
Integrative Transcriptomic Meta-Analysis Reveals Risk Signatures and Immune Infiltration Patterns in High-Grade Serous Ovarian Cancer

 , ,
, ,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
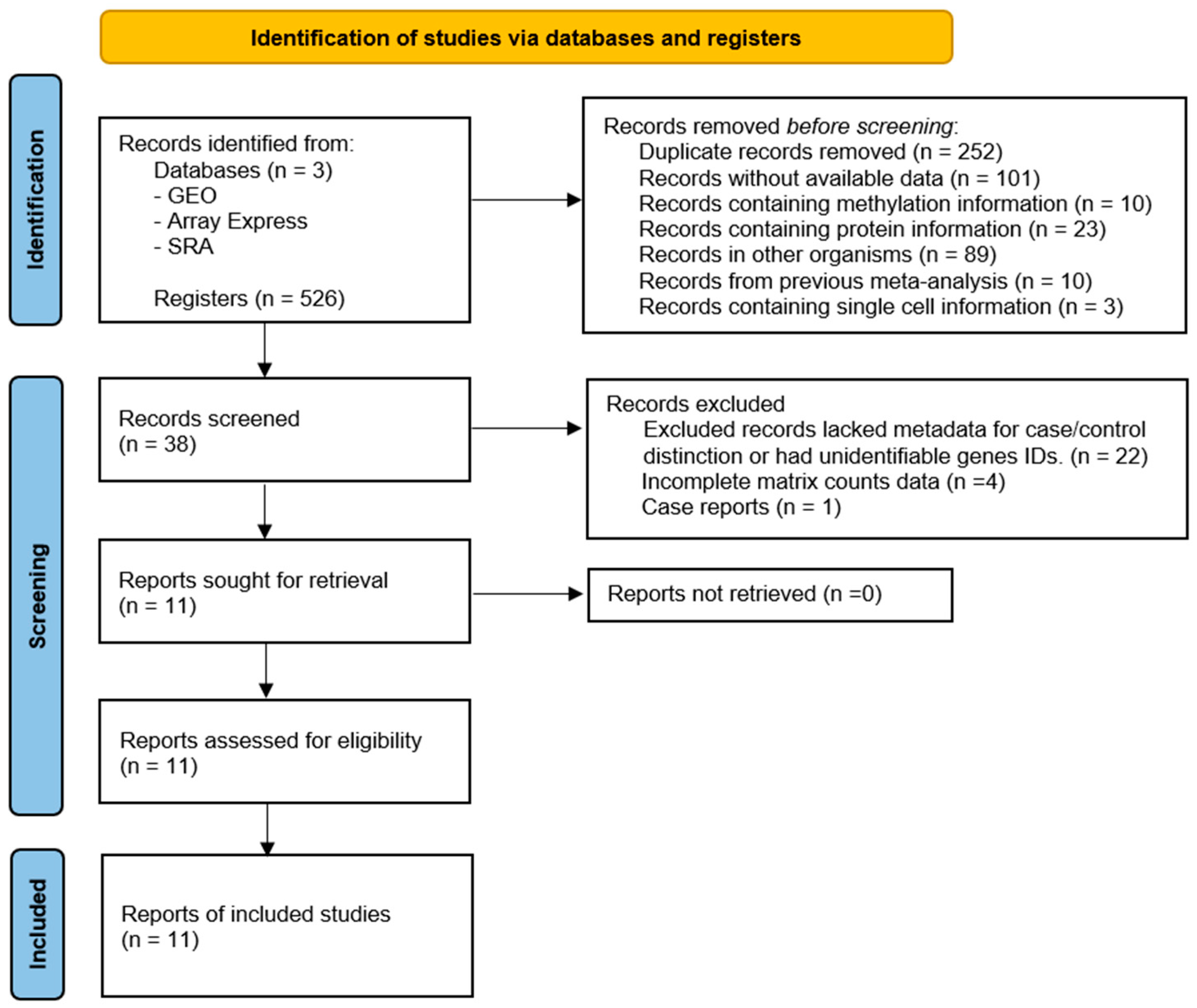
2.1. Search Strategy and Information Sources
2.2. Meta-Analysis of Differentially Expressed Genes
2.3. Functional Enrichment Analysis
2.4. Protein-Protein Interaction (PPI)
2.5. Cox Regression Analysis of DEGs
2.6. Survival Analysis
2.7. Estimation of Immune and Stromal Infiltration
2.8. Analysis of Immunotherapy-Associated Gene Expression
2.9. Convergence Analysis
2.10. Single Cell Analysis
3. Results
3.1. Selection of Gene Expression Datasets
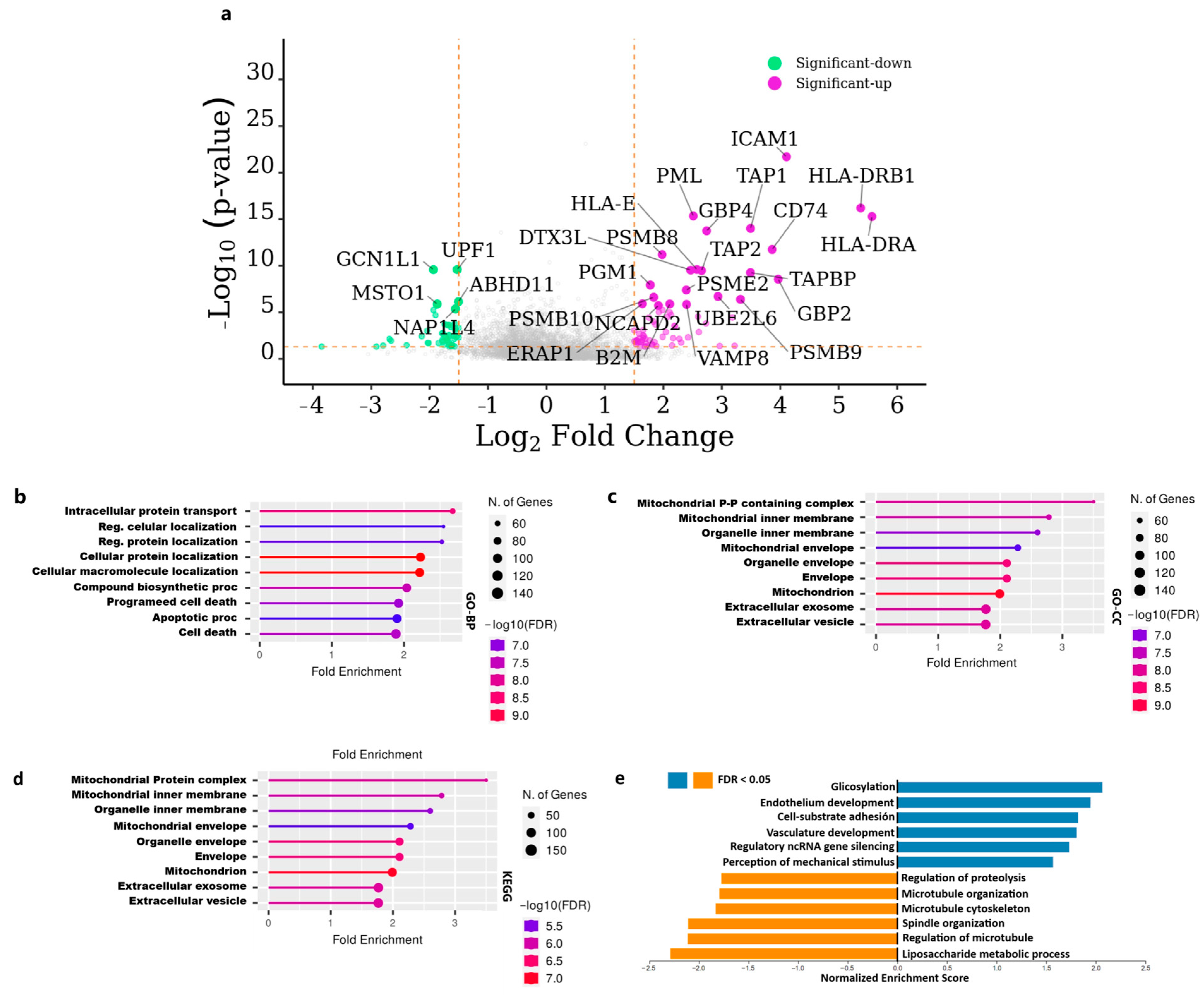
3.2. Functional Enrichment and Pathway Analysis of Differentially Expressed Genes
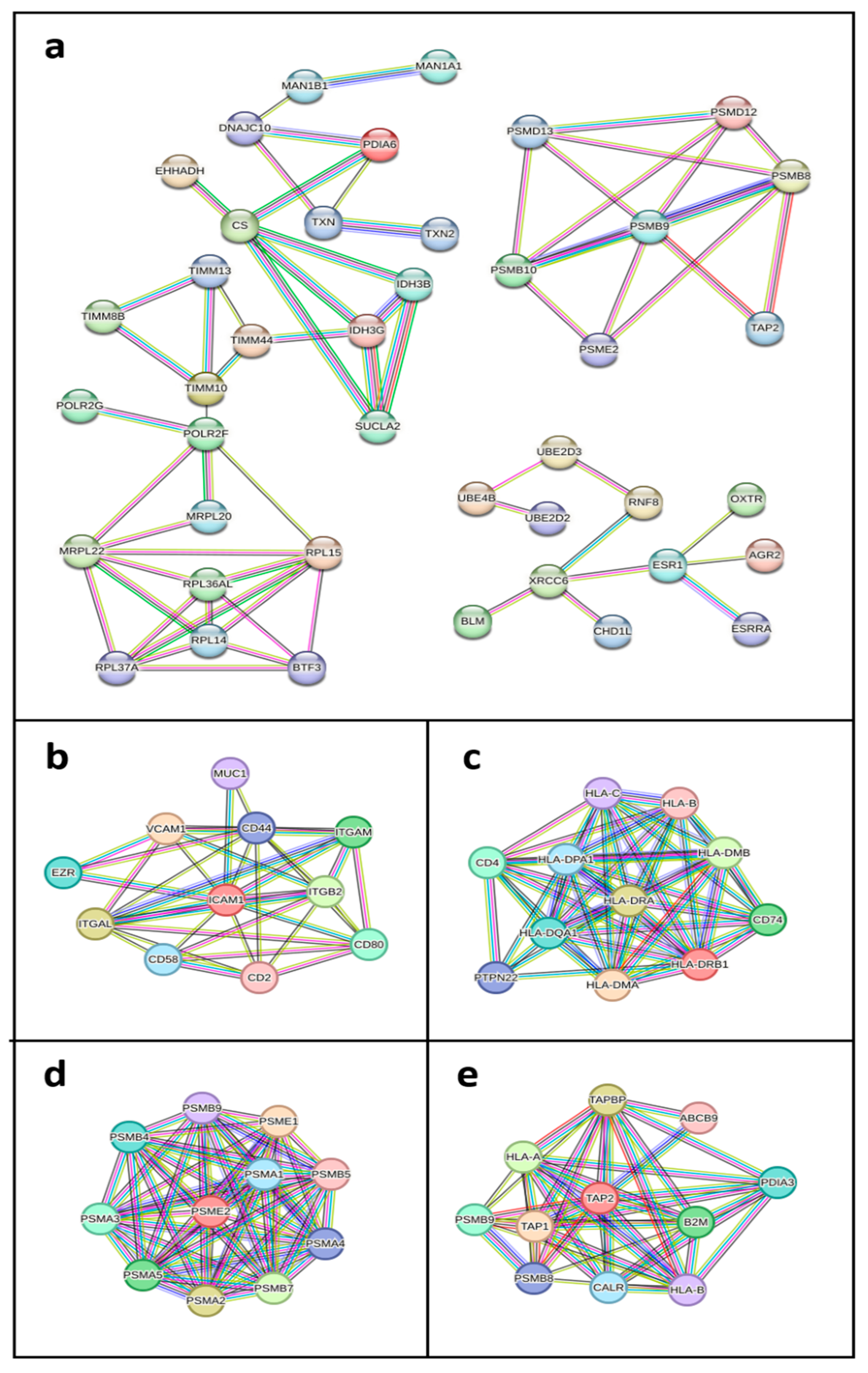
3.3. Protein-Protein Interaction Networks of Key Differentially Expressed Genes in HGSOC
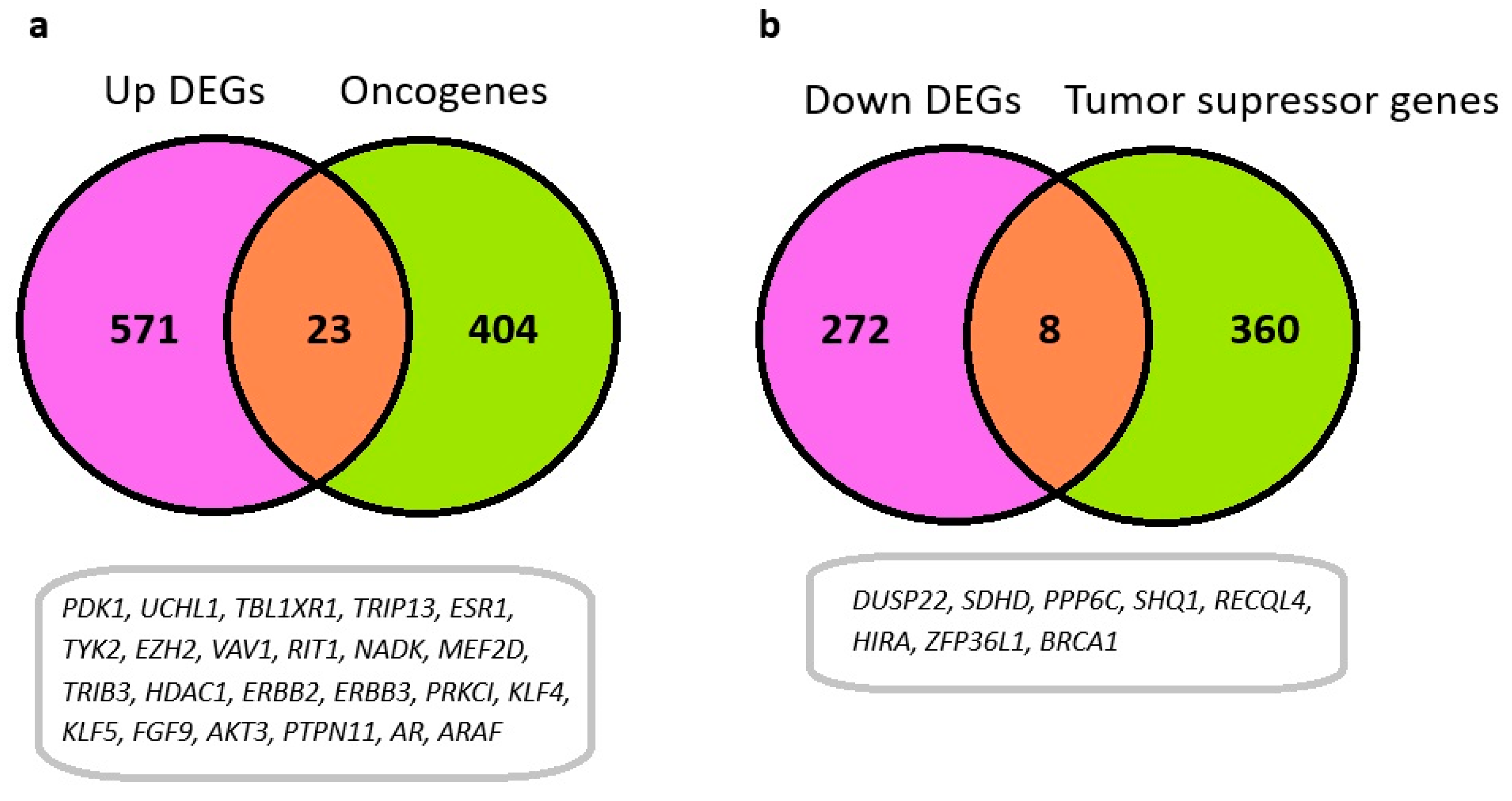
3.4. Identification of Oncogenes and Tumor Suppressor Genes Among Differentially Expressed Genes in HGSOC
3.5. Survival Analysis of Key Upregulated Genes and Risk Stratification in Ovarian Cancer
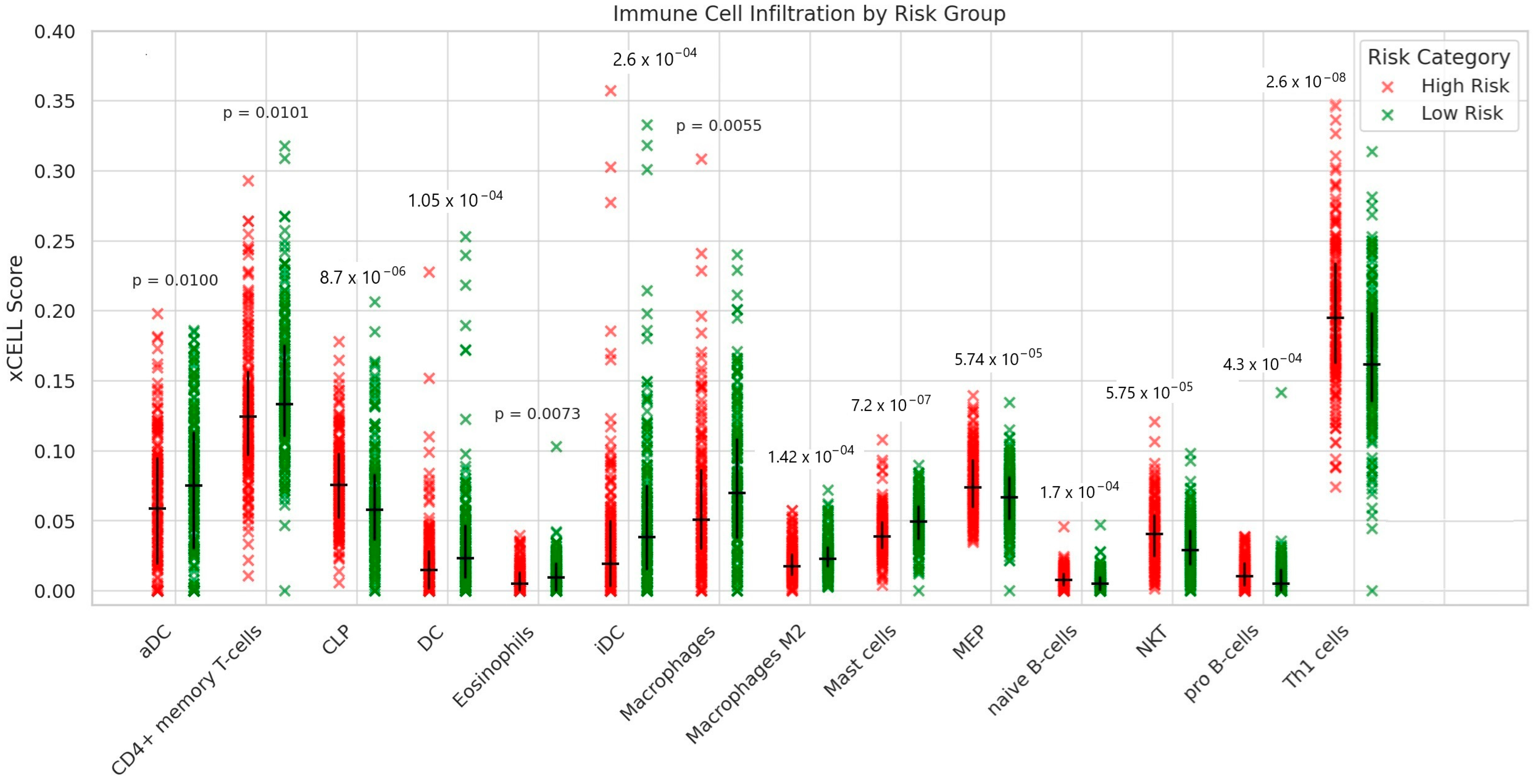
3.6. Estimation of Immune Cell Infiltration Between High- and Low-Risk Ovarian Cancer Patients
Differential Expression of Immunotherapy-Associated Genes
3.7. Single-Cell Analysis Reveals Cellular Expression Patterns of Survival Associated Genes and Dysregulation of Extracellular Matrix Pathways in HGSOC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HGSOC | High Grade Serous Ovarian Cancer |
| DEGs | Differentially Expressed Genes |
| GEO | Gene Expression Omnibus |
| NCBI | National Center of Biotechnology Information |
| SRA | Sequence Read Archive |
| RNA-seq | RNA Sequencing |
| PCA | Principal Component Analysis |
| UMAP | Uniform Manifold Approximation and Projection |
| CNVs | Copy Number Variations |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GSEA | Gene Set Enrichment Analysis |
| MHC | Major histocompatibility Complex |
| aDC | Activated Dendritic Cells |
| DC | Dendritic Cells |
| CLP | Common lymphoid progenitors |
| ECM | Extracellular Matrix |
| iDC | Immature Dendritic Cells |
| MEP | Megakaryocyte-erythroid Progenitors |
| NKT | Natural Killer T-Cells |
| FDR | False Discovery Rate |
