Abstract
Pain is a multifactorial phenomenon involving neuronal, immune, and glial components. Annexin A1 (AnxA1), a glucocorticoid-regulated protein with pro-resolving properties, has emerged as a critical modulator of pain. Present in both peripheral and central compartments, AnxA1 acts through the formyl peptide receptor FPR2/ALX to regulate immune responses, modulate nociceptive signaling, and promote tissue homeostasis. Its mimetic peptide, Ac2–26, has demonstrated robust antinociceptive effects in various pain models, including those induced by inflammation, tissue injury, viral infection, and opioid exposure. AnxA1 modulates cytokine expression, inhibits pro-nociceptive pathways such as TRPV1 and CXCL12/CXCR4, and reprograms macrophages. In the central nervous system, it attenuates neuroinflammation and central sensitization. Notably, AnxA1 can exhibit context-dependent effects, contributing to either the resolution or exacerbation of inflammation. This review examines the molecular mechanisms by which AnxA1 bridges the immune and nervous system pathways, highlighting its therapeutic potential in pain management.
1. Introduction
Pain is a complex sensory and emotional experience involving neuronal, immune, and glial components, whose persistence in acute and chronic inflammatory contexts represents a significant therapeutic challenge. Unlike classical anti-inflammatory agents, endogenous pro-resolving mediators actively contribute to restoring tissue homeostasis. Among these mediators, Annexin A1 (AnxA1) stands out for its dual role in immune modulation and signal transduction [1,2].
AnxA1 is widely distributed in various cellular compartments, including the cytosol, nucleoplasm, plasma membrane, endosomes, endoplasmic reticulum, and even nuclear lamina. It is also present in the extracellular space, often associated with extracellular vesicles such as exosomes and microvesicles released by leukocytes and other cells. This structural and functional versatility underlies its role as a transducer of calcium-dependent (Ca2+) signals, particularly in specialized cellular regions like focal adhesions and microvilli, where it modulates vesicle trafficking, activates membrane receptors like FPR2/ALX, and participates in intracellular signaling cascades, including the PI3K/Akt and PLC-γ pathways [3].
In the central nervous system (CNS), AnxA1 plays crucial roles in both neurons and glial cells under both physiological and pathological conditions. Under homeostatic conditions, AnxA1 secreted by microglia contributes to neuronal maintenance by promoting the phagocytosis of apoptotic neurons through its interaction with phosphatidylserine exposed on the outer membrane leaflet, as previously detailed [4].
Crucially, these diverse actions of AnxA1 extend to the modulation of pain pathways. Several experimental studies have demonstrated that AnxA1, along with its N-terminal mimetic peptide Ac2–26, exerts potent antinociceptive effects in various pain models, including those induced by inflammation, tissue injury, viral infection, and opioid-induced hyperalgesia [5,6,7,8,9]. These effects are primarily mediated through the activation of the FPR2/ALX receptor, leading to the inhibition of pro-inflammatory signaling pathways, such as CXCL12/CXCR4 and TRPV1, the modulation of cytokine expression (e.g., TNF-α and IL-1β), and the promotion of resolution through macrophage reprogramming in the dorsal root ganglion (DRG). It is essential to note that AnxA1’s influence is not limited to peripheral mechanisms; it also acts centrally, in pain-processing regions such as the spinal cord, where it contributes to the regulation of neuronal excitability and attenuates central sensitization processes mediated by glial cells [4,10].
This review aims to shed light on the multiple roles of AnxA1 in both the periphery and the central nervous system, highlighting its relevance as an endogenous and exogenous mediator of inflammation resolution and pain modulation.
2. AnxA1 in Acute and Chronic Inflammatory Pain
AnxA1 is a versatile protein with multiple actions in different inflammatory pain scenarios. This protein has been the subject of several studies on acute and chronic pain, demonstrating significant roles in resolving inflammation and alleviating pain (Figure 1).
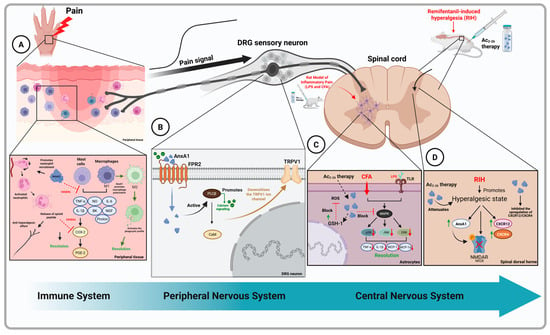
Figure 1.
