; Chick, J.M.; et al. Selective inhibitors of JAK1 targeting an isoform-restricted allosteric cysteine. Nat. Chem. Bio. 2022, 18, 1388–1398. [Google Scholar] [CrossRef]
- Gao, Y.; Lan, L.; Wang, C.; Wang, Y.; Shi, L.; Sun, L. Selective JAK1 inhibitors and the therapeutic applications thereof: A patent review (2016–2023). Expert Opin. Ther. Pat. 2025, 35, 181–195. [Google Scholar] [CrossRef]
- Puca, P.; Del Gaudio, A.; Iaccarino, J.; Blasi, V.; Coppola, G.; Laterza, L.; Lopetuso, L.R.; Colantuono, S.; Gasbarrini, A.; Scaldaferri, F. Cancer Risk in IBD Patients Treated with JAK Inhibitors: Reassuring Evidence from Trials and Real-World Data. Cancers 2025, 17, 735. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr No | Compounds | CDocker Energy (kcal/mol) | CDocker Interaction Energy (kcal/mol) |
|---|---|---|---|
| 1 | CHEMBL4165344 | −49.6512 | −64.7590 |
| 2 | CHEMBL4170168 | −48.5137 | −63.0943 |
| 3 | CHEMBL4286867 | −46.3802 | −57.7018 |
| 4 | CHEMBL4169802 | −46.0797 | −69.0932 |
| 5 | CHEMBL4162254 | −42.8768 | −56.2747 |
| 6 | CHEMBL4172110 | −41.9531 | −58.2818 |
| 7 | CHEMBL2208033 | −39.4211 | −55.7272 |
| 8 | CHEMBL4168767 | −38.889 | −60.4567 |
| 9 | CHEMBL4168707 | −36.9968 | −58.7271 |
| 10 | Momelotinib | −36.3542 | −52.5442 |
| 11 | CHEMBL1079690 | −36.1376 | −49.2426 |
| 12 | Ruxolitinib | 3.63757 | −38.4951 |
| Compounds | Interacting Residues | Binding Distances |
|---|---|---|
| CHEMBL4165344 | Pro933, Phe860 | 1.96Å, 2.05Å |
| CHEMBL4170168 | Lys943, Gln853 Asn981 | 2.62Å, 2.50Å 2.05Å, 2.30Å |
| CHEMBL4286867 | Asp994, Pro933 Leu855 | 2.61Å, 2.82Å 2.12Å |
| CHEMBL4169802 | Leu932 Lys882, Pro933 Leu855, Lys943 Arg938 | 2.24Å, 2.09Å 2.91Å, 2.52Å 2.64Å, 2.34Å 2.10Å |
| CHEMBL4162254 | Pro933, Lys943 Leu932 | 2.64Å, 1.74Å 2.01Å, 2.23Å |
| CHEMBL4172110 | Lys943 Asn981 | 2.12Å, 2.02Å 2.09Å |
| CHEMBL2208033 | Asp994 Pro933 Leu932 Asn981 | 1.84Å 2.60Å 2.06Å, 2.41Å 2.76Å, 1.79Å |
| CHEMBL4168767 | Leu932 Pro933, Lys943 | 2.16Å, 2.52Å 2.90Å, 2.32Å |
| CHEMBL4168707 | Gln853, Pro933 Phe860, Asn859 | 2.56Å, 2.50Å 2.14Å, 2.42Å |
| CHEMBL1079690 | Gly993 | 2.91Å |
| Momelotinib | Pro933 Leu932 | 2.49Å 2.04Å, 2.19Å |
| Compounds | Mean ± SD | Stability |
|---|---|---|
| CHEMBL4165344 | 0.55 ± 0.30 | 0.768 |
| CHEMBL4170168 | 0.65 ± 0.10 | 0.905 |
| CHEMBL4286867 | 0.53 ± 0.06 | 0.944 |
| CHEMBL4169802 | 0.50 ± 0.04 | 0.961 |
| CHEMBL4162254 | 0.30 ± 0.04 | 0.964 |
| CHEMBL4172110 | 0.51 ± 0.11 | 0.903 |
| CHEMBL2208033 | 0.22 ± 0.04 | 0.965 |