| GO | Gene Ontology |
| BP | Biological Process |
| MF | Molecular Function |
| CC | Cellular Component |
| WebGestalt | WEB-based Gene SeT AnaLysis |
| PPI | Protein Protein Interaction |
| STRING | Search Tool for the Retrieval of Interacting Genes |
References
- Elsherif, S.B.; Bhosale, P.R.; Lall, C.; Menias, C.O.; Itani, M.; Butler, K.A.; Ganeshan, D. Current Update on Malignant Epithelial Ovarian Tumors. Abdom. Radiol. 2021, 46, 2264–2280. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial Ovarian Cancer: Evolution of Management in the Era of Precision Medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef]
- Govindarajan, M.; Wohlmuth, C.; Waas, M.; Bernardini, M.Q.; Kislinger, T. High-Throughput Approaches for Precision Medicine in High-Grade Serous Ovarian Cancer. J. Hematol. Oncol. 2020, 13, 134. [Google Scholar] [CrossRef]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A. Molecular Classification of Cancer: Class Discovery and Class Prediction by Gene Expression Monitoring. Science (1979) 1999, 286, 531–537. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. Review The Cancer Genome Atlas (TCGA): An Immeasurable Source of Knowledge. Contemp. Oncol./Współczesna Onkol. 2015, 2015, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Hollis, R.L.; Gourley, C. Genetic and Molecular Changes in Ovarian Cancer. Cancer Biol. Med. 2016, 13, 236. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Levanon, K.; Drapkin, R. Modeling High-Grade Serous Ovarian Carcinogenesis from the Fallopian Tube. Proc. Natl. Acad. Sci. USA 2011, 108, 7547–7552. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness Revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Spentzos, D.; Karlan, B.Y.; Taniguchi, T.; Fountzilas, E.; Francoeur, N.; Levine, D.A.; Cannistra, S.A. Gene Expression Profile of BRCA Ness That Correlates with Responsiveness to Chemotherapy and with Outcome in Patients with Epithelial Ovarian Cancer. J. Clin. Oncol. 2010, 28, 3555–3561. [Google Scholar] [CrossRef]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.; Shih, I. TP53 Mutations in Serous Tubal Intraepithelial Carcinoma and Concurrent Pelvic High-grade Serous Carcinoma—Evidence Supporting the Clonal Relationship of the Two Lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Fung-Leung, W.-P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and Microarray in Transcriptome Profiling of Activated T Cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, A.; Mondry, A.; Holmes, C.C.; Altman, D.G. Key Issues in Conducting a Meta-Analysis of Gene Expression Microarray Datasets. PLoS Med. 2008, 5, e184. [Google Scholar] [CrossRef]
- Shabalin, A.A.; Tjelmeland, H.; Fan, C.; Perou, C.M.; Nobel, A.B. Merging Two Gene-Expression Studies via Cross-Platform Normalization. Bioinformatics 2008, 24, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, D.A.; Moreno, Y.; Petro, J.L.; Forero, D.A.; Vargas-Molina, S.; Odriozola-Martínez, A.; Orozco, C.A.; Stout, J.R.; Rawson, E.S.; Kreider, R.B. A Bioinformatics-Assisted Review on Iron Metabolism and Immune System to Identify Potential Biomarkers of Exercise Stress-Induced Immunosuppression. Biomedicines 2022, 10, 724. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A Visual Analytics Platform for Comprehensive Gene Expression Profiling and Meta-Analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef]
- DerSimonian, R.; Laird, N. Meta-Analysis in Clinical Trials. Control Clin. Trials 1986, 7, 177–188. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M. Gene Set Knowledge Discovery with Enrichr. J. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- The Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res. 2021, 49, D325–D334. [CrossRef]
- Zhang, B.; Kirov, S.; Snoddy, J. WebGestalt: An Integrated System for Exploring Gene Sets in Various Biological Contexts. Nucleic Acids Res. 2005, 33, W741–W748. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New Approach for Understanding Genome Variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2. 0 for Analysis of Tumor-Infiltrating Immune Cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. XCell: Digitally Portraying the Tissue Cellular Heterogeneity Landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef]
- Oliveros, J.C. Venny (v2.1.0), An Interactive Tool for Comparing Lists with Venn’s Diagrams. BioinfoGP, CNB: Madrid, Spain, 2007. [Google Scholar]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Olbrecht, S.; Busschaert, P.; Qian, J.; Vanderstichele, A.; Loverix, L.; Van Gorp, T.; Van Nieuwenhuysen, E.; Han, S.; Van den Broeck, A.; Coosemans, A. High-Grade Serous Tubo-Ovarian Cancer Refined with Single-Cell RNA Sequencing: Specific Cell Subtypes Influence Survival and Determine Molecular Subtype Classification. Genome Med. 2021, 13, 1–30. [Google Scholar] [CrossRef]