Immunological, sensory modulatory, and neuroprotective effects of Annexin A1. Pain perception begins in the periphery, where immune cells are recruited to the site of tissue injury, initiating an inflammatory response. Mast cells and macrophages become activated, releasing pro-inflammatory mediators such as TNF-α, IL-1β, and IL-6. Additionally, levels of nitric oxide (NO), bradykinin (BK), nerve growth factor (NGF), and protons are elevated, contributing to the sensitization of nociceptors. In response to inflammatory stimuli, endogenous levels of AnxA1 increase, and neutrophils migrating from the bloodstream to the inflamed tissue also secrete AnxA1. This protein exerts anti-hyperalgesic effects by inhibiting cyclooxygenase-2 (COX-2), thereby reducing prostaglandin synthesis, a key contributor to inflammatory pain. Furthermore, neutrophils promote the tonic release of opioid peptides, which act directly on peripheral nerve endings to modulate pain (A). The nociceptive signal is transmitted from peripheral nerve endings via sensory neurons in the dorsal root ganglia (DRG) to the spinal cord. Within the DRG, AnxA1 activates the FPR2/ALX receptor, triggering the Gi/o signaling pathway and increasing intracellular calcium levels. This rise in Ca2+ is mediated by phospholipase C beta (PLCβ) and phosphoinositide 3-kinase (PI3K), which stimulate calcium release from the endoplasmic reticulum. Calcium, in turn, activates calmodulin (CaM), leading to desensitization of the TRPV1 channel and attenuation of nociceptive transmission (B). Exogenous administration of the AnxA1-derived peptide Ac2–26 inhibits the MAPK signaling pathway in astrocytes activated by lipopolysaccharide (LPS) and complete Freund’s adjuvant (CFA), thus promoting the resolution of neuroinflammation. This treatment also upregulates GSH-1 expression, which helps suppress the production of reactive oxygen species (ROS) (C). Finally, AnxA1 reduces hyperalgesia in the dorsal horn of the spinal cord in a model of remifentanil-induced hyperalgesia (RIH) by downregulating the CXCR12/CXCR4 signaling axis and inhibiting phosphorylation of the NR2B subunit of the NMDA receptor, further contributing to pain relief (D). Created with Biorender.
Previous studies [11] showed the potential role of ANXA1 in interacting with FPR2/ALX receptors, demonstrating that it modulates inflammatory pain by inhibiting pro-inflammatory cytokines, such as TNF-α, IL-1β, IL-16, and the production of prostaglandins, such as Cox-2 and PGE2, thereby reducing nociceptor sensitization. It also highlights the role of ANXA1 in decreasing the trans-endothelial migration of neutrophils to the inflamed site by blocking the FPR2/ALX pathway, reducing local inflammation, and promoting the release of opioid peptides by neutrophils. It also shows that ANXA1 can reduce peripheral nociceptive transmission by suppressing the excitability of sensory neurons [11]
Subsequently, in studies in rats subjected to carrageenan- and bradykinin-induced hyperalgesia, the co-administration of Ac2–26 and dexamethasone reduced nociceptive behaviors while suppressing the expression of inflammatory cytokines (TNF-α, IL-1β, and IL-6) and the enzyme COX-2 [6]. This suggests that AnxA1 potentiates glucocorticoid-mediated anti-inflammatory pathways, enhancing pain control.
Similarly, in human third molar extraction, a clinical model of acute tissue injury, elevated AnxA1 expression was associated with increased levels of IL-6 and neutrophil-attracting chemokines (CXCL1, CXCL2), supporting its regulatory role in acute nociceptive inflammation [5]. These findings highlight AnxA1 as both a marker and modulator of acute inflammatory responses in humans.
In visceral pain, particularly bladder pain syndrome (BPS), AnxA1 might contribute to the development of BPS [12]. In vitro experiments demonstrated that knockdown of AnxA1 in TEU-2 cells by siRNA led to lower cell survival after exposure to the bacterial toxin streptolysin. With this, the authors show that the downregulation of AnxA1 in patients with BPS can decrease cell survival, contributing to impaired kidney function [12].
Animal models of CFA-induced inflammation have further supported the antinociceptive actions of Ac2–26. The intrathecal administration of Ac2–26 attenuated opioid-induced hyperalgesia, thereby improving mechanical allodynia and thermal hyperalgesia [7]. This effect is related to modulation of the NMDA receptor, which is crucial in nociceptive transmission, as well as reduced gene expression of the chemokine CXCL12 and its receptor, and inhibition of phosphorylation of the NMDA NR2B subreceptor. The peptide also stimulated the endogenous production of AnxA1 in the dorsal horn of the spinal cord, reinforcing its protective role in neuroinflammatory pain. In another experimental model of inflammatory pain induced by CFA, the Ac2–26 peptide showed similar effects in reducing allodynia and thermal hyperalgesia [8]. These effects were associated with inhibition of astrocyte activation and decreased expression of pro-inflammatory cytokines and chemokines such as TNF-α, IL-1β, MCP-1 and MIP-1α, evidencing the central anti-inflammatory action of the peptide. Together, these models validate the central and peripheral roles of AnxA1 in resolving inflammatory pain.
In infection-driven pain, particularly in Chikungunya virus (CHIKV) infection, prophylactic Ac2–26 treatment reduced edema, neutrophil infiltration, and mechanical hypernociception. These effects were absent in AnxA1- or FPR2-deficient mice, indicating that the AnxA1-FPR2/ALX axis is essential for limiting infection-associated inflammation and pain [13].
Further evidence of AnxA1’s role in pain modulation comes from models of gouty arthritis induced by monosodium urate crystals in C57BL/6 mice. Treatment with the therapeutic peptide P140 significantly reduced hypernociception, neutrophil influx, and levels of pro-inflammatory cytokines in intra-articular and subcutaneous models. In vitro, P140 increased the production of AnxA1, thereby increasing its therapeutic potential in modulating inflammation associated with hypernociception [14].
In addition, other studies have highlighted the therapeutic potential of natural compounds and pro-resolution mediators in controlling inflammatory pain, reinforcing the importance of the LXA4-FPR2/ALX axis in the resolution of inflammation and analgesia. One of these studies evaluated the effects of the crude hydroalcoholic extract of Casearia sylvestris (HCE-CS), lipoxin A4 (LXA4), and synthetic analogues in a murine model of chronic post-ischemic pain (CPIP), which simulates complex regional pain syndrome type I (CRPS-I). Treatment with HCE-CS or FPR2/ALX receptor agonists significantly reduced mechanical hyperalgesia. This effect was reversed by the antagonist WRW4, indicating that the FPR2/ALX axis participates in this antinociceptive effect. When evaluating the role of AnxA1, it was observed that its absence did not affect the induced hyperalgesia, suggesting that the anti-hyperalgesic impact of the extract primarily occurs through the activation of the FPR2/ALX receptor, possibly mediated by LXA4 [15].
In the model of persistent inflammatory hyperalgesia induced by CFA, the percutaneous electrical stimulation of the vagus nerve (pVNS) modulated pain through pro-resolving effects mediated by the FPR2/ALX receptor. pVN stimulation promoted a significant reduction in the levels of the inflammatory cytokines IL-1β and IL-6, as well as increasing the expression of AnxA1 and CD86 in the animals’ paws. It was observed that this pro-resolving effect is associated with the polarization of macrophages from the M1 to M2 phenotype, suggesting an immunological reprograming favorable to the control of inflammatory pain via Anxa1-FPR2/ALX [9].
These studies collectively illustrate AnxA1’s consistent role in dampening inflammatory responses and pain through both endogenous and pharmacologic pathways. They also introduce the concept of FPR2/ALX as a converging node for multiple pro-resolving mediators.
3. AnxA1 and Pain Pathways: Molecular Interactions and Signal Transduction
AnxA1 is a glucocorticoid-regulated protein with well-established roles in the resolution of inflammation [16]. Its relevance in pain physiology has been illuminated through studies demonstrating its expression in both peripheral sensory neurons and spinal circuits, as well as in immune cells such as macrophages and T lymphocytes [17]. AnxA1 exerts its effects predominantly through the FPR2/ALX, triggering downstream signaling cascades that influence both neuronal excitability and immune cell function [2].
Proteomic analyses were conducted on the DRG of C57BL/6J mice subjected to plantar incision to investigate the mechanisms involved in postoperative pain. As a result, 44 differentially expressed proteins were identified, primarily associated with immune and inflammatory pathways, such as the MAPK/ERK signaling pathway, as well as proteins linked to metabolic regulation and cellular stress. Additionally, through orthogonal analyses and bioinformatics approaches, the AnxA1 protein was identified as a promising target for the development of non-opioid analgesics [18].
In models of inflammatory pain, such as those induced by carrageenan or CFA, the exogenous administration of AnxA1 or its mimetic peptide Ac2–26 has been shown to attenuate hyperalgesia, with evidence suggesting modulation of transient receptor potential vanilloid 1 (TRPV1) activity via PLCβ–CaM-dependent pathways in DRG neurons [10]. At the central level, ANXA1 inhibits opioid-induced hyperalgesia by blocking the CXCL12/CXCR4–NMDA receptor axis in the spinal cord, underscoring its importance in counteracting pain sensitization beyond the periphery [7]. In viral infection models, such as CHIKV, AnxA1’s immunomodulatory role further supports its potential as a therapeutic target in infection-associated pain [13]. AnxA1 is also implicated in adaptive immune responses, where it modulates CD4+ T-cell polarization through the HA/CD44–Akt/mTOR pathway, contributing to the control of autoimmune inflammation and its associated nociceptive outcomes [19].
AnxA1 and its derived peptide Ac2–26 play a key role in resolving inflammation and selectively modulating inflammatory pain through formyl peptide receptor FPR signaling. Evaluating the effects of AnxA1 and its peptide Ac2–26 on pain modulation in mice, the mimetic peptide of AnxA1 selectively reduced inflammatory pain in the second phase of the formalin test, without affecting acute or thermal pain responses (e.g., hot plate, tail-flick), showing stimulus-specific action. Mechanistically, the FPR agonist fMLP mimicked Ac2–26 effects, while the FPR antagonist Boc-1 blocked them, confirming that formyl peptide receptors (FPRs) mediate the antinociceptive action. Both the peripheral and central administration of Ac2–26 were effective, though central effects still depended on peripheral inflammation. Shorter fragments (Ac2–12, Ac2–6) lacked efficacy, indicating that the full Ac2–26 sequence is necessary for FPR engagement. Overall, the findings suggest that AnxA1-FPR signaling can selectively inhibit inflammatory pain without impairing protective nociception, supporting the development of safer, targeted analgesics based on pro-resolving pathways [20].
3.1. AnxA1-FPR2 Axis in Dorsal Root Ganglia and Inflammatory Pain
Compelling evidence supports the anti-nociceptive role of AnxA1 in the modulation of inflammatory pain, particularly through its interaction with the FPR2/ALX within the DRG of rats. Using a well-established model of peripheral inflammatory pain, male Sprague Dawley rats received intraplantar injections of CFA, leading to localized hindpaw inflammation and robust mechanical and thermal hyperalgesia. The study then explored whether the intrathecal administration of AnxA1 mimetics or FPR2/ALX ligands could reverse this nociceptive sensitization [17].
Immunofluorescence and Western blot analyses confirmed that both AnxA1 and its receptor FPR2/ALX are constitutively expressed in the L4/L5 DRG, localized in both neurons and satellite glial cells. Following CFA-induced inflammation, AnxA1 expression was significantly upregulated in the DRG bilaterally by day 7, indicating a systemic compensatory mechanism that dampens inflammation and nociception. Notably, this increase in AnxA1 was accompanied by a potent anti-nociceptive effect when rats were treated intrathecally with Ac2–26, a pharmacologically active peptide derived from the N-terminal domain of AnxA1, or with BML-111, a synthetic FPR2/ALX agonist. Both treatments significantly attenuated mechanical and thermal hyperalgesia in a dose-dependent manner. Importantly, the specificity of this pathway was further confirmed using the FPR2/ALX antagonist Boc-1. When administered alone, Boc-1 exacerbated CFA-induced pain behaviors, indicating the tonic engagement of the endogenous AnxA1-FPR2/ALX axis in restraining inflammatory nociception. Moreover, co-administration of Boc-1 with Ac2–26 abrogated the peptide’s anti-nociceptive effects, underscoring the receptor-dependent nature of this signaling mechanism. Interestingly, Boc-1 also led to a significant downregulation of AnxA1 expression in the DRG, further suggesting a regulatory loop in which receptor activity modulates AnxA1 availability and function [17].
Beyond behavioral outcomes, molecular assessments revealed that both Ac2–26 and BML-111 enhanced AnxA1 protein levels in DRG tissue, suggesting a positive feedback mechanism that could amplify resolution signals under conditions of chronic inflammation. Together, these findings delineate a clear functional role for AnxA1 in dampening inflammatory pain through FPR2/ALX activation, supporting its potential as a molecular target for novel analgesic therapies, particularly in conditions characterized by persistent nociceptive input and peripheral sensitization [21].
In addition, in the context of medication overuse headache (MOH) associated with triptans in a murine model of chronic migraine induced by nitroglycerin (GTN) combined with repeated use of rizatriptan (RIZ), it was observed that animals undergoing combined treatment with GTN+RIZ exhibited a significant increase in ANXA1 expression, mainly in microglia of the spinal trigeminal caudal nucleus (SPVC), mainly in the cytoplasm, showing that its antinociceptive action depends on the activation of FPR1/FPR2 receptors. ANXA1 was also observed in SPVC microglia. Furthermore, treatment with Ac2–26 resulted in a reduction in light-induced allodynia, an effect that was reversed by the FPR antagonist (Boc-MLF TFA), in addition to reducing the expression of c-Fos after painful stimulation in SPVC neurons, indicating a direct effect on trigeminal pain pathways [22]
The molecular mechanisms by which Anxa1 modulates nociception in DRG neurons have been explored, with a focus on its interaction with the TRPV1 ion channel. AnxA1 exerts anti-inflammatory and pro-resolving effects primarily through activation of the FPR2/ALX. This study aimed to elucidate how AnxA1 influences TRPV1 activity and nociceptive signaling [10]. Using a combination of behavioral assays, calcium imaging, patch-clamp electrophysiology, immunofluorescence, and co-immunoprecipitation techniques, the researchers demonstrated that AnxA1 plays a critical role in modulating TRPV1-mediated nociception. Mice with conditional knockout of AnxA1−/− exhibited heightened sensitivity to noxious stimuli, including heat and chemical irritants such as capsaicin and formalin. Specifically, AnxA1−/− mice showed significantly reduced withdrawal latencies in thermal nociception tests and increased pain behaviors in chemical-induced nociception models [10].
DRG neurons from AnxA1−/− mice displayed enhanced capsaicin-induced calcium influx, increased TRPV1 currents, and elevated neuronal firing rates, indicating heightened excitability. Conversely, treatment with the ANXA1 mimetic peptide Ac2–26 in wild-type DRG neurons resulted in a robust increase in intracellular calcium levels, activation of phospholipase C beta (PLCβ), and promotion of the calmodulin (CaM)-TRPV1 interaction. These effects were attenuated by the FPR2 antagonist Boc2, suggesting that ANXA1 exerts its modulatory effects through the FPR2/ALX receptor pathway. Further analysis revealed that Ac2–26 treatment enhanced the co-localization and interaction between CaM and TRPV1, as evidenced by immunofluorescence and co-immunoprecipitation assays. This interaction is known to lead to TRPV1 desensitization, thereby reducing nociceptive signaling. Electrophysiological recordings confirmed that Ac2–26 decreased capsaicin-evoked TRPV1 currents, an effect reversed by co-application of Boc2. In vivo, the intrathecal administration of Ac2–26 in AnxA1−/− mice reduced formalin-induced pain behaviors and increased thermal pain thresholds in a complete Freund’s adjuvant (CFA)-induced inflammatory pain model. These anti-nociceptive effects were abolished by Boc2, further confirming the involvement of the FPR2/ALX pathway. Taken together, the study elucidates a novel mechanism by which AnxA1 modulates TRPV1 activity and nociception in DRG neurons. Through the FPR2/ALX receptor, ANXA1 activates the PLCβ-Ca2-CaM signaling cascade, leading to the desensitization of TRPV1 and the attenuation of nociceptive signaling. These findings provide valuable insights into the molecular underpinnings of pain modulation and suggest potential therapeutic targets for treating inflammatory pain [10].
It was also shown that AnxA1−/− mice had significantly increased responses to the acetic acid-induced writhing test, indicating greater susceptibility to acute nociceptive pain compared to AnxA1+/+ mice, regardless of sex. Furthermore, it was shown that, in AnxA1−/− mice, PGE2 was specifically increased in the spinal cord, but not in other brain areas, suggesting that ANXA1 modulates pain by negatively influencing PGE2 synthesis. Also, observing the effect of Ac2–26, it was shown that it resulted in analgesia and reduced spinal PGE2 levels in both AnxA1−/− and AnxA1+/+ animals. In AnxA1−/− mice, there was also an increase in the expression of cPLA2, COX-1, COX-2, and COX-3 in the spinal cord, correlating with the increase in PGE2 [23].
The neuroimmune mechanisms underlying persistent pain in rheumatoid arthritis (RA) were investigated, focusing on the role of macrophages within the DRG. Recognizing that many RA patients experience chronic pain independent of active joint inflammation, the researchers sought to elucidate the cellular and molecular interactions underlying this phenomenon. Using the K/BxN serum transfer model of inflammatory arthritis in mice, the study demonstrated that, even after the resolution of joint inflammation, mechanical hypersensitivity persisted, indicating ongoing pain. In addition, a critical role for pro-resolving lipid mediators, particularly those produced by 12/15-lipoxygenase (Alox15), in modulating this pain response. To delve deeper, they generated conditional knockout (cKO) mice lacking Alox15 specifically in CX3CR1+ monocytes/macrophages. These cKO mice exhibited exacerbated mechanical hypersensitivity compared to wild-type controls, correlating with increased infiltration of neutrophils into the DRG. The study revealed a novel neuron–macrophage–neutrophil circuit: nociceptive neurons released extracellular vesicles containing arachidonic acid, which macrophages converted into leukotriene B4 via 5-lipoxygenase. This leukotriene B4 acted as a chemoattractant for neutrophils. Subsequently, the apoptosis of these neutrophils should have promoted macrophage-mediated efferocytosis and the production of antinociceptive mediators like lipoxin A4. However, in cKO mice, this efferocytosis process was defective, leading to sustained inflammation and pain. Importantly, the study found that the intrathecal administration of a MerTK-activating antibody in both wild-type and cKO mice attenuated persistent hypersensitivity. This treatment polarized DRG macrophages toward a proresolving phenotype, enhancing the production of lipoxin A4 and restoring the resolution of inflammation [24].
3.2. AnxA1 in Non-Neuronal Pain-Related Immune Mechanisms
The hyaluronan (HA)/CD44 signaling axis plays a pivotal role in driving proinflammatory T-helper cell responses, with implications for chronic pelvic pain and inflammation in autoimmune prostatitis. A murine model of chronic prostatitis/chronic pelvic pain syndrome CP/CPPS showed that the hyaluronan (HA)/CD44 axis contributes to T-helper cell polarization in autoimmune prostatitis (EAP). CD44, highly expressed on CD4+ T cells from CP/CPPS individuals, binds HA to promote Th1 differentiation via a signaling cascade involving Annexin A1 (AnxA1), Akt, and mTOR. Disruption of this axis, through CD44 silencing, HA synthesis inhibition (4-MU), or AnxA1 knockdown, suppressed Th1 responses, reduced prostate inflammation, and alleviated pain in EAP mice. Mechanistically, HA stimulation enhanced Th1 differentiation and activated Akt/mTOR signaling, which was blocked by Akt inhibitors (MK-2206 and everolimus). CD44 physically interacted with AnxA1, stabilizing its expression and sustaining Akt/mTOR activity. These findings define a novel HA/CD44–AnxA1–Akt/mTOR pathway driving Th1-mediated inflammation and suggest it as a potential therapeutic target for CP/CPPS [19].
Beyond its role in autoimmune prostatitis, AnxA1 also contributes to the regulation of acute inflammatory responses during viral infections, such as Chikungunya virus (CHIKV). The AnxA1-FPR2/ALX signaling axis plays a regulatory role in acute inflammation triggered by CHIKV infection. As an arthritogenic alphavirus, CHIKV induces a typically self-limiting illness characterized by joint pain and polyarthralgia. The immune responses during the acute phase of CHIKV infection are critical in determining disease progression and resolution. Genetic and pharmacological approaches elucidate the function of the AnxA1-FPR2/ALX pathway. Mice genetically deficient in AnxA1 or FPR2/ALX exhibited exacerbated inflammatory responses upon CHIKV infection, characterized by increased neutrophil accumulation and augmented tissue damage at the infection site. These findings suggest that the absence of AnxA1 or its receptor leads to uncontrolled inflammation during CHIKV infection. Conversely, treating wild-type mice with the AnxA1-mimetic peptide Ac2–26 resulted in a significant reduction in neutrophil infiltration, decreased local concentrations of inflammatory mediators, and diminished mechanical hypernociception and paw edema induced by CHIKV infection. Notably, alterations in viral load were mild in both genetic deletion and treatment scenarios, indicating that the AnxA1-FPR2/ALX pathway primarily modulates the host’s inflammatory response rather than directly affecting viral replication [13]. In addition to infectious and autoimmune conditions, AnxA1 also modulates neuroinflammatory responses in pain-related contexts such as opioid-induced hyperalgesia.
AnxA1 mitigates remifentanil-induced hyperalgesia (RIH) in rats by modulating the spinal CXCL12/CXCR4 axis and NR2B-containing NMDA receptor activation. Remifentanil elevated AnxA1, CXCL12/CXCR4, and NR2B phosphorylation, contributing to pain hypersensitivity. The intrathecal administration of AnxA1-mimetic Ac2–26 reduced hyperalgesia, downregulated CXCL12/CXCR4 signaling, and inhibited NR2B phosphorylation. These effects were supported by CXCR4 antagonism and reversed by exogenous CXCL12, positioning AnxA1 as a potential therapeutic target for opioid-induced hyperalgesia [7]. Collectively, these studies highlight the central role of AnxA1-centered signaling pathways across diverse inflammatory and pain-related conditions, underscoring its potential as a versatile therapeutic target for modulating immune responses and alleviating chronic inflammation and hyperalgesia.
4. Conclusions and Future Challenges in Annexin A1 Research in Pain
The insights presented in this review show a growing body of evidence that positions AnxA1 as a central mediator in the resolution of inflammation and modulation of pain. Traditionally viewed through the lens of glucocorticoid-mediated anti-inflammatory pathways, AnxA1 is now recognized as a key interface between the immune and nervous systems. Its capacity to engage both peripheral and central mechanisms of nociceptive control underscores its therapeutic potential in a wide range of pain conditions, from acute inflammation to chronic pain.
The efficacy of AnxA1 and its mimetic peptide Ac2–26 in attenuating inflammatory hyperalgesia, while preserving physiological nociceptive responses, is particularly noteworthy. This selective modulation contrasts with the broader, often undesirable, suppression of protective pain seen with conventional analgesics, such as opioids or NSAIDs. Studies employing models such as formalin and CFA-induced inflammation have repeatedly demonstrated that AnxA1 operates through the FPR2/ALX, initiating signaling cascades that culminate in desensitization of TRPV1 channels via PLCβ–CaM pathways. These mechanisms contribute to the attenuation of nociceptive neuron excitability, providing molecular evidence for the sensory-modulating effects of AnxA1.
Beyond neuronal targets, AnxA1 influences macrophage function within DRG, promoting efferocytosis and tissue homeostasis. This role is exemplified in rheumatoid arthritis models, where defective resolution programs in DRG macrophages correlate with persistent pain despite the absence of joint inflammation. Restoration of resolution via MerTK activation or AnxA1 mimetics supports the concept that chronic pain is not merely a result of ongoing inflammation, but also of failed inflammatory resolution. Furthermore, AnxA1 activity extends into the adaptive immune compartment. In autoimmune prostatitis, the HA/CD44–ANXA1–Akt/mTOR axis has been shown to skew CD4+ T-cell responses toward a Th1 phenotype, revealing that ANXA1 also modulates pain through T cell-mediated inflammatory pathways. These insights broaden the scope of AnxA1 beyond classical innate immune regulation, positioning it as a bridge between matrix-derived cues, intracellular signaling, and chronic pain pathophysiology.
Importantly, studies in viral infection models, including CHIKV-induced arthropathy, emphasize the role of AnxA1 in curbing excessive neutrophilic responses. The worsened disease phenotype observed in AnxA1 or FPR2/ALX knockout mice further highlights the protective function of this pathway in resolving infection-associated inflammation and pain.
Together, these findings delineate AnxA1 as a multifaceted modulator of pain through its immunoregulatory, neuroprotective, and pro-resolving actions. The convergence of data from pharmacological interventions, knockout models, and diverse pain paradigms strengthens the rationale for targeting the AnxA1-FPR2/ALX axis in clinical pain management. Unlike current analgesics, therapies based on this axis may provide effective relief while preserving protective nociception and reducing long-term adverse effects.
Despite growing evidence supporting the role of AnxA1 in pain modulation, the current literature is still relatively limited. There are few studies that directly address the involvement of glial cells in AnxA1-mediated pathways. Given the fundamental role of astrocytes and microglia in neuroinflammation and pain chronicity, the lack of mechanistic insights into how AnxA1 interacts with glial signaling represents a significant gap. This limitation restricts our ability to fully characterize the neuroimmunology interaction underlying nociceptive control and highlights the need for experimental designs that specifically question glial contributions.
Furthermore, although AnxA1 has been investigated in various pain models including inflammatory, neuropathic, and infection-related conditions, the total number of studies remains limited compared to the vast landscape of pain research. To our knowledge, few reports have systematically evaluated the long-term outcomes, receptor specificity, or therapeutic window of AnxA1-based interventions in chronic pain states. This highlights the need to expand the evidence base with more comprehensive preclinical and translational studies. Explicit recognition of these limitations not only provides readers with the appropriate context but also points to important opportunities for future research, including the development of targeted approaches that integrate glial biology and pathophysiology in the exploration of the AnxA1-FPR2/ALX axis.
Author Contributions
L.P.d.S.F.: Writing—original draft, Investigation, Data curation, Conceptualization. D.D.d.S.: Writing—original draft, investigation; R.P.L.: Writing—original draft, investigation; J.M.S.: Writing—original draft, Investigation; C.D.G.: Writing—review and editing, Supervision, Investigation. All authors have read and agreed to the published version of the manuscript.
Funding
Work in our laboratory is funded by Fundação de Amparo à Pesquisa do Estado de Sao Paulo (FAPESP) (grant no. 2022/02327-6). L.P.d.S.F. and D.D.d.S. were supported by the FAPESP scholarships (2021/00270-4 and 2023/16282-7, respectively). R.P.L. was supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES—Finance code 001) scholarship. C.D.G. is a researcher fellow of the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- D’Acquisto, F.; Perretti, M.; Flower, R.J. Annexin-A1: A pivotal regulator of the innate and adaptive immune systems. Br. J. Pharmacol. 2008, 155, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.H.; Solito, E. Annexin A1: Uncovering the Many Talents of an Old Protein. Int. J. Mol. Sci. 2018, 19, 1045. [Google Scholar] [CrossRef] [PubMed]
- de Souza Ferreira, L.P.; da Silva, R.A.; Gil, C.D.; Geisow, M.J. Annexin A1, A2, A5, and A6 involvement in human pathologies. Proteins 2023, 91, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- de Souza Ferreira, L.P.; da Silva, R.A.; Borges, P.P.; Xavier, L.F.; Scharf, P.; Sandri, S.; Oliani, S.M.; Farsky, S.H.P.; Gil, C.D. Annexin A1 in neurological disorders: Neuroprotection and glial modulation. Pharmacol. Ther. 2025, 267, 108809. [Google Scholar] [CrossRef]
- Wang, X.M.; Hamza, M.; Wu, T.X.; Dionne, R.A. Upregulation of IL-6, IL-8 and CCL2 gene expression after acute inflammation: Correlation to clinical pain. Pain 2009, 142, 275–283. [Google Scholar] [CrossRef]
- Ferreira, S.H.; Cunha, F.Q.; Lorenzetti, B.B.; Michelin, M.A.; Perretti, M.; Flower, R.J.; Poole, S. Role of lipocortin-1 in the anti-hyperalgesic actions of dexamethasone. Br. J. Pharmacol. 1997, 121, 883–888. [Google Scholar] [CrossRef]
- Li, T.; Wang, H.; Wang, J.; Chen, Y.; Yang, C.; Zhao, M.; Wang, G.; Yang, Z. Annexin 1 inhibits remifentanil-induced hyperalgesia and NMDA receptor phosphorylation via regulating spinal CXCL12/CXCR4 in rats. Neurosci. Res. 2019, 144, 48–55. [Google Scholar] [CrossRef]
- Luo, Z.; Wang, H.; Fang, S.; Li, L.; Li, X.; Shi, J.; Zhu, M.; Tan, Z.; Lu, Z. Annexin-1 Mimetic Peptide Ac2-26 Suppresses Inflammatory Mediators in LPS-Induced Astrocytes and Ameliorates Pain Hypersensitivity in a Rat Model of Inflammatory Pain. Cell. Mol. Neurobiol. 2020, 40, 569–585. [Google Scholar] [CrossRef]
- Salm, D.C.; Horewicz, V.V.; Tanaka, F.; Ferreira, J.K.; de Oliveira, B.H.; Maio, J.M.B.; Donatello, N.N.; Ludtke, D.D.; Mazzardo-Martins, L.; Dutra, A.R.; et al. Electrical Stimulation of the Auricular Branch Vagus Nerve Using Random and Alternating Frequencies Triggers a Rapid Onset and Pronounced Antihyperalgesia via Peripheral Annexin A1-Formyl Peptide Receptor 2/ALX Pathway in a Mouse Model of Persistent Inflammatory Pain. Mol. Neurobiol. 2023, 60, 2889–2909. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, S.; Ke, X.; Yi, Y.; Yu, H.; Yu, D.; Li, Q.; Shang, Y.; Lu, Y.; Pei, L. The mechanism of Annexin A1 to modulate TRPV1 and nociception in dorsal root ganglion neurons. Cell Biosci. 2021, 11, 167. [Google Scholar] [CrossRef]
- Chen, L.; Lv, F.; Pei, L. Annexin 1: A glucocorticoid-inducible protein that modulates inflammatory pain. Eur. J. Pain 2014, 18, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskaya, K.; Babiychuk, E.B.; Draeger, A.; Burkhard, F.C. Down-regulation of annexin A1 in the urothelium decreases cell survival after bacterial toxin exposure. J. Urol. 2013, 190, 325–333. [Google Scholar] [CrossRef] [PubMed]
- de Araújo, S.; de Melo Costa, V.R.; Santos, F.M.; de Sousa, C.D.F.; Moreira, T.P.; Gonçalves, M.R.; Félix, F.B.; Queiroz-Junior, C.M.; Campolina-Silva, G.H.; Nogueira, M.L.; et al. Annexin A1-FPR2/ALX Signaling Axis Regulates Acute Inflammation during Chikungunya Virus Infection. Cells 2022, 11, 2717. [Google Scholar] [CrossRef] [PubMed]
- Galvão, I.; Mastrippolito, D.; Talamini, L.; Aganetti, M.; Rocha, V.; Verdot, C.; Mendes, V.; de Oliveira, V.L.S.; Braga, A.D.; Martins, V.D.; et al. The Therapeutic Effect of Phosphopeptide P140 Attenuates Inflammation Induced by Uric Acid Crystals in Gout Arthritis Mouse Model. Cells 2022, 11, 3907. [Google Scholar] [CrossRef]
- Piovezan, A.P.; Batisti, A.P.; Benevides, M.L.A.C.; Turnes, B.L.; Martins, D.F.; Kanis, L.; Duarte, E.C.W.; Cavalheiro, A.J.; Bueno, P.C.P.; Seed, M.P.; et al. Hydroalcoholic crude extract of Casearia sylvestris Sw. reduces chronic post-ischemic pain by activation of pro-resolving pathways. J. Ethnopharmacol. 2017, 204, 179–188. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Vago, J.P.; Teixeira, M.M.; Sousa, L.P. Annexin A1 and the Resolution of Inflammation: Modulation of Neutrophil Recruitment, Apoptosis, and Clearance. J. Immunol. Res. 2016, 2016, 8239258. [Google Scholar] [CrossRef]
- Pei, L.; Zhang, J.; Zhao, F.; Su, T.; Wei, H.; Tian, J.; Li, M.; Shi, J. Annexin 1 exerts anti-nociceptive effects after peripheral inflammatory pain through formyl-peptide-receptor-like 1 in rat dorsal root ganglion. Br. J. Anaesth. 2011, 107, 948–958. [Google Scholar] [CrossRef]
- Pogatzki-Zahn, E.M.; Gomez-Varela, D.; Erdmann, G.; Kaschube, K.; Segelcke, D.; Schmidt, M. A proteome signature for acute incisional pain in dorsal root ganglia of mice. Pain 2021, 162, 2070–2086. [Google Scholar] [CrossRef]
- Chen, J.; Meng, J.; Li, X.; Liu, Y.; Jin, C.; Zhang, L.; Hao, Z.; Chen, X.; Zhang, M.; Liang, C. HA/CD44 Regulates the T Helper 1 Cells Differentiation by Activating Annexin A1/Akt/mTOR Signaling to Drive the Pathogenesis of EAP. Front. Immunol. 2022, 13, 875412. [Google Scholar] [CrossRef]
- Pieretti, S.; Di Giannuario, A.; De Felice, M.; Perretti, M.; Cirino, G. Stimulus-dependent specificity for annexin 1 inhibition of the inflammatory nociceptive response: The involvement of the receptor for formylated peptides. Pain 2004, 109, 52–63. [Google Scholar] [CrossRef]
- Vieira, C.; Salm, D.C.; Horewicz, V.V.; Ludtke, D.D.; Emer, A.A.; Koerich, J.F.; Mazzardo, G.; Elias, S.; Moré, A.O.O.; Mazzardo-Martins, L.; et al. Electroacupuncture decreases inflammatory pain through a pro-resolving mechanism involving the peripheral annexin A1-formyl peptide receptor 2/ALX-opioid receptor pathway. Pflugers Arch. 2021, 473, 683–695. [Google Scholar] [CrossRef]
- Gong, Z.; Yang, C.; Dai, W.; Miao, S.; Liu, Y.; Jiao, Z.; Li, B.; Xie, W.; Zhao, W.; Han, X.; et al. Annexin A1 exerts analgesic effect in a mouse model of medication overuse headache. iScience 2023, 26, 108153. [Google Scholar] [CrossRef]
- Ayoub, S.S.; Yazid, S.; Flower, R.J. Increased susceptibility of annexin-A1 null mice to nociceptive pain is indicative of a spinal antinociceptive action of annexin-A1. Br. J. Pharmacol. 2008, 154, 1135–1142. [Google Scholar] [CrossRef]
- Oggero, S.; Voisin, M.B.; Picco, F.; Huerta, M.; Cecconello, C.; Burgoyne, T.; Perretti, M.; Malcangio, M. Activation of proresolving macrophages in dorsal root ganglia attenuates persistent arthritis pain. Proc. Natl. Acad. Sci. USA 2025, 122, e2416343122. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).