| CHEMBL4168767 | 0.38 ± 0.10 | 0.908 |
| CHEMBL4168707 | 0.49 ± 0.09 | 0.917 |
| CHEMBL1079690 | 0.42 ± 0.08 | 0.923 |
| Momelotinib | 0.21 ± 0.05 | 0.951 |
| Sr No | Compound | Interaction Energy (kcal/mol) | ||
|---|---|---|---|---|
| Coul-SR | LJ-SR | Total Energy | ||
| 1 | CHEMBL4165344 | −15.9192 | −43.1047 | −59.0239 |
| 2 | CHEMBL4170168 | −14.2580 | −41.9567 | −56.2147 |
| 3 | CHEMBL4286867 | −16.3347 | −51.2077 | −67.5424 |
| 4 | CHEMBL4169802 | −26.9632 | −46.8829 | −73.8461 |
| 5 | CHEMBL4162254 | −36.3054 | −42.8738 | −79.1792 |
| 6 | CHEMBL4172110 | −11.7477 | −41.2447 | −52.9924 |
| 7 | CHEMBL2208033 | −47.9883 | −32.0189 | −80.0072 |
| 8 | CHEMBL4168767 | −18.0870 | −37.1288 | −55.2158 |
| 9 | CHEMBL4168707 | −27.7299 | −38.4469 | −66.1769 |
| 10 | CHEMBL1079690 | −12.9653 | −37.1334 | −50.0987 |
| 11 | Momelotinib | −26.7285 | −36.7803 | −63.5088 |
| Sr | Compounds | ΔG(TOTAL) | Standard Deviation |
|---|---|---|---|
| 1 | CHEMBL4165344 | −19.88 | 5.84 |
| 2 | CHEMBL4170168 | −23.55 | 5.96 |
| 3 | CHEMBL4286867 | −28.25 | 6.66 |
| 4 | CHEMBL4169802 | −29.91 | 5.28 |
| 5 | CHEMBL4162254 | −28.71 | 4.81 |
| 6 | CHEMBL4172110 | −18.83 | 5.80 |
| 7 | CHEMBL2208033 | −26.45 | 4.99 |
| 8 | CHEMBL4168767 | −25.89 | 4.98 |
| 9 | CHEMBL4168707 | −24.15 | 6.49 |
| 10 | CHEMBL1079690 | −20.32 | 6.58 |
| 11 | Momelotinib | −24.17 | 3.99 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasir, M.; Park, J.; Choe, J.; Han, J.-H.; Han, E.-T.; Park, W.S.; Chun, W. Structure-Guided Identification of JAK2 Inhibitors: From Similarity to Stability and Specificity. Future Pharmacol. 2025, 5, 66. https://doi.org/10.3390/futurepharmacol5040066
Yasir M, Park J, Choe J, Han J-H, Han E-T, Park WS, Chun W. Structure-Guided Identification of JAK2 Inhibitors: From Similarity to Stability and Specificity. Future Pharmacology. 2025; 5(4):66. https://doi.org/10.3390/futurepharmacol5040066
Chicago/Turabian StyleYasir, Muhammad, Jinyoung Park, Jongseon Choe, Jin-Hee Han, Eun-Taek Han, Won Sun Park, and Wanjoo Chun. 2025. "Structure-Guided Identification of JAK2 Inhibitors: From Similarity to Stability and Specificity" Future Pharmacology 5, no. 4: 66. https://doi.org/10.3390/futurepharmacol5040066
APA StyleYasir, M., Park, J., Choe, J., Han, J.-H., Han, E.-T., Park, W. S., & Chun, W. (2025). Structure-Guided Identification of JAK2 Inhibitors: From Similarity to Stability and Specificity. Future Pharmacology, 5(4), 66. https://doi.org/10.3390/futurepharmacol5040066