- Tone, A.A.; Begley, H.; Sharma, M.; Murphy, J.; Rosen, B.; Brown, T.J.; Shaw, P.A. Gene Expression Profiles of Luteal Phase Fallopian Tube Epithelium from BRCA Mutation Carriers Resemble High-Grade Serous Carcinoma. Clin. Cancer Res. 2008, 14, 4067–4078. [Google Scholar] [CrossRef]
- Yoshihara, K.; Tajima, A.; Komata, D.; Yamamoto, T.; Kodama, S.; Fujiwara, H.; Suzuki, M.; Onishi, Y.; Hatae, M.; Sueyoshi, K. Gene Expression Profiling of Advanced-stage Serous Ovarian Cancers Distinguishes Novel Subclasses and Implicates ZEB2 in Tumor Progression and Prognosis. Cancer Sci. 2009, 100, 1421–1428. [Google Scholar] [CrossRef]
- Tung, C.S.; Mok, S.C.; Tsang, Y.; Zu, Z.; Song, H.; Liu, J.; Deavers, M.T.; Malpica, A.; Wolf, J.K.; Lu, K.H. PAX2 Expression in Low Malignant Potential Ovarian Tumors and Low-Grade Ovarian Serous Carcinomas. Mod. Pathol. 2009, 22, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Bowen, N.J.; Walker, L.D.; Matyunina, L.V.; Logani, S.; Totten, K.A.; Benigno, B.B.; McDonald, J.F. Gene Expression Profiling Supports the Hypothesis That Human Ovarian Surface Epithelia Are Multipotent and Capable of Serving as Ovarian Cancer Initiating Cells. BMC Med. Genom. 2009, 2, 71. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.C.; Bonome, T.; Vathipadiekal, V.; Bell, A.; Johnson, M.E.; Park, D.-C.; Hao, K.; Yip, D.K.P.; Donninger, H.; Ozbun, L. A Gene Signature Predictive for Outcome in Advanced Ovarian Cancer Identifies a Survival Factor: Microfibril-Associated Glycoprotein 2. Cancer Cell 2009, 16, 521–532. [Google Scholar] [CrossRef]
- Shahab, S.W.; Matyunina, L.V.; Mezencev, R.; Walker, L.D.; Bowen, N.J.; Benigno, B.B.; McDonald, J.F. Evidence for the Complexity of MicroRNA-Mediated Regulation in Ovarian Cancer: A Systems Approach. PLoS ONE 2011, 6, e22508. [Google Scholar] [CrossRef]
- Bonome, T.; Levine, D.A.; Shih, J.; Randonovich, M.; Pise-Masison, C.A.; Bogomolniy, F.; Ozbun, L.; Brady, J.; Barrett, J.C.; Boyd, J. A Gene Signature Predicting for Survival in Suboptimally Debulked Patients with Ovarian Cancer. Cancer Res. 2008, 68, 5478–5486. [Google Scholar] [CrossRef] [PubMed]
- King, E.R.; Tung, C.S.; Tsang, Y.T.M.; Zu, Z.; Lok, G.T.M.; Deavers, M.T.; Malpica, A.; Wolf, J.K.; Lu, K.H.; Birrer, M.J. The Anterior Gradient Homolog 3 (AGR3) Gene Is Associated with Differentiation and Survival in Ovarian Cancer. Am. J. Surg. Pathol. 2011, 35, 904–912. [Google Scholar] [CrossRef]
- Elgaaen, B.V.; Olstad, O.K.; Sandvik, L.; Ødegaard, E.; Sauer, T.; Staff, A.C.; Gautvik, K.M. ZNF385B and VEGFA Are Strongly Differentially Expressed in Serous Ovarian Carcinomas and Correlate with Survival. PLoS ONE 2012, e46317. [Google Scholar] [CrossRef]
- Yeung, T.-L.; Leung, C.S.; Wong, K.-K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. TGF-β Modulates Ovarian Cancer Invasion by Upregulating CAF-Derived Versican in the Tumor Microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef]
- Wu, R.; Hendrix-Lucas, N.; Kuick, R.; Zhai, Y.; Schwartz, D.R.; Akyol, A.; Hanash, S.; Misek, D.E.; Katabuchi, H.; Williams, B.O. Mouse Model of Human Ovarian Endometrioid Adenocarcinoma Based on Somatic Defects in the Wnt/β-Catenin and PI3K/Pten Signaling Pathways. Cancer Cell 2007, 11, 321–333. [Google Scholar] [CrossRef]
- Shi, J.; Yang, Y.; Chen, F.; Zhou, L.; Wei, H.; Dong, F.; Wang, X.; Shan, Y.; Chen, T. RPL36A Activates ERK Pathway and Promotes Colorectal Cancer Growth. Transl. Oncol. 2025, 51, 102170. [Google Scholar] [CrossRef]
- Wang, M.; He, T.; Meng, D.; Lv, W.; Ye, J.; Cheng, L.; Hu, J. BZW2 Modulates Lung Adenocarcinoma Progression through Glycolysis-Mediated IDH3G Lactylation Modification. J. Proteome Res. 2023, 22, 3854–3865. [Google Scholar] [CrossRef] [PubMed]
- Spentzos, D.; Levine, D.A.; Ramoni, M.F.; Joseph, M.; Gu, X.; Boyd, J.; Libermann, T.A.; Cannistra, S.A. Gene Expression Signature with Independent Prognostic Significance in Epithelial Ovarian Cancer. J. Clin. Oncol. 2004, 22, 4700–4710. [Google Scholar] [CrossRef]
- Denkert, C.; Budczies, J.; Darb-Esfahani, S.; Györffy, B.; Sehouli, J.; Könsgen, D.; Zeillinger, R.; Weichert, W.; Noske, A.; Buckendahl, A. A Prognostic Gene Expression Index in Ovarian Cancer—Validation across Different Independent Data Sets. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2009, 218, 273–280. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, S.-M.; Jin, Y.-L.; Xiong, G.-W.; Wang, P.; Snijders, A.M.; Mao, J.-H.; Zhang, X.-W.; Hang, B. A Robust Gene Expression Prognostic Signature for Overall Survival in High-Grade Serous Ovarian Cancer. J. Oncol. 2019, 2019, 3614207. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, J.; Simon, R. Gene Expression–Based Prognostic Signatures in Lung Cancer: Ready for Clinical Use? J. Natl. Cancer Inst. 2010, 102, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Millstein, J.; Budden, T.; Goode, E.L.; Anglesio, M.S.; Talhouk, A.; Intermaggio, M.P.; Leong, H.S.; Chen, S.; Elatre, W.; Gilks, B. Prognostic Gene Expression Signature for High-Grade Serous Ovarian Cancer. Ann. Oncol. 2020, 31, 1240–1250. [Google Scholar] [CrossRef]
- Jing, J.; Wang, B.; Liu, P. The Functional Role of SEC23 in Vesicle Transportation, Autophagy and Cancer. Int. J. Biol. Sci. 2019, 15, 2419. [Google Scholar] [CrossRef]
- Gerashchenko, T.; Skitchenko, R.; Korobeynikova, A.; Kuanysheva, K.; Khozyainova, A.; Vorobiev, R.; Rodionov, E.; Miller, S.; Topolnitsky, E.; Shefer, N. Whole-Exome Sequencing Reveals an Association of Rs112065068 in TGOLN2 Gene with Distant Metastasis of Non-Small Cell Lung Cancer. Gene 2024, 920, 148507. [Google Scholar] [CrossRef]
- Zhang, F.; Ren, G.; Lu, Y.; Jin, B.; Wang, J.; Chen, X.; Liu, Z.; Li, K.; Nie, Y.; Wang, X. Identification of TRAK1 (Trafficking Protein, Kinesin-Binding 1) as MGb2-Ag: A Novel Cancer Biomarker. Cancer Lett. 2009, 274, 250–258. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial Metabolism and Cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Yarden, Y. Endocytosis and Cancer. Cold Spring Harb. Perspect. Biol. 2013, 5, a016949. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, J.S.; Skelding, K.A. The Multi-Functional Calcium/Calmodulin Stimulated Protein Kinase (CaMK) Family: Emerging Targets for Anti-Cancer Therapeutic Intervention. Pharmaceuticals 2019, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian Cancer. Nat. Rev. Dis. Primers 2016, 2, 16061. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Knapp, S.; Sood, A.K.; Bast, R.C. Targeting Therapies in Cancer: Opportunities in Ovarian Cancer. In Gynaecological Cancers: Biology and Therapeutics; Cambridge University Press: Cambridge, UK, 2011; pp. 167–182. [Google Scholar]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B. Novel Molecular Subtypes of Serous and Endometrioid Ovarian Cancer Linked to Clinical Outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef]
- Onodera, Y.; Nam, J.-M.; Horikawa, M.; Shirato, H.; Sabe, H. Arf6-Driven Cell Invasion Is Intrinsically Linked to TRAK1-Mediated Mitochondrial Anterograde Trafficking to Avoid Oxidative Catastrophe. Nat. Commun. 2018, 9, 2682. [Google Scholar] [CrossRef]
- Yang, Q.; Jiang, W.; Hou, P. Emerging Role of PI3K/AKT in Tumor-Related Epigenetic Regulation. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2019; Volume 59, pp. 112–124. [Google Scholar]
- Sparano, J.A.; Gray, R.J.; Makower, D.F.; Pritchard, K.I.; Albain, K.S.; Hayes, D.F.; Geyer, C.E., Jr.; Dees, E.C.; Goetz, M.P.; Olson, J.A., Jr. Adjuvant Chemotherapy Guided by a 21-Gene Expression Assay in Breast Cancer. N. Engl. J. Med. 2018, 379, 111–121. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M. Specific Recruitment of Regulatory T Cells in Ovarian Carcinoma Fosters Immune Privilege and Predicts Reduced Survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Hwang, W.-T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic Significance of Tumor-Infiltrating T Cells in Ovarian Cancer: A Meta-Analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef]
- Kurozumi, S.; Fujii, T.; Matsumoto, H.; Inoue, K.; Kurosumi, M.; Horiguchi, J.; Kuwano, H. Significance of Evaluating Tumor-Infiltrating Lymphocytes (TILs) and Programmed Cell Death-Ligand 1 (PD-L1) Expression in Breast Cancer. Med. Mol. Morphol. 2017, 50, 185–194. [Google Scholar] [CrossRef]
- Jacquelot, N.; Seillet, C.; Vivier, E.; Belz, G.T. Innate Lymphoid Cells and Cancer. Nat. Immunol. 2022, 23, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wan, G.; Wang, Y.; Chen, W.; Guan, J.-Z. The Role of Erythrocytes and Erythroid Progenitor Cells in Tumors. Open Life Sci. 2022, 17, 1641–1656. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A.R. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author/Year | NCBI GEO | Cases (n = 291) | Controls (n = 96) | Platform | PMID |
|---|---|---|---|---|---|
| Tone AA, et al., 2008 | GSE10971 | 13 | 24 | Affymetrix Human Genome U133 Plus 2.0 | 18593983 [30] |
| Yoshihara K, et al., 2009 | GSE12470 | 43 | 10 | Agilent-012097 Human 1A Microarray (V2) | 19486012 [31] |
| Tung CS et al., 2009 | GSE14001 | 10 | 3 | Affymetrix Human Genome U133 Plus 2.0 | 19525924 [32] |
| Bowen NJ et al., 2009 | GSE14407 | 12 | 12 | Affymetrix Human Genome U133 Plus 2.0 | 20040092 [33] |
| Mok SC, et al., 2009 | GSE18520 | 53 | 10 | Affymetrix Human Genome U133 Plus 2.0 | 19962670 [34] |
| Shahab S, et al., 2011 | GSE23391 | 3 | 5 | Affymetrix U133 Plus 2.0 3’ expression | 21811625 [35] |
| Bonome T, et al., 2008 | GSE26712 | 107 | 10 | Affymetrix Human Genome U133A | 18593951 [36] |
| King ER, et al., 2011 | GSE27651 | 22 | 6 | Affymetrix Human Genome U133 Plus 2.0 | 21451362 [37] |
| Elgaaen BV, et al., 2012 | GSE36668 | 8 | 4 | Affymetrix Human Genome U133 Plus | 23029477 [38] |
| Yeung TL, et al., 2013 | GSE40595 | 32 | 8 | Affymetrix human genome U133 Plus 2.0 | 23824740 [39] |
| Wu R et al., 2007 | GSE6008 | 41 | 4 | Affymetrix HG_U133A array | 23824740 [40] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Suárez, P.D.; Zambrano-O, Y.T.; Mejía-Garcia, A.; Tsao, H.M.; Lopez-Kleine, L.; Bonilla, D.A.; Combita, A.L.; Parra-Medina, R.; Lopez-Correa, P.; Serrano-G, S.J.; et al. Integrative Transcriptomic Meta-Analysis Reveals Risk Signatures and Immune Infiltration Patterns in High-Grade Serous Ovarian Cancer. Immuno 2025, 5, 23. https://doi.org/10.3390/immuno5030023
Morales-Suárez PD, Zambrano-O YT, Mejía-Garcia A, Tsao HM, Lopez-Kleine L, Bonilla DA, Combita AL, Parra-Medina R, Lopez-Correa P, Serrano-G SJ, et al. Integrative Transcriptomic Meta-Analysis Reveals Risk Signatures and Immune Infiltration Patterns in High-Grade Serous Ovarian Cancer. Immuno. 2025; 5(3):23. https://doi.org/10.3390/immuno5030023
Chicago/Turabian StyleMorales-Suárez, Paula D., Yina T. Zambrano-O, Alejandro Mejía-Garcia, Hsuan Megan Tsao, Liliana Lopez-Kleine, Diego A. Bonilla, Alba L. Combita, Rafel Parra-Medina, Patricia Lopez-Correa, Silvia J. Serrano-G, and et al. 2025. "Integrative Transcriptomic Meta-Analysis Reveals Risk Signatures and Immune Infiltration Patterns in High-Grade Serous Ovarian Cancer" Immuno 5, no. 3: 23. https://doi.org/10.3390/immuno5030023
APA StyleMorales-Suárez, P. D., Zambrano-O, Y. T., Mejía-Garcia, A., Tsao, H. M., Lopez-Kleine, L., Bonilla, D. A., Combita, A. L., Parra-Medina, R., Lopez-Correa, P., Serrano-G, S. J., Rodriguez, J. L., & Orozco, C. A. (2025). Integrative Transcriptomic Meta-Analysis Reveals Risk Signatures and Immune Infiltration Patterns in High-Grade Serous Ovarian Cancer. Immuno, 5(3), 23. https://doi.org/10.3390/immuno5030023