Abstract
Carbon tetrachloride (CCl4) is an efficient but highly toxic solvent, used in households and commercially in the industry under regulatory surveillance to ensure safety at the working place and to protect the workers’ health. However, acute unintentional or intentional intoxications by CCl4 may rarely occur and are potentially life-threatening. In this review article, therapy options are discussed that are based on a literature review of traditional poisoning cases and the clinical experience with 16 patients with acute poisoning by CCl4. Among various therapy options, the CO2-induced hyperventilation therapy will be considered in detail as the most promising approach. This special therapy was developed because only around 1% of the intoxicating CCl4 is responsible for the liver injury after conversion to toxic radicals via microsomal cytochrome P450 2E1 whereas 99% of the solvent will leave the body unchanged by exhalation. Therefore, to enhance CCl4 elimination through the lungs, CO2 is added to the inspiration air at a flow rate of 2–3 L min−1 in order to achieve hyperventilation with a respiratory volume of 25–30 L min−1. Under this therapy, the clinical course was favorable in 15/16 patients, corresponding to 93.8%. In essence, patients with acute CCl4 intoxication should be treated by forced ventilation.
1. Introduction
Carbon tetrachloride (CCl4) is known for its hepatotoxic potency and was previously often used as effective solvent and cleaning agent in industrial manufactories, households, dry-cleaning textile laundries, in fire extinguishers, as a precursor of refrigerants or rocket propellant, and also appreciated in humans as an effective anthelmintic chemical for treating ankylostomiasis [1,2,3,4,5,6]. Ingested as a chemical drug to treat helminths diseases, CCl4 had a long tradition and was classified as a typical drug with reference to more than 30,000 treatments in humans without marked symptoms at doses of 1–4 mL as described in 1923 [1]. Based on its chemical structure, CCl4 is a typical halogenated or more specifically a chlorinated hydrocarbon molecule also called tetrachloromethan, because all 4 hydrogen atoms of methane are substituted by chloride atoms closely tied with the single C atom in the center [2,3,4,5,6]. These are perfect conditions for an efficient solvent that keeps other chemical products in solution without reacting with these. CCl4 is continuously used in experimental models of reproducible liver injury which explains the large number of scientific reports on studies of liver injury caused by this chemical [2,3,4,5,6,7,8]. Additional information on the cascade of events leading to CCl4-mediated acute liver injury was also provided by experiments comparing liver enzyme activities such as GDH (glutamate dehydrogenase), ALP (alkaline phosphatase), ALT (alanine aminotransferase), or AST (aspartate aminotransferase), in the serum with levels of CCl4 in blood, liver and fat, using the rapid head space method of gas chromatography (GC) [8], with details described in earlier publications [9,10].
Liver injury observed in CCl4 poisoning is due to toxic metabolites of CCl4 rather than to the parent chemical [2,3,4,5,6,7,11], catalyzed by the hepatic microsomal cytochrome P450 (CYP), especially by its isoenzyme CYP2E1 [5,6] that is also a component of the hepatic microsomal ethanol-oxidizing system (MEOS) involved in the hepatic metabolism of ethanol [12,13,14]. The CYP2E1 content and MEOS activity are inducible following prolonged alcohol use [12], and even a single dose of ethanol is sufficient to induce MEOS activity [15]. Triggered by CYP2E1, chronic alcohol consumption predisposes to experimental liver injury by CCl4 [11] and explains acute hepatic interactions between alcohol and CCl4 [16]. In patients acutely intoxicated by CCl4, alcohol is a major issue whether ingested concomitantly or used chronically before.
Understanding CCl4-related molecular mechanisms is prerequisite to efficiently treat patients acutely intoxicated from CCl4 and to develop new therapy options. Because most of the CCl4 taken up by the body will be eliminated by expiration through the lungs, a therapy of forced ventilation had been developed to enhance toxin removal. Additional efforts must be directed to reduce microsomal production of toxic metabolites derived from CCl4 by searching for appropriate inhibitors of microsomal drug metabolizing enzymes, in addition to cimetidine that is presently used for this purpose.
This article provides an overview on recent developments of treatment modalities for patients with acute intoxications by carbon tetrachloride and discusses the use of the CO2-induced hyperventilation in 16 patients. These present cases were compared to historical cases published from time to time since 1922.
2. Literature Search
A computerized search of the Medline database was used with the following two search terms: human acute carbon tetrachloride intoxication case reports, providing around 831,000 hits, and human acute carbon tetrachloride liver injury case reports, providing around 654,000 hits. The yield of assessable cases was poor, due to interfering experimental reports and limited use of CCl4, being largely been removed from the market, replaced by other less toxic solvents, or used under regulatory occupational restrictions.
3. Historical Cases of CCl4 Poisoning and Liver Injury Caused by CCl4
Clinical features of human CCl4-induced liver injury and CCl4 poisoning in general have been well described in a variety of publications [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33]. Most of these clinical details described are expected as transferable from results obtained in animal studies [2,3,4,5,6]. The impact on human toxicity was well assessable only with the development of analytical methods and its introduction in clinical practice. For instance, the valid detection of liver injury by blood analysis was established at a time when serum activities of transaminases such as AST or ALT could be measured. Reviewing the traditional cases of patients with CCl4 intoxication provides a good clinical insight in this toxic disease. Originally used as an anthelminthic drug without knowledge of its hepatotoxic and nephrotoxic properties, CCl4 was then appreciated for many purposes as an effective solvent in household and industry but it was mostly used without any safety measures to protect human health because CCl4 was viewed as harmless chemical. Indeed, one of the earliest, still vague description of liver injury goes back to a publication in the second volume of the BMJ in 1922 [17], followed by CCl4 poisoning in an occupational context [18]. Surprisingly, historical reports provided little new ideas how to accelerate CCl4 removal out of the intoxicated body, nor were there any significant suggestions how to decrease the high lethality rate associated with acute CCl4 intoxications, except perhaps considering hemodialysis to treat renal failure in severe intoxications. Details of selected historical cases are presented for a broad overview (Table 1).

Table 1.
Selection of historical cases of liver injury by acute CCl4 poisoning.
Some results of the historical reports are of major clinical interest or remain an actual issue (Table 1). For instance, suggestions for treatment included hyperbaric oxygen therapy, hypothermia, oral use of calcium lactate, a diet high in carbohydrates, intravenous administration of dextrose and calcium gluconate, rectal application of dextrose, and the use of N-acetylcysteine. Only a few of these therapy options are based on experimental studies, rarely verified in patients with CCl4 intoxication. Key points of the historical cases are presented in condensed form for a quick clinical overview (Table 2).
Table 2.
Summary of historical data on CCl4 intoxication.
4. Actual Patients with CCl4 Poisoning and CO2-Induced Hyperventilation
As opposed to historical reports on previous patients, who did not receive a specific therapy directed against CCl4 as the poisonous chemical (Table 1 and Table 2) [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33], some actual recommendations how to diagnose and treat patients intoxicated by acute ingestion or inhalation of CCl4 or other aliphatic halogenated hydrocarbons have been presented in a recent report [34]. Essential diagnostic requirements at admission and during the clinical course are now outlined in a tabular listing (Table 3), which should be followed in a setting of an intensive care unit and at best within a center with special experience with these intoxications as described [34]. The diagnostic workup of the patient includes among many other essentials also the determination of CCl4 in the blood by GC [10] or in the exhalation air by the Draegerunknown-tube® system (DTS) supplied by Draeger, Lübeck in Germany [34,35]. Details of using the DTS are provided online [35] and in the legend of Table 3. In addition to diagnostic recommendations for clinical practice in acute CCl4 intoxication (Table 3), special attention is placed on therapy aspects with focus on forced ventilation achieved as CO2-induced hyperventilation to accelerate CCl4 removal via the lungs (Table 4 and Table 5) [34], supplemented by data on the acid-base balance essential for clinical treatment (Table 6). These recommendations result from previous experience as outlined in various publications [10,36,37,38,39,40,41,42].
Table 3.
Diagnosis of liver injury by acute CCl4 intoxication.
Table 4.
Therapy of adult patients with acute CCl4 intoxication.
Table 5.
CO2-induced hyperventilation therapy for acute CCl4 intoxication.
Table 6.
Acid-base balance under CO2-induced hyperventilation.
The hyperventilation therapy was applied in 16 patients with acute CCl4 intoxication, their case narratives help characterize the specific toxic disease caused by CCl4 and are presented including some selected clinical details (Table 7). Among the overall 16 patients with acute CCl4 poisoning, for 13 patients CCl4 was the only poison confronting the patients whereas in 3 other patients CCl4 was causative as mixture with other aliphatic halogenated hydrocarbons. Apart from a few younger patients, most of the 16 patients were adults with an age of up to 70 years. The male: female ratio was 10:6. Intoxication occurred via inhalation (n = 7) or ingestion (n = 9), by intention (n = 10) or unintentionally (n = 6). The duration of the hyperventilation therapy was variable in the 13 patients who had been intoxicated by CCl4 alone, with a longer treatment in those with an intentional intoxication as compared to those who were intoxicated unintentionally (156.0 ± 32.7 h vs. 87.0 ± 13.4 h; p < 0.01). For most patients who ingested CCl4, the approximate amount of the swallowed toxin could fairly well be documented. However, only part of the ingested CCl4 will exert its toxic property in the body because vomiting was a common feature in many patients and gastro-intestinal lavage likely contributes to reduce the quantity of CCl4 in the body.
Table 7.
Clinical details of 16 patients with acute CCl4 intoxication treated with the CO2-induced hyperventilation.
CCl4 may cause abnormal liver tests (LTs) at a variable extent as evidenced by increased serum activities of AST, ALT, and GDH, but time of peak occurrence depends on the route and duration of toxin uptake (Table 7). Facilitating rapid toxin absorption through the bronchial mucosa, inhalation of CCl4 at intoxicating amounts leads to variably increased serum activities of AST, ALT, and GDH found already at admission in 3 patients with a subsequent rapid decline (Table 7, cases 11–13). In one of the 3 patients (case 11), AST was higher than ALT, but GDH was only little increased at hospital admission (Figure 1). Interestingly, electron microscopy of a liver tissue specimen obtained on day 5 after admission and at the day when hyperventilation has been discontinued (Figure 1) revealed toxic injury of mitochondria that appeared swollen associated with a reduction of their cristae, and deposits of bile pigments (Figure 2). In the 2 other patients intoxicated by CCl4 inhalation (cases 12 and 13) (Table 7), the serum activity of AST was higher compared to ALT (Figure 3 and Figure 4), but GDH activities were extremely high with 1534 U/L in case 12 (Figure 3) and with 4746 U/L in case 13 (Figure 4). In the latter patient, this high GDH activity reflects severe toxicity towards the liver and is in line with severe renal toxicity with serum creatinine values of 14.5 mg/dL on day 5 and subsequent requirement of hemodialysis due to renal failure (Table 7, case 13). It seems that the first hit after CCl4 intoxication is directed to the liver followed by the kidneys.
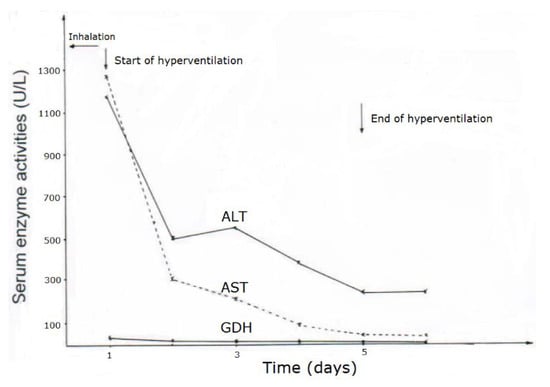
Figure 1.
Patient 11 intoxicated by inhalation of CCl4 (unknown amounts), presenting serum activities of ALT, AST, and GDH under CO2-induced hyperventilation therapy. Abbreviations: ALT, Alanine aminotransferase; AST, Aspartate aminotransferase; GDH, Glutamate dehydrogenase.
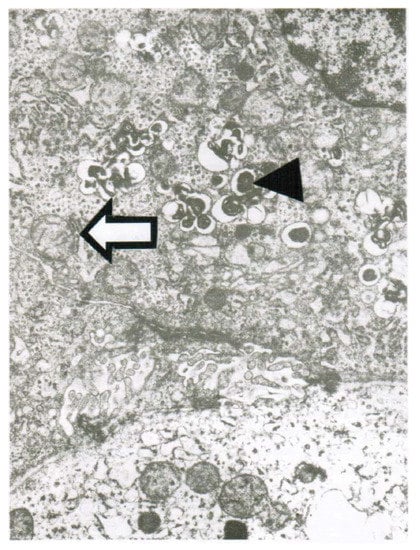
Figure 2.
Patient 11 with intoxication of CCl4 by inhalation (unknown amounts): Liver tissue specimen for electron microscopy (18,500-fold magnification) was obtained on day 5 after admission. In addition to abundant bile pigments (◄), as sign of major subcellular injury liver mitochondria are slightly swollen and their cristae are reduced (⇦).
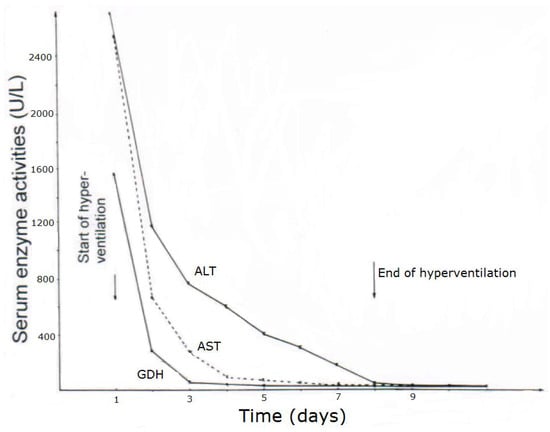
Figure 3.
Patient 12 with CCl4 intoxication by inhalation (unknown amounts). Serum activities of ALT, AST, and GDH after intoxication and during CO2-induced hyperventilation therapy. Abbreviations: ALT, Alanine aminotransferase; AST, Aspartate aminotransferase; GDH, Glutamate dehydrogenase.
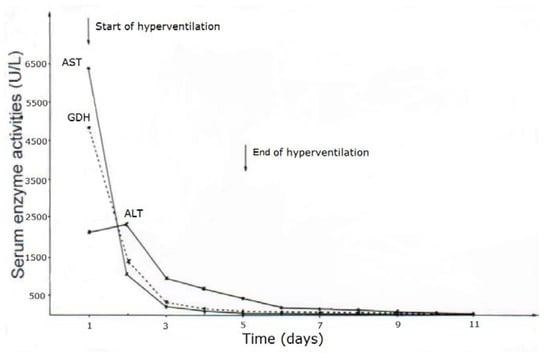
Figure 4.
Patient 13 with CCl4 intoxication by inhalation (unknown amounts) and serum activities of AST, ALT, and GDH following poisoning and under CO2-induced hyperventilation therapy. Abbreviations: ALT, Alanine aminotransferase; AST, Aspartate aminotransferase; GDH, Glutamate dehydrogenase.
As opposed to CCl4 intoxication by inhalation, case analyses showed that acute ingestion of CCl4 with its delayed gastrointestinal absorption results in abnormal LTs only during the hospital stay and therefore usually between days 4 and 5 after intoxication, shown as example for 2 patients (Table 7, cases 1 and 5), with details presented for patient 1 (Figure 5) and patient 5 (Figure 6). In many cases, there is only a minimal or moderate increase of LTs that is associated with normal values of total bilirubin, a parameter of liver function as opposed to AST and ALT reflecting LTs and therefore diagnostic parameters of liver injury but not those of liver function (Table 7). Following intoxications with high amounts of ingested CCl4 in rare cases, total bilirubin starts to increase at the day after ingestion with possibly high values in the further course together with clinical features of jaundice, signifying severe disturbances of liver functions. Except for severe intoxications where serum creatinine values start to increase on day 2 after intoxication, enhanced serum levels of creatinine or emerging renal failure were rarely observed (Table 7), likely as a consequence of daily creatinine measurements, assessing fluid balance, and early initiation of forced diuresis using intravenous electrolytes and furosemide in line with recommendations (Table 4).
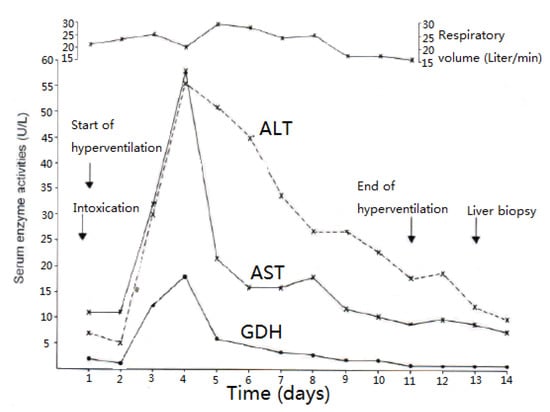
Figure 5.
Patient 1 after ingestion 30 mL CCl4 and serum activities of AST, ALT, and GDH under CO2-induced hyperventilation therapy. Abbreviations: ALT, Alanine aminotransferase; AST, Aspartate aminotransferase; GDH, Glutamate dehydrogenase.
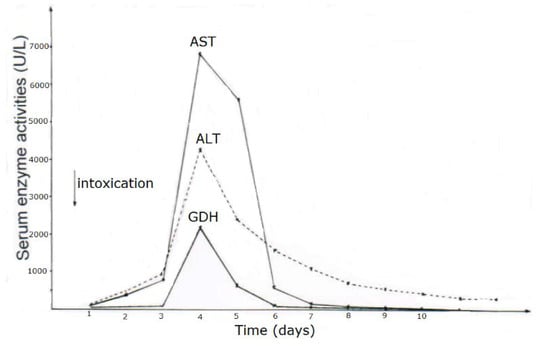
Figure 6.
Patient 5 with ingestion of 100 mL CCl4 and serum activities of AST, ALT, and GDH during CO2-induced hyperventilation therapy for 10 days. Abbreviations: ALT, Alanine aminotransferase; AST, Aspartate aminotransferase; GDH, Glutamate dehydrogenase.
Obtained shortly after cessation of the forced ventilation therapy and at times of normal or near normal serum AST and ALT activities, liver histology was devoid of severe confluent liver cell necroses but showed occasionally moderate steatosis and signs of remnant liver injury such as inflammatory cells or rarely single liver cell necrosis (Table 7). However, at the same time electron microscopy data still indicate severe liver injury especially related to mitochondria, shown for 2 patients (Figure 7 and Figure 8) with case narratives presented earlier (Table 7, cases 2 and 5). Several weeks or months after discharge patients and their doctors have been contacted to assess the further clinical course. Data provided for some patients indicated that the CCl4-liver injury was self-limited without major health of liver problems. However, in severe intoxications the risk of a life-threatening course will remain if a point of no return is achieved.
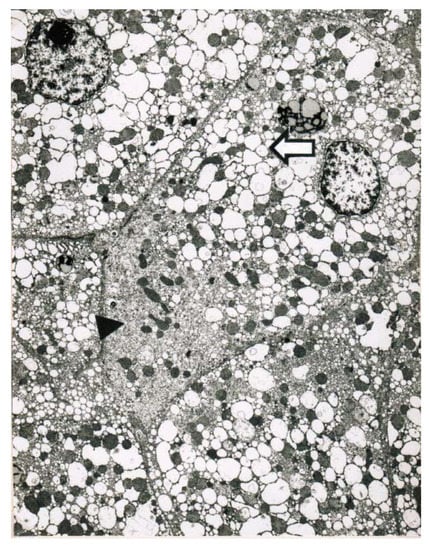
Figure 7.
Patient 5 with poisoning by CCl4 ingestion (50 mL) and liver tissue specimen obtained on day 14 after intoxication for electron microscopy (5200-fold magnification). Key features include a striking proliferation of the smooth endoplasmic reticulum of the hepatocyte (►) with close by injured mitochondria, and in addition to a pronounced dilatation of the smooth endoplasmic reticulum presenting as dilated cisterns (⇦).
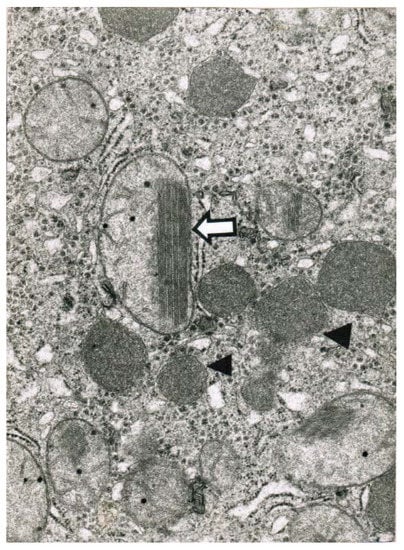
Figure 8.
Patient 2 with CCl4 ingestion (10–20 mL) showed 4 days after termination of the CO2-induced hyperventilation therapy by electron microscopy (32,000-fold magnification) a reduction and disorganization of hepatic mitochondria, some of which contained crystalline inclusion bodies. A mega-mitochondria revealed loss of cristae and a pronounced crystalline inclusion (⇦). In other parts there is an increase of lysosomes (◄).
Regrettably, the clinical course was lethal for one patient (Table 7, case 9) among the 16 patients (Table 7), which corresponds to a lethality rate of 6.3%. In this patient (case 9), liver transplantation was considered but declined due to contraindications including pneumonia, multi-organ failure and still detectable CCl4 in the blood. Although information on the patients with CCl4 intoxication is available, the present data are limited and do not allow defining the lethal dose of CCl4 in the cohort of assessed patients due to confounding variables of vomiting, gastro-intestinal lavage, and forced ventilation, which differently contribute to toxin removal.
5. Clinical and Experimental Challenges of CO2-Induced Hyperventilation
5.1. Clinical Data
CO2-induced hyperventilation therapy was initially suggested as a therapy option for intoxications by trichloroethylene, a commonly used aliphatic halogenated hydrocarbon in France, and clinical and experimental evidence of efficacy was concomitantly provided [37]. This new therapy aimed to accelerate the elimination of volatile solvents through increased exhalation and created interest in Germany at hospitals of the Heinrich Heine University in Düsseldorf [38,39], first applied in 110 children to treat intoxications by various aliphatic hydrocarbons as summarized in 1979 [39] and later also used in adults with initial results on 13 patients published in 1977 [38]. Details of the historical background on the experience in Düsseldorf have been summarized recently [34], with related clinical and experimental data provided in additional publications [8,9,10,16,36,40,41,42,43,44,45].
Based on the initial study of F. Pebay-Peyroula and A. M. Nicaise published in 1970 [37], it became increasingly more clear that in future patients with acute intoxications by aliphatic halogenated hydrocarbons should be treated by CO2-induced hyperventilation because additional supporting evidence was provided that this approach can enhance toxin removal via the lungs in patients [10,37,38,39]. Under these conditions it appeared unethical to conduct a randomized clinical trial (RCT), similar to other treatment conditions with the purpose to evaluate efficacy. Any RCT would require homogeneity of 2 cohorts, one receiving the CO2-induced hyperventilation therapy and the other one receiving some kind of a control therapy, hardly to be defined and open for future discussions. Cohort homogeneity is poorly achievable and uncertain, because the causative hydrocarbons vary from one patient to the other, and variability expands to the amount ingested or inhaled. Confounding variables include acute or previous chronic alcohol use and as to whether the patients did experience vomiting, decreasing the amount of toxins that could be injurious. Consequently, there have been no good arguments to conduct such RCT to verify efficacy. Instead, additional studies were carried to systematically evaluate best conditions applying the CO2-induced hyperventilation and to establish the impact of this therapy on toxin elimination.
Of clinical interest are data on the acid-base balance analyzed under clinical CO2-induced hyperventilation in 5 patients who had ingested CCl4 in various amounts (Table 6). The results show some variability among the assessed patients and ask for continuous re-analysis when this therapy is used. In another set of a clinical study, the effect of a variable CO2 flow min−1 on the respiratory volume min−1 was assessed in 2 patients (Figure 9). One of the patients appears as a good responder by reaching a respiratory volume of 25–30 L min−1 or slightly above with a CO2 flow of 1.5–4.0 L min−1. In the other patient and rarely in additional patients, the response is poor, because the desired respiratory volume was not achieved with CO2 at a flow rate of 3 L min−1 but required a somewhat higher flow rate of 4–5 L min−1. However, increasing the flow rate above 5 L min−1 is unsuccessful (Figure 9) and not recommended. Consequently, in all patients the initial flow rate of CO2 should be 2–3 L min−1, to be adjusted and titrated according the obtained respiratory minute volume.
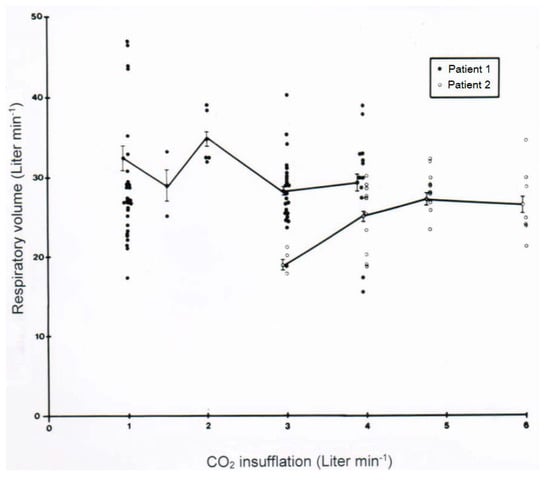
Figure 9.
Effect of a variable CO2 flow min−1 on the respiratory volume min−1 was assessed in 2 patients.
With a newly described rapid analytical method using the head space technique of GC, a quick quantitative determination of CCl4 in the blood of patients intoxicated by CCl4 became feasible and allowed an improved clinical management of these patients [10]. For instance, in a patient intoxicated by ingested CCl4 and treated with CO2-induced hyperventilation, CCl4 disposal via the lungs was quantitatively assessed in relation to the respiratory minute volume (Figure 10). The results clearly show that the amount of CCl4 eliminated is dependent on the respiratory minute volume achieved.
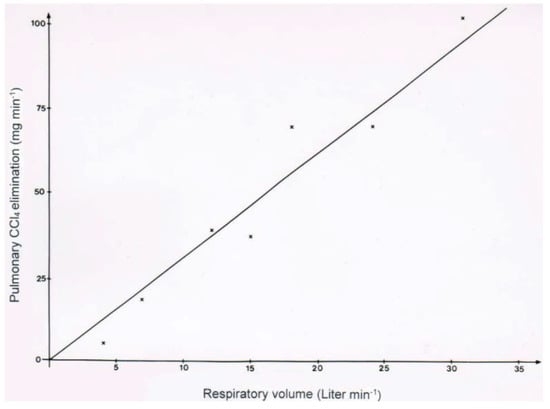
Figure 10.
In a patient intoxicated by CCl4 ingestion and treated with CO2-induced hyperventilation, CCl4 disposal via the lungs was quantitatively assessed in relation to the respiratory minute volume, showing that the amount of CCl4 eliminated is dependent on the respiratory minute volume achieved.
In two patients with CCl4 intoxication by ingestion, blood levels of CCl4 were determined under various ventilation conditions (Figure 11 and Figure 12). Intermittent discontinuation of the CO2-induced hyperventilation led to a striking increase of CCl4 levels in the blood, whereas its re-introduction reduces CCl4 to levels achieved under the previous hyperventilation regimen (Figure 11). In another patient intoxicated by oral use of CCl4, blood levels of CCl4 declined under the hyperventilation therapy and reached a plateau that lasted for around 16 days before a striking increase of blood levels was observed, due to an impaired CCl4 elimination via the lungs as a consequence of an emerging pneumonia with fatal outcome (Figure 12).
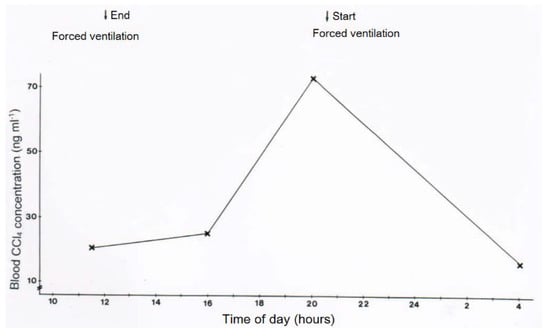
Figure 11.
In a patient with CCl4 intoxication by ingestion, blood levels of CCl4 were determined with or without forced ventilation induced by CO2. Blood CCl4 levels increased when forced ventilation was terminated and decreased again with reinstitution of forced ventilation.
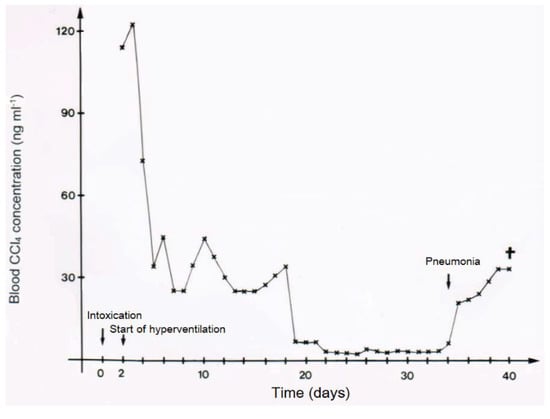
Figure 12.
In another patient (case 9) intoxicated by oral use of 50 mL CCl4, blood levels of CCl4 declined under the hyperventilation therapy and reached a plateau that lasted for around 16 days before a striking increase of blood levels was observed, due to impaired CCl4 elimination via the lungs as a consequence of an emerging pneumonia with fatal outcome, reproduced with permission from [10]. Copyright Springer, 1983.
Follow-up data were provided by physicians or patients for only 5/12 patients following acute CCl4 intoxication by ingestion or inhalation. Requests had been confined to serum activities of ALT, AST, and GGT, all of which remained in the normal range or normalized within 7–10 days after discharge, considering 3 patients who had already normal values at discharge, and the two other patients with slightly increased values at discharge. In all five patients, all values remained in the normal range also for 3 to 7 years after intoxication. Based on the results of this small cohort, there is no evidence of long-term hepatic sequelae, such as vanishing bile duct syndrome, in patients who experienced acute CCl4 intoxications.
Clinical evidence suggests the efficacy of CO2-induced hyperventilation by accelerating CCl4 removal via the lungs (Figure 9, Figure 10, Figure 11 and Figure 12). However, these data do not allow the firm conclusion that this therapy reduces the overall lethality rate in patients with CCl4 intoxication [36], which was estimated at 28–35% until 1953 and at 17% thereafter until 1965 due to increased use of dialysis devices [25], but with 25% this rate was somewhat higher in a study from Düsseldorf published in 1969 without applying the new CO2-induced hyperventilation therapy [25]. These figures compare with the lethality rate of 6.7% observed with 16 patients treated with forced ventilation (Table 7), but such comparisons of various clinical cohorts with their case variabilities are uncertain and open for discussion. Another approach using studies in animal models provides additional supporting data.
5.2. Experimental Results
CO2-induced hyperventilation is an essential part of the overall new therapeutic approach for acute poisonings by CCl4 in humans (Table 3, Table 4, Table 5 and Table 6), and animal studies are suggestive of its efficacy [43,44,45]. First of all, basic knowledge is essential on the distribution of orally applied CCl4 in animals that mimic conditions of intoxicated humans (Figure 13) [8]. In this rat model, CCl4 administered by gavage is found within 3 h in the liver and the blood, and with higher CCl4 amounts in the fat around 6 h after gavage. CCl4 levels then decline, more quickly in the blood and the liver as compared to the fat (Figure 13). In other studies comparing the time course of CCl4 levels in the blood with serum activities of liver enzymes, peak levels of CCl4 are found at 3 h after gavage and activity peaks of ALT and AST then between 12 and 24 h after gavage, followed by GDH and ALP with a peak at 48 and 72 h [8]. Compared to CCl4 poisonings by ingestion in humans (Table 7, Figure 5 and Figure 6), changes of serum enzyme activities occur earlier in the animals [8]. The CCl4-induced rise of serum ALT activity is associated with a corresponding decline of ALT activity in the liver [8], indicating that the liver is likely the origin of the increased ALT activity in the serum. Under these experimental conditions, other enzyme activities in the liver are also reduced [8], again likely due to an increased efflux out of the liver into the blood.
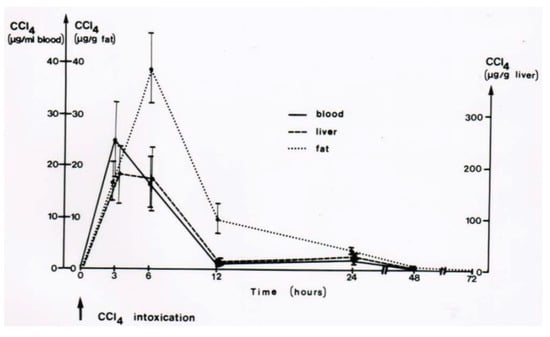
Figure 13.
In this rat model, CCl4 administered by gavage is found within 3 h in the liver and the blood, and with higher CCl4 amounts in the fat around 6 h after gavage. CCl4 levels then decline, more quickly in the blood and the liver as compared to the fat. Figure reproduced with permission of the publisher from a previous report [8]. Copyright Elsevier, 1983.
For experimental hyperventilation and the question of efficacy, female rats received 2.5 mL CCl4 per kg body weight as mixture with olive oil (1:1) and applied by gavage. Half of the animals were placed in a chamber ventilated by air in which part of the nitrogen was substituted by CO2 that caused an increase of the respiratory frequency by 50%. The other half of the CCl4-treated animals were kept in another chamber and had access to a similar gas mixture that did not contain CO2 [43,44]. The lethal dose (LD) was determined as LD50 for 4 days, with 3.6 ± 0.5 mL CCl4 per kg body weight for the non-hyperventilated animals versus 10.5 ± 3.0 mL CCl4 per kg body weight for the hyperventilated animals (Figure 14) [44,45]. The difference was statistically significant using the chi-square test (p = 0.015). These results led to the conclusion that experimental CO2-induced hyperventilation is effective in reducing short-term lethality due to acute CCl4 intoxication by gavage [44]. Of interest are other comparative studies of experimental CO2-induced hyperventilation versus lacking hyperventilation on CCl4 levels [43]. These levels are all reduced under conditions of experimental hyperventilation, not only in the blood (Figure 15), but also in the liver (Figure 16), and the fat (Figure 17) [43]. Experimental CO2-induced hyperventilation also ameliorated liver injury by CCl4 as assessed by liver histology [44]. In comparison to non-hyperventilated animals showing pronounced centrilobular necroses and signs of steatosis, hyperventilated animals presented only few signs of liver injury. In addition, experimental CO2-induced hyperventilation partially prevented the increase in serum activities of AST, ALT, and GDH [43,44]. These experimental data substantiate the beneficial effect of CO2-induced hyperventilation on CCl4-induced liver injury and related lethality.
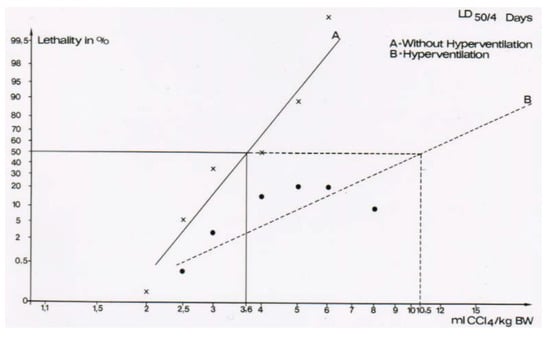
Figure 14.
The lethal dose (LD) was determined as LD50 for 4 days, with 3.6 ± 0.5 mL CCl4 per kg body weight for the non-hyperventilated animals versus 10.5 ± 3.0 mL CCl4 per kg body weight for the hyperventilated animals. The difference was statistically significant using the chi-square test (p = 0.015), reproduced with permission from [44,45]. Copyright Wiley, 1982 and Springer, 1982.
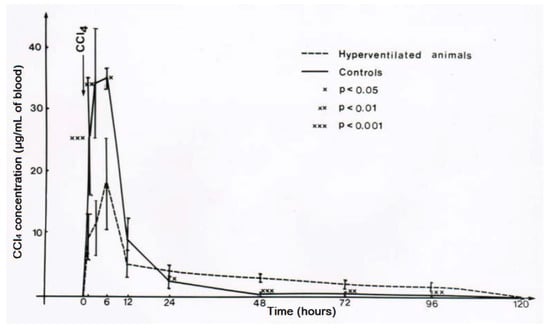
Figure 15.
Experimental hyperventilation leads to a reduction of CCl4 levels in the blood, reproduced with permission from [43]. Copyright Springer, 1983.
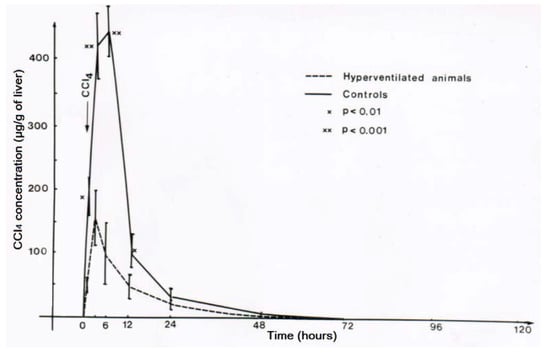
Figure 16.
Experimental hyperventilation reduces CCl4 levels in the liver, reproduced with permission from [43]. Copyright Springer, 1983.
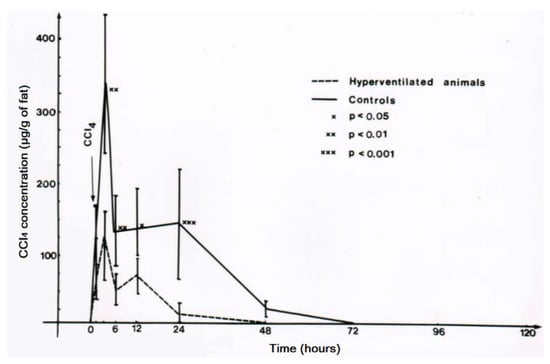
Figure 17.
Experimental hyperventilation reduces CCl4 in the fat tissue where it is soluble and can be quantified, reproduced with permission from [43]. Copyright Springer, 1983.
6. CCl4 and Hepatic Microsomal CYP 2E1
6.1. Carbon Tetrachloride
CCl4 is soluble in fat where it can be quantified (Figure 13 and Figure 17) [8,43], but with 0.08 g/100 mL water it is virtually insoluble in water [41,46]. For clinical purposes it is important that renal CCl4 excretion per hour is low with <0.6% of the dose taken up, conditions not recommending forced diuresis for increasing renal CCl4 excretion although this approach early applied is nephroprotective [41]. Conversely, total CCl4 excretion in breath after 1 h is as much as 33% of the dose taken up [40,46], and this is why in CCl4 intoxication forced ventilation is recommended to accelerate CCl4 removal via the lungs.
Agreement exists that only around 1% of the incorporated CCl4 is responsible for liver injury while 99% thereof will leave the body unchanged via the lungs (Figure 18) [29,47]. This requires additional efforts to minimize toxic effects by CCl4 and its metabolites at the microsomal level.
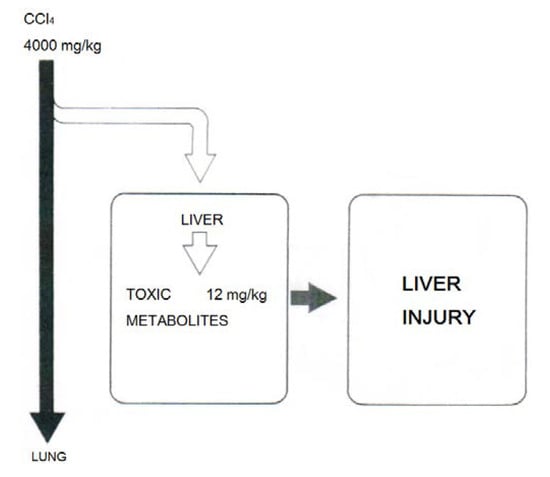
Figure 18.
Experimental data suggest that only around 1% of the incorporated CCl4 is responsible for liver injury while 99% thereof will leave the body unchanged via the lungs, reproduced with permission from [47]. Copyright Schattauer, 1975.
6.2. Cytochrome P450 2E1
There is also consensus that hepatic microsomal CYP2E1 is the preferred isoenzyme of CYP responsible for the conversion of CCl4 to toxic intermediates [5,6,12,48,49,50,51,52] in analogy to other toxins such as vinyl chloride and dimethylnitrosamine (Figure 19), requiring NADPH-cytochrome 450 reductase, NADPH + H+ as reducing equivalent, and O2 (Figure 20), with various steps involving CYP using CCl4 as substrate and other aliphatic halogenated hydrocarbons (Figure 21). Since experimental administration of CCl4 drastically decreased CY2E1, CYP2B, CYP3A2, CYP2C11, and CYP1A2 mRNA and protein expressions [53], CYP isoenzymes others than CYP2E1 may be involved in catalyzing CCl4.
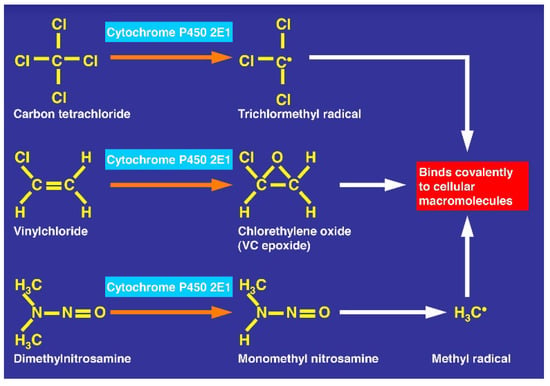
Figure 19.
CCl4 metabolized by cytochrome P450 2E1 similarly to other toxins such as vinyl chloride and dimethylnitrosamine.
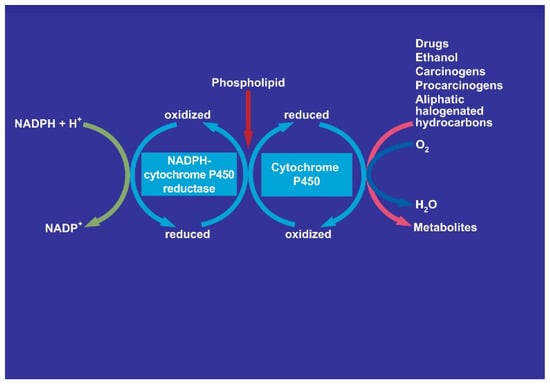
Figure 20.
Involvement of cytochrome P450 in the microsomal metabolism of various substrates including aliphatic halogenated hydrocarbons with carbon tetrachloride as example. The NADPH-cytochrome P450 uses NADPH + H+ and will itself be reduced, allowing the cytochrome P450 to be transferred from the oxidized state to the reduced state. The overall reactions needs also molecular oxygen and phospholipids.
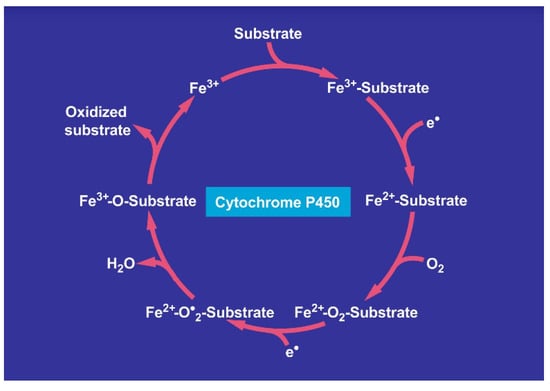
Figure 21.
Cycle involving cytochrome P450 for the metabolism of various substrates including carbon tetrachloride.
6.2.1. Molecular Oxygen
Molecular oxygen is required in the liver cell for the microsomal NADPH-dependent degradation of CCl4 (Figure 20 and Figure 21), a process that leads to a variety of intermediates including free radicals and ROS [54]. Referring to anecdotal reports, the suggestion has been made that hyperbaric oxygen treatment may ameliorate CCl4 hepatotoxicity in humans and animals [29]. However, the role of oxygen in liver injury by CCl4 is still a matter of debate [29,54]. With the CO2-induced hyperventilation, pO2 values are achieved in the blood that are well within the normal range (Table 6) and are likely achievable in the liver, but it has not been investigated whether this is protective or injurious to the liver. This uncertainty was also discussed for hyperbaric oxygen [29]. Although hyperbaric oxygen treatment with focus on the injury at the microsomal level may be an option for human CCl4 intoxications, its superiority over the CO2-induced hyperventilation targeting at acceleration of toxin removal has not been established.
6.2.2. Destabilization and Suicidal Inactivation
CCl4 causes a rapid decrease in CYP2E1 [55,56]. Mechanism-based inactivation of cytochrome P450 can result in the chemical modification of the heme, the protein, or both as a result of covalent binding of modified heme to the protein. Circumstantial evidence suggests that the inactivation of P4502E1 by CCl4 is a result of P4502E1 proteolysis from the microsomal membrane. While there are many potential pathways for protein degradation, the loss of P4502E1 was associated with increased formation of high molecular weight microsomal ubiquitin conjugates. The formation of ubiquitin-conjugated microsomal protein, which correlates with P4502E1 loss, suggests that ubiquitination may represent a proteolytic signal for the rapid and selective proteolysis of certain unstable conformations of P4502E1 from the endoplasmic reticulum [55]. In other studies using quantum chemical calculations, the anaerobic metabolism of CCl4 by P450 enzymes was investigated [56]. It was found that the substrate CCl4 might undergo one or two subsequent one-electron reductions to generate different reactive metabolites, trichloromethyl radical (˙CCl3) and dichlorocarbene (:CCl2) respectively. Meanwhile, it was the reduced ferrous heme complex rather than the unreduced ferric heme complex that could directly achieve such reductions. Based on the formation of the former reactive metabolite, a further one-electron reduction could take place with the assistance of a proton to yield the latter reactive species, i.e., a further reductive dechloridation of ˙CCl3 could take place. In addition, the ˙CCl3 species was capable of binding covalently to the meso-carbon atom of the prosthetic group, leading to the suicidal destruction of P450 enzymes. Whereas the :CCl2 species with CO as its hydrolysis product was involved in the CCl4-dependent reversible P450 inhibition, it was not significantly involved in the CCl4-dependent irreversible P450 destruction. It is obvious that the reductive metabolism of CCl4 to reactive intermediates by P450 enzymes is an essential prerequisite for its toxicity [56]. Of importance in the clinical context, such destructed CYP isoenzymes are inactive for another injurious metabolic attack by another CCl4 molecule. A cascade of events leading to liver injury by CCl4 has been described in detail recently [34].
6.2.3. Down-Regulation by Glucose
Short-term treatment with 400 g glucose per day is recommended in CCl4 intoxication (Table 4) [36] This suggestion is based on the high dosed glucose treatment of acute intermittent porphyria [57] in order to downregulate the hepatic ALA synthase activity, reducing thereby the formation of δ-aminolevulinic acid (ALA) [58,59] and heme as the precursor of the hemoprotein CYP [59]. Indeed, the existence of this ALA-dependent pathway has been verified by studies on incorporation of radioactive-delta-aminolevulinic acid into microsomal CYP [60].
6.2.4. Inhibition by Cimetidine
Cimetidine was included as pharmacotherapy in the treatment recommendations for CCl4 intoxication (Table 4), based on experimental studies showing that this drug reduced liver injury and lethality in animals with CCl4 poisoning [61]. This ameliorating effect is likely due to an inhibitory effect of CYP isoenzymes including CYP2E1 (Figure 20) due to substrate competition of cimetidine versus CCl4. Comparative studies are needed to prove whether other injectable drugs or compounds are to be preferred in future cases, but this question is outside the focus of this review. Cimetidine could also inhibit CYP2E1 present in the kidneys [62] possibly responsible for initiating renal injury due to CCl4 degradation and leading to renal failure. Therefore, cimetidine may have a dual effect protecting the liver and kidneys in CCl4 intoxication. Apart from the therapy using cimetidine, more important is likely providing a priory the patient with sufficient electrolyte infusions combined with furosemide to achieve forced diuresis preventing renal insufficiency by CCl4 (Table 4).
6.2.5. Up-Regulation by Alcohol
CCl4 and ethanol share with CYP2E1 a common metabolic pathway (Figure 19, Figure 20 and Figure 21) [5,6] that is induced by chronic alcohol consumption [12]. This induction is viewed as risk factor for liver injury by acute CCl4 intoxication in animals, associated with increased covalent binding of 14CCl4 metabolites to microsomal protein in vitro and an increased metabolism of 14CCl4 to 14CO2 [11]. These changes likely occur also in those patients with a history of chronic alcohol consumption prior to the acute CCl4 intoxication (Table 1 and Table 7). In animal studies, concomitant acute application of ethanol with CCl4 reduces initially the liver injury by CCl4 but this is offset later on and replaced by potentiation of liver injury [16]. In other experimental studies, CCl4 levels were analyzed under various conditions in animals receiving intragastrically either CCl4 alone or combined with ethanol [63]. Three hours after experimental gavage, CCl4 levels were higher in the blood, the liver, and fat in the group of animals receiving CCl4 combined with ethanol as compared to those treated with CCl4 alone. Transferring these results to patients who acutely ingested CCl4 simultaneously with ethanol, their risk seems to be increased because they may experience initially higher CCl4 levels triggered by ethanol.
Hepatic microsomal cytochrome P450 (CYP), especially its isoenzyme CYP2E1, is also a component of the hepatic microsomal ethanol-oxidizing system (MEOS) involved in the hepatic metabolism of ethanol (Figure 20) [12,13,14]. The CYP2E1 content and MEOS activity are both inducible following prolonged alcohol use [12], whereas for MEOS activity even a single dose of ethanol is sufficient for its induction [15]. It was therefore not unexpected that chronic alcohol consumption predisposes to liver injury by CCl4 [11]. Other experimental studies focused on the effect of an acute dose of ethanol on the hepatotoxicity due to a single dose of CCl4 [16]. Under clinical aspects, alcohol acutely ingested or used chronically before is a major issue in patients acutely intoxicated by CCl4.
On a molecular basis, alcoholic liver injury is due to acetaldehyde (C2H4O) generated for instance via MEOS from ethanol (C2H5OH) as its first oxidation product and due to various reactive O2-species (ROS) [64]. These include Ethoxy radical CH3CH2O, Hydroxyethyl radical CH3C(.)HOH, Acetyl radical CH3CHO., Singlet radical 1O2, Superoxide radical HO.2, Hydrogen peroxide H2O2, Hydroxyl radical HO•, Alkoxyl radical RO., and Peroxyl radical ROO•. Some of these radicals are generated also during CCl4 decomposition by CYP 2E1. Because radical formation determines liver injury by ethanol [64] and CCl4 [5,6,54], both chemicals follow some type cascade of liver injury events as discussed for CCl4 [34] and ethanol [64], a hazardous combination for patients with an alcohol problem who are acutely intoxicated by CCl4.
6.2.6. Preexisting Liver Disease
A crucial question remains as to whether preexisting liver disease may have an impact on acute liver injury by CCl4 in humans. Cytochrome P450 isoenzymes 2E1, 2D6, 1A2 and 2C19 contents decline with increasing hepatic disease severity, but their activities were differently affected [65]. For instance, CYP2E1 activity was only lost in patients with decompensated cirrhosis. In the actual cohort with acute CCl4 intoxication, decompensated cirrhosis was not diagnosed in any of the patients at admission (Table 7). Therefore, the crucial question raised above remains unresolved regarding severe liver diseases in humans such as decompensated cirrhosis due to lack of clinical evidence. Theoretically, this question can be studied using animal models of prolonged CCl4 application to achieve cirrhosis [66,67,68]. Among these, a good approach is the animal model in which both variation and level of critical damage are monitored by the daily weight change of the rat in response to intragastric carbon tetrachloride given during light halothane/oxygen anesthesia; the response each time being used to calibrate the subsequent dose of carbon tetrachloride to fit the individual rat [68]. The method is effective in producing cirrhosis with ascites in about 75% of rats after 8–10 doses of carbon tetrachloride.
In addition, acute liver injury by CCl4 will presumably be attenuated by mild pre-existing liver diseases such as nonalcoholic fatty liver disease (NAFLD) or alcoholic fatty liver (AFL) due to increased CCl4 metabolism via CYP2E1, the isoenzyme commonly found with increased contents and enzymatic activities in both, NAFLD and AFL [69].
7. Liver Transplantation
Published data on liver transplantation in patients with acute liver failure due to CCl4 poisoning are not available, with the exception of a single patient who was treated by forced ventilation and received an orthotopic liver transplantation [70] but died from aspergillus sepsis after re-transplantation of the liver together with a kidney transplantation. This patient also experienced rhabdomyolysis that has never been described in previous cases. The recommendation is provided to delay transplantation until most of the toxin has been eliminated in order to prevent fatal graft damage. Considering a potential liver transplantation will remain a case by case decision.
8. Summarized Considerations of CCl4 Poisoning for Clinical Practice
Due to its highly toxic properties, CCl4 has been abandoned as a solvent from the market in many countries or is still used in the industry under strict regulatory surveillance. Nevertheless, acute intoxications by CCl4 still occur in humans who incorporated it by ingestion or inhalation causing serious health problems by organ injury including the liver and kidneys. Historical cases are valuable to understand previously limited treatment (Table 1 and Table 2), but progress has been made regarding pathogenesis, diagnosis and treatment of this potentially deleterious CCl4 poisoning (Table 3, Table 4, Table 5, Table 6 and Table 7).
It is now clear that the hepatic microsomal CYP 2E1 plays an important pathogenetic role for bioactivation of CCl4 to toxic radicals for initiating liver injury, requiring two strategies aiming to early eliminate CCl4 by gastro-intestinal lavage and hyperventilation and also to reduce microsomal toxification. Progress has been made in the clinical setting by measurements of CCl4 levels in poisoned patients to establish the diagnosis. Care must be taken for various complications that may occur during clinical treatment and have to be diagnosed in time (Table 8). These include disturbances of coagulation, renal injury and failure, respiratory insufficiency, and cardiac arrhythmias. Finally, treatment approaches are now much better defined (Table 4 and Table 5), and the clinical features are clearly described (Table 8).
Table 8.
Clinical characteristics of CCl4 liver injury.
In most patients intoxicated by CCl4, three phases are clinically apparent whereby the second phase is the interval phase in between with little or no symptoms (Table 8). CCl4 intoxication is a serious clinical issue as most centers may not well be prepared treating these patients when admitted, unless the required devices for the CO2-induced hyperventilation therapy are available locally and quickly at hands during the patient’s transfer to the center [34]
9. Conclusions
Acute intoxications by CCl4 are clinical challenges due to the associated high lethality rate, conditions that require quick initiation of therapy strategies to enhance CCl4 elimination via (1) gastro-intestinal lavage to clear the intestinal tract from ingested CCl4 and via (2) forced ventilation induced by CO2 to accelerate CCl4 removal as unchanged chemical via the lungs. As compared to CCl4, which in itself is not toxic, its metabolites generated from CCl4 via the hepatic microsomal cytochrome P450 2E1 represent toxic radicals that attack cellular structures including proteins and phospholipids leading to apoptosis and cell necrosis. To reduce these toxic events at the microsomal levels, two approaches are beneficial, (1) high doses of glucose should be applied intravenously to downregulate cytochrome P450 levels, and (2) the intravenous application of drugs such as cimetidine with the potency to inhibit cytochrome P450 functions and thereby reducing the conversion of CCl4 to toxic radicals. Whereas these two approaches primarily help reduce liver injury, CCl4-related renal injury must be circumvented by forced diuresis, keeping in mind that this approach does not help accelerating CCl4 removal through the kidneys. It is obvious that various strategies are applicable to reduce the deleterious effects of CCl4.
Acknowledgments
There was no funding of this commissioned article. The author is grateful to Hartmut Frenzel, Institute of Pathology of the Heinrich Heine University of Düsseldorf (Germany) for providing analyses of light microscopy of liver specimens of some of the patients and for analyzing and providing the electron microscopy figures.
Conflicts of Interest
The author declares no conflict of interest.
References
- Smillie, W.G.; Pessao, S.B. Treatment of hookworm disease with carbon tetrachloride. Am. J. Hyg. 1923, 3, 35–45. [Google Scholar] [CrossRef]
- Plaa, G.L. Chlorinated methanes and liver injury: Highlights of the past 50 years. Ann. Rev. Pharmacol. Toxicol. 2000, 40, 43–65. [Google Scholar] [CrossRef] [PubMed]
- Recknagel, R.O.; Glende, E.A., Jr.; Dolak, J.A.; Waller, R.L. Mechanisms of carbon tetrachloride toxicity. Pharmacol. Ther. 1989, 43, 139–154. [Google Scholar] [CrossRef]
- Weber, L.W.D.; Boll, M.; Stampfl, A. Hepatotoxicity and mechanism of action of haloalkanes: Carbon tetrachloride as a toxicological model. Crit. Rev. Toxicol. 2003, 33, 105–136. [Google Scholar] [CrossRef] [PubMed]
- Stoyanovsky, D.A.; Cederbaum, A.I. Metabolism of carbon tetrachloride to trichloromethyl radical: An ESR and HPLC-EC study. Chem. Res. Toxicol. 1999, 12, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Raucy, J.L.; Kraner, J.C.; Lasker, J.M. Bioactivation of halogenated hydrocarbons by cytochrome P4502E1. Crit. Rev. Toxicol. 1993, 23, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Al-Rasheed, N.M.; Fadda, L.M.; Ali, H.M.; Abdel Baky, N.A.; El-Orabi, N.F.; Al-Rasheed, N.M.; Yacoub, H.I. New mechanism in the modulation of carbon tetrachloride hepatotoxicity in rats using different natural antioxidants. Toxicol. Mech. Methods 2016, 26, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Vierke, W.; Goldermann, L. Carbon tetrachloride (CCl4) levels and serum activities of liver enzymes following acute CCl4 intoxication. Toxicol. Lett. 1983, 17, 175–180. [Google Scholar] [CrossRef]
- Vierke, W.; Gellert, J.; Teschke, R. Head-space gas chromatographic analysis for rapid quantitative determination of carbon tetrachloride in blood and liver of rats. Arch. Toxicol. 1982, 51, 91–99. [Google Scholar] [CrossRef]
- Goldermann, L.; Gellert, J.; Teschke, R. Quantitative assessment of carbon tetrachloride levels in human blood by head-space gas chromatography: Application in a case of suicidal carbon tetrachloride intoxication. Intensive Care Med. 1983, 9, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Hasumura, Y.; Teschke, R.; Lieber, C.S. Increased carbon tetrachloride hepatotoxicity, and its mechanism, after chronic ethanol consumption. Gastroenterology 1974, 66, 226–234. [Google Scholar]
- Lieber, C.S. Cytochrome P-4502E1: Its physiological and pathological role. Physiol. Rev. 1997, 77, 517–544. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Matsuzaki, S.; Ohnishi, K.; DeCarli, L.M.; Lieber, C.S. Microsomal ethanol oxidizing system (MEOS): Current status of its characterization and its role. Alcohol. Clin. Exp. Res. 1977, 1, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Ohnishi, K.; Hasumura, Y.; Lieber, C.S. Hepatic microsomal ethanol oxidizing system: Isolation and reconstitution. In Microsomes and Drug Oxidations; Ullrich, V., Roots, I., Hildebrandt, A., Estabrook, R.W., Conney, A.H., Eds.; Pergamon Press: Oxford, UK, 1977; pp. 103–110. [Google Scholar]
- Petersen, D.R.; Atkinson, N.; Hjelle, