The Role of Genetics in Risk Stratification of Thoracic Aortic Aneurysm Dissection

 , , ,
, , ,
Abstract
1. Introduction—Thoracic Aortic Aneurysms
2. Current Genetic Risk Stratification Strategies
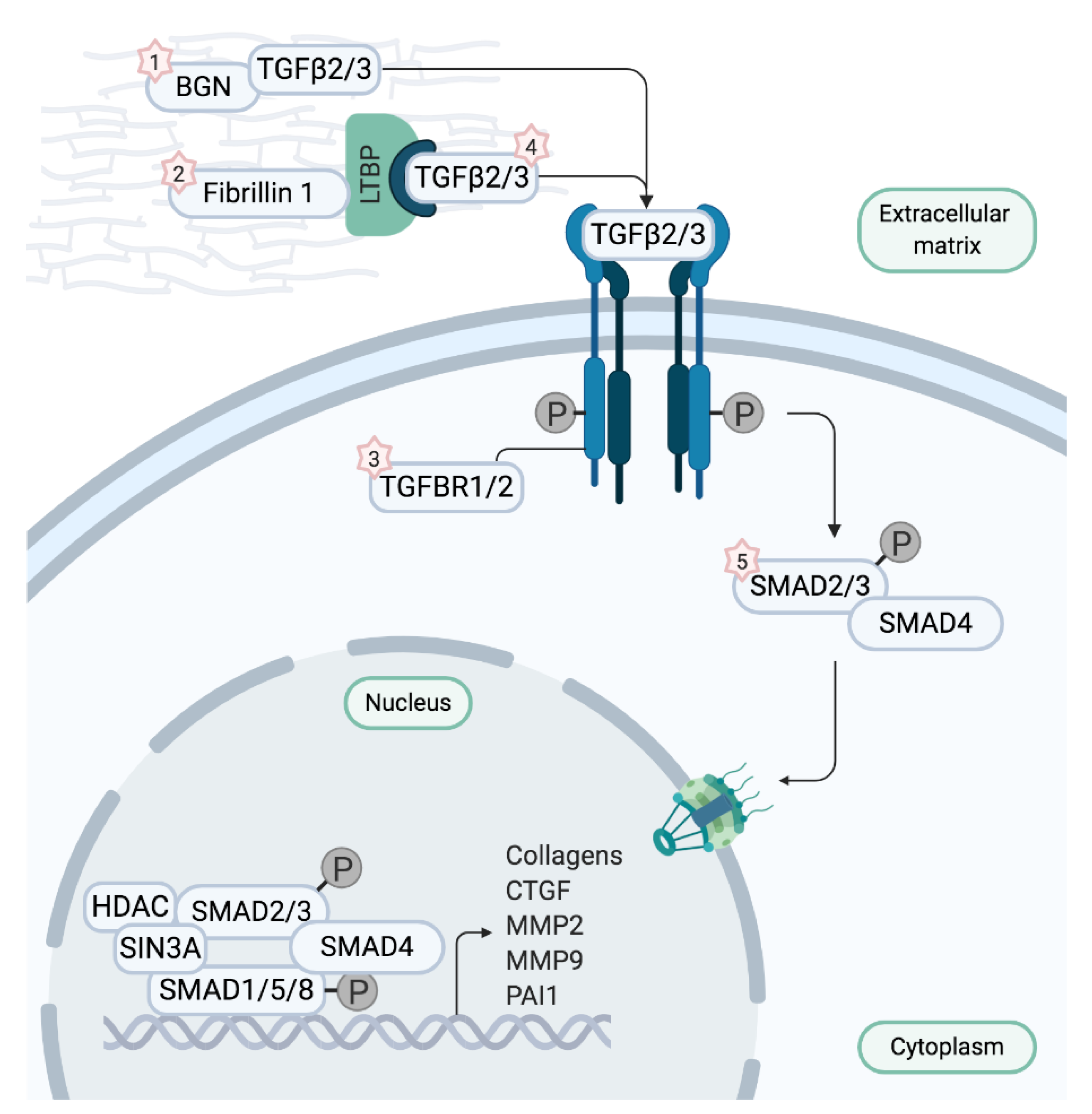
2.1. Stratification by Affected Genes: Marfan Syndrome and Loeys–Dietz Syndrome
2.2. Stratification by Gender: Meester-Loeys Syndrome and Bicuspid Valve Associated TAA
2.3. Stratification by Variant Type: Vascular Ehlers–Danlos Syndrome, Haploinsufficiency and Dominant Negative Mutations
2.3.1. Effect of Variant Types on the Presentation of Vascular Ehlers–Danlos Syndrome
2.3.2. Haploinsufficiency and Dominant-Negative FBN1 Mutations
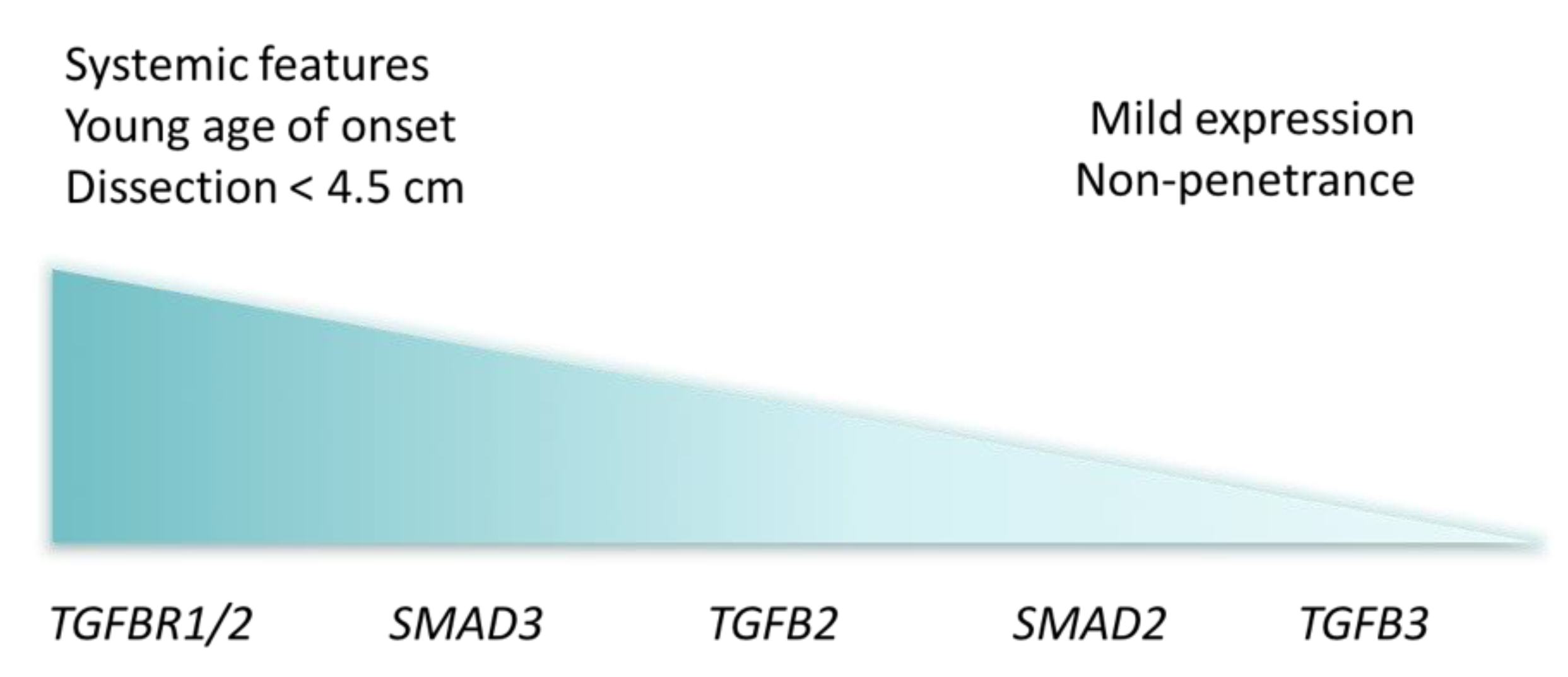
2.4. Stratification by Variant Location: From Low-Penetrant Familial Thoracic Aortic Aneurysms to Severe Early Onset Syndrome
3. Genetic Modifiers
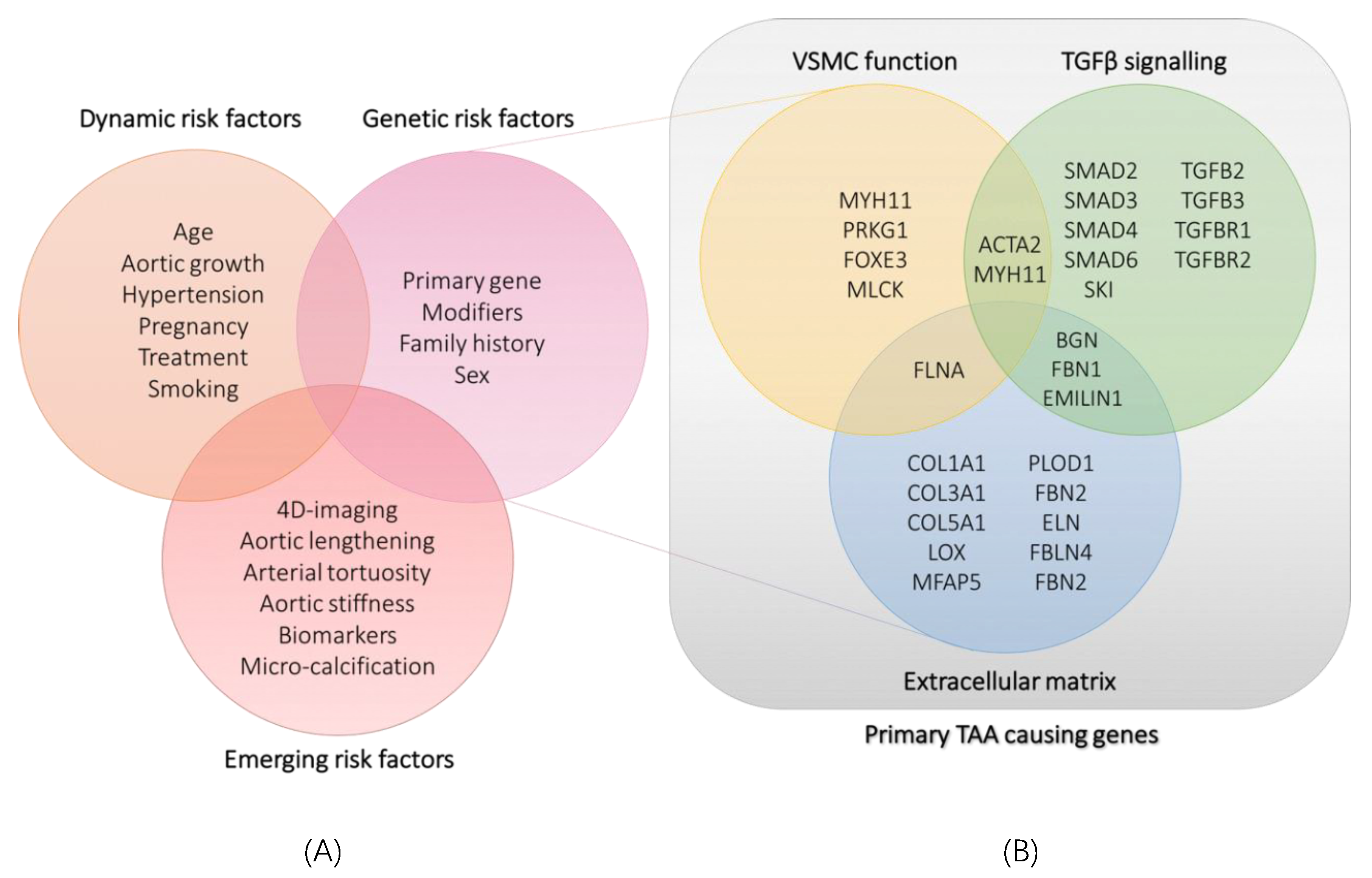
4. Emerging Non-Genetic Risk Determinants
5. Towards Gene-Tailored Management
Author Contributions
Funding
Conflicts of Interest
References
- Verstraeten, A.; Luyckx, I.; Loeys, B. Aetiology and management of hereditary aortopathy. Nat. Rev. Cardiol. 2017, 14, 197–208. [Google Scholar] [CrossRef]
- Lindsay, M.E.; Dietz, H.C. The genetic basis of aortic aneurysm. Cold Spring Harb. Perspect. Med. 2014, 4, a015909. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Regalado, E. Heritable Thoracic Aortic Disease Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E., Jr.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010, 121, e266–e369. [Google Scholar]
- Pape, L.A.; Tsai, T.T.; Isselbacher, E.M.; Oh, J.K.; O’Gara, P.T.; Evangelista, A.; Fattori, R.; Meinhardt, G.; Trimarchi, S.; Bossone, E.; et al. Aortic diameter >= 5.5 cm is not a good predictor of type A aortic dissection - Observations from the international registry of acute aortic dissection (IRAD). Circulation 2007, 116, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- MacCarrick, G.; Black, J.H.; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C. Loeys-Dietz syndrome: A primer for diagnosis and management. Genet. Med. 2014, 16, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Pepin, M.G.; Schwarze, U.; Rice, K.M.; Liu, M.; Leistritz, D.; Byers, P.H. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet. Med. 2014, 16, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Judge, D.P.; Dietz, H.C. Marfan’s syndrome. Lancet 2005, 366, 1965–1976. [Google Scholar]
- Van Laer, L.; Dietz, H.; Loeys, B. Loeys-Dietz syndrome. Adv. Exp. Med. Biol. 2014, 802, 95–105. [Google Scholar]
- Waters, K.M.; Rooper, L.M.; Guajardo, A.; Halushka, M.K. Histopathologic differences partially distinguish syndromic aortic diseases. Cardiovasc. Pathol. 2017, 30, 6–11. [Google Scholar]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef]
- Loeys, B.L.; Dietz, H.C. Loeys-Dietz Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Hostetler, E.M.; Regalado, E.S.; Guo, D.C.; Hanna, N.; Arnaud, P.; Muino-Mosquera, L.; Callewaert, B.L.; Lee, K.; Leal, S.M.; Wallace, S.E.; et al. SMAD3 pathogenic variants: Risk for thoracic aortic disease and associated complications from the Montalcino Aortic Consortium. J. Med. Genet. 2019, 56, 252–260. [Google Scholar] [CrossRef] [PubMed]
- van de Laar, I.M.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.A.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Bertoli-Avella, A.M.; Gillis, E.; Morisaki, H.; Verhagen, J.M.A.; de Graaf, B.M.; van de Beek, G.; Gallo, E.; Kruithof, B.P.T.; Venselaar, H.; Myers, L.A.; et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 2015, 65, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I.; et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634. [Google Scholar] [CrossRef]
- Micha, D.; Guo, D.C.; Hilhorst-Hofstee, Y.; van Kooten, F.; Atmaja, D.; . Overwater, E.; Cayami, F.K.; Regalado, E.S.; van Uffelen, R.; Venselaar, H.; et al. SMAD2 Mutations Are Associated with Arterial Aneurysms and Dissections. Hum. Mutat. 2015, 36, 1145–1149. [Google Scholar] [CrossRef]
- Marsili, L.; Overwater, E.; Hanna, N.; Baujat, G.; Baars, M.J.H.; Boileau, C.; Bonneau, D.; Brehin, A.C.; Capri, Y.; Cheung, H.Y.; et al. Phenotypic spectrum of TGFB3 disease-causing variants in a Dutch-French cohort and first report of a homozygous patient. Clin. Genet. 2020, 97, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Muhlstadt, K.; De Backer, J.; von Kodolitsch, Y.; Kutsche, K.; Muino Mosquera, L.; Brickwedel, J.; Girdauskas, E.; Mir, T.S.; Mahlmann, A.; Tsilimparis, N.; et al. Case-matched Comparison of Cardiovascular Outcome in Loeys-Dietz Syndrome versus Marfan Syndrome. J. Clin. Med. 2019, 8, 2079. [Google Scholar]
- Meester, J.A.; Vandeweyer, G.; Pintelon, I.; Lammens, M.; Van Hoorick, L.; De Belder, S.; Waitzman, K.; Young, L.; Markham, L.W.; Vogt, J.; et al. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet. Med. 2017, 19, 386–395. [Google Scholar] [CrossRef]
- Rodriguez-Palomares, J.F.; Dux-Santoy, L.; Guala, A.; Kale, R.; Maldonado, G.; Teixido-Tura, G.; Galian, L.; Huguet, M.; Valente, F.; Gutierrez, L.; et al. Aortic flow patterns and wall shear stress maps by 4D-flow cardiovascular magnetic resonance in the assessment of aortic dilatation in bicuspid aortic valve disease. J. Cardiovasc. Magn. Reson. 2018, 20, 28. [Google Scholar] [CrossRef]
- Hope, M.D.; Sigovan, M.; Wrenn, S.J.; Saloner, D.; Dyverfeldt, P. MRI hemodynamic markers of progressive bicuspid aortic valve-related aortic disease. J. Magn. Reson. Imaging 2014, 40, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Guzzardi, D.G.; Barker, A.J.; van Ooij, P.; Malaisrie, S.C.; Puthumana, J.J.; Belke, D.D.; Mewhort, H.E.; Svystonyuk, D.A.; Kang, S.; Verma, S.; et al. Valve-Related Hemodynamics Mediate Human Bicuspid Aortopathy: Insights From Wall Shear Stress Mapping. J. Am. Coll. Cardiol. 2015, 66, 892–900. [Google Scholar]
- Lee, J.; Shen, M.; Parajuli, N.; Oudit, G.Y.; McMurtry, M.S.; Kassiri, Z. Gender-dependent aortic remodelling in patients with bicuspid aortic valve-associated thoracic aortic aneurysm. J. Mol. Med. 2014, 92, 939–949. [Google Scholar] [PubMed]
- Kong, W.K.; Regeer, M.V.; Ng, A.C.; McCormack, L.; Poh, K.K.; Yeo, T.C.; Shanks, M.; Parent, S.; Enache, R.; Popescu, B.A.; et al. Sex Differences in Phenotypes of Bicuspid Aortic Valve and Aortopathy: Insights From a Large Multicenter, International Registry. Circ. Cardiovasc. Imaging 2017, 10, 2079. [Google Scholar]
- Kuivaniemi, H.; Ryer, E.J.; Elmore, J.R.; Hinterseher, I.; Smelser, D.T.; Tromp, G. Update on abdominal aortic aneurysm research: From clinical to genetic studies. Scientifica 2014, 2014, 564734. [Google Scholar]
- Steckmeier, B. Epidemiology of aortic disease: Aneurysm, dissection, occlusion. Radiologe 2001, 41, 624–632. [Google Scholar] [CrossRef]
- Carlson, M.; Silberbach, M. Dissection of the aorta in Turner syndrome: Two cases and review of 85 cases in the literature. BMJ Case Rep. 2009, 2009, bcr0620091998. [Google Scholar] [CrossRef]
- Frank, M.; Albuisson, J.; Ranque, B.; Golmard, L.; Mazzella, J.M.; Bal-Theoleyre, L.; Fauret, A.L.; Mirault, T.; Denarie, N.; Mousseaux, E.; et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur. J. Hum. Genet. 2015, 23, 1657–1664. [Google Scholar] [CrossRef]
- Kuivaniemi, H.; Tromp, G. Type III collagen (COL3A1): Gene and protein structure, tissue distribution, and associated diseases. Gene 2019, 707, 151–171. [Google Scholar] [CrossRef]
- Franken, R.; den Hartog, A.W.; Radonic, T.; Micha, D.; Maugeri, A.; van Dijk, F.S.; Meijers-Heijboer, H.E.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; et al. Beneficial Outcome of Losartan Therapy Depends on Type of FBN1 Mutation in Marfan Syndrome. Circ. Cardiovasc. Genet. 2015, 8, 383–388. [Google Scholar] [CrossRef]
- Byers, P.H. Determination of the molecular basis of Marfan syndrome: A growth industry. J. Clin. Investig. 2004, 114, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Franken, R.; Groenink, M.; de Waard, V.; Feenstra, H.M.; Scholte, A.J.; van den Berg, M.P.; Pals, G.; Zwinderman, A.H.; Timmermans, J.; Mulder, B.J. Genotype impacts survival in Marfan syndrome. Eur. Heart J. 2016, 37, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Franken, R.; Teixido-Tura, G.; Brion, M.; Forteza, A.; Rodriguez-Palomares, J.; Gutierrez, L.; Garcia Dorado, D.; Pals, G.; Mulder, B.J.; Evangelista, A. Relationship between fibrillin-1 genotype and severity of cardiovascular involvement in Marfan syndrome. Heart 2017, 103, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- den Hartog, A.W.; Franken, R.; van den Berg, M.P.; Zwinderman, A.H.; Timmermans, J.; Scholte, A.J.; de Waard, V.; Spijkerboer, A.M.; Pals, G.; Mulder, B.J.; et al. The effect of losartan therapy on ventricular function in Marfan patients with haploinsufficient or dominant negative FBN1 mutations. Neth. Heart J. 2016, 24, 675–681. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guo, D.C.; Papke, C.L.; Tran-Fadulu, V.; Regalado, E.S.; Avidan, N.; Johnson, R.J.; Kim, D.H.; Pannu, H.; Willing, M.C.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 2009, 84, 617–627. [Google Scholar] [CrossRef]
- van de Laar, I.; Arbustini, E.; Loeys, B.; Bjorck, E.; Murphy, L.; Groenink, M.; Kempers, M.; Timmermans, J.; Roos-Hesselink, J.; Benke, K.; et al. European reference network for rare vascular diseases (VASCERN) consensus statement for the screening and management of patients with pathogenic ACTA2 variants. Orphanet J. Rare Dis. 2019, 14, 264. [Google Scholar] [CrossRef]
- Regalado, E.S.; Mellor-Crummey, L.; De Backer, J.; Braverman, A.C.; Ades, L.; Benedict, S.; Bradley, T.J.; Brickner, M.E.; Chatfield, K.C.; Child, A.; et al. Clinical history and management recommendations of the smooth muscle dysfunction syndrome due to ACTA2 arginine 179 alterations. Genet. Med. 2018, 20, 1206–1215. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Ostergaard, J.R.; Ala-Kokko, L.M.; Khan, N.; Grange, D.K.; Mendoza-Londono, R.; Bradley, T.J.; Olney, A.H.; Ades, L.; Maher, J.F.; et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am. J. Med. Genet. A 2010, 152, 2437–2443. [Google Scholar] [CrossRef]
- Georgescu, M.M.; Pinho Mda, C.; Richardson, T.E.; Torrealba, J.; Buja, L.M.; Milewicz, D.M.; Raisanen, J.M.; Burns, D.K. The defining pathology of the new clinical and histopathologic entity ACTA2-related cerebrovascular disease. Acta Neuropathol. Commun. 2015, 3, 81. [Google Scholar] [CrossRef]
- Drumm, M.L.; Konstan, M.W.; Schluchter, M.D.; Handler, A.; Pace, R.; Zou, F.; Zariwala, M.; Fargo, D.; Xu, A.; Dunn, J.M.; et al. Genetic modifiers of lung disease in cystic fibrosis. N. Engl. J. Med. 2005, 353, 1443–1453. [Google Scholar] [CrossRef]
- Aubart, M.; Gazal, S.; Arnaud, P.; Benarroch, L.; Gross, M.S.; Buratti, J.; Boland, A.; Meyer, V.; Zouali, H.; Hanna, N.; et al. Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. Eur. J. Hum. Genet. 2018, 26, 1759–1772. [Google Scholar] [CrossRef] [PubMed]
- Landis, B.J.; Schubert, J.A.; Lai, D.; Jegga, A.G.; Shikany, A.R.; Foroud, T.; Ware, S.M.; Hinton, R.B. Exome Sequencing Identifies Candidate Genetic Modifiers of Syndromic and Familial Thoracic Aortic Aneurysm Severity. J. Cardiovasc. Transl. Res. 2017, 10, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Emerel, L.; Thunes, J.; Kickliter, T.; Billaud, M.; Phillippi, J.A.; Vorp, D.A.; Maiti, S.; Gleason, T.G. Predissection-derived geometric and distensibility indices reveal increased peak longitudinal stress and stiffness in patients sustaining acute type A aortic dissection: Implications for predicting dissection. J. Thorac. Cardiovasc. Surg. 2019, 158, 355–363. [Google Scholar] [CrossRef]
- Selamet Tierney, E.S.; Levine, J.C.; Sleeper, L.A.; Roman, M.J.; Bradley, T.J.; Colan, S.D.; Chen, S.; Campbell, M.J.; Cohen, M.S.; De Backer, J.; et al. Influence of Aortic Stiffness on Aortic-Root Growth Rate and Outcome in Patients With the Marfan Syndrome. Am. J. Cardiol. 2018, 121, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Franken, R.; El Morabit, A.; de Waard, V.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; Marquering, H.; Planken, N.R.; Zwinderman, A.H.; Mulder, B.J.; et al. Increased aortic tortuosity indicates a more severe aortic phenotype in adults with Marfan syndrome. Int. J. Cardiol. 2015, 194, 7–12. [Google Scholar] [CrossRef]
- Heuts, S.; Adriaans, B.P.; Gerretsen, S.; Natour, E.; Vos, R.; Cheriex, E.C.; Crijns, H.; Wildberger, J.E.; Maessen, J.G.; Schalla, S.; et al. Aortic elongation part II: The risk of acute type A aortic dissection. Heart 2018, 104, 1778–1782. [Google Scholar] [CrossRef]
- Mahadevia, R.; Barker, A.J.; Schnell, S.; Entezari, P.; Kansal, P.; Fedak, P.W.; Malaisrie, S.C.; McCarthy, P.; Collins, J.; Carr, J.; et al. Bicuspid aortic cusp fusion morphology alters aortic three-dimensional outflow patterns, wall shear stress, and expression of aortopathy. Circulation 2014, 129, 673–682. [Google Scholar] [CrossRef]
- Wanga, S.; Hibender, S.; Ridwan, Y.; van Roomen, C.; Vos, M.; van der Made, I.; van Vliet, N.; Franken, R.; van Riel, L.A.; Groenink, M.; et al. Aortic microcalcification is associated with elastin fragmentation in Marfan syndrome. J. Pathol. 2017, 243, 294–306. [Google Scholar] [CrossRef]
- Marshall, L.M.; Carlson, E.J.; O’Malley, J.; Snyder, C.K.; Charbonneau, N.L.; Hayflick, S.J.; Coselli, J.S.; Lemaire, S.A.; Sakai, L.Y. Thoracic aortic aneurysm frequency and dissection are associated with fibrillin-1 fragment concentrations in circulation. Circ. Res. 2013, 113, 1159–1168. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disease | MIM | Gene | Mode of Inheritance (% De Novo) | Key Features | Vascular Involvement |
|---|---|---|---|---|---|
| Marfan | #154700 | FBN1 | AD (30%) | Ectopia lentis—skeletal overgrowth | Sinus of Valsalva aneurysm/dissection |
| LDS1 | #609192 | TGFBR1 | AD (25%) | Hypertelorism, bifid uvula/cleft palate, arterial tortuosity | Widespread aortic and arterial aneurysm/dissection |
| LDS2 | #610168 | TGFBR2 | |||
| LDS3 | #613795 | SMAD3 | |||
| LDS4 | #614816 | TGFB2 | |||
| LDS5 | #615582 | TGFB3 | |||
| vEDS | #130050 | COL3A1 | AD (50%) | Thin skin—joint hypermobility | Predominant dissection of middle-sized arteries |
| MRLS | #300989 | BGN | XL | Hypertelorism, skeletal findings | Aortic aneurysm |
| FTAA6 | #611788 | ACTA2 | AD (rare) | Levido reticularis, iris flocculi | Ascending aortic aneurysm |
| MSMDS | #613834 | ACTA2-R179 | AD (100%) | Mydriasis, white matter densities, bowel dysmotility, hypotonic bladder | Persistent ductus arteriosus, aortic aneurysm |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues Bento, J.; Meester, J.A.N.; Luyckx, I.; Verstraeten, A.; Loeys, B.L. The Role of Genetics in Risk Stratification of Thoracic Aortic Aneurysm Dissection. Hearts 2020, 1, 50-61. https://doi.org/10.3390/hearts1020007
Rodrigues Bento J, Meester JAN, Luyckx I, Verstraeten A, Loeys BL. The Role of Genetics in Risk Stratification of Thoracic Aortic Aneurysm Dissection. Hearts. 2020; 1(2):50-61. https://doi.org/10.3390/hearts1020007
Chicago/Turabian StyleRodrigues Bento, Jotte, Josephina A.N. Meester, Ilse Luyckx, Aline Verstraeten, and Bart L. Loeys. 2020. "The Role of Genetics in Risk Stratification of Thoracic Aortic Aneurysm Dissection" Hearts 1, no. 2: 50-61. https://doi.org/10.3390/hearts1020007
APA StyleRodrigues Bento, J., Meester, J. A. N., Luyckx, I., Verstraeten, A., & Loeys, B. L. (2020). The Role of Genetics in Risk Stratification of Thoracic Aortic Aneurysm Dissection. Hearts, 1(2), 50-61. https://doi.org/10.3390/hearts1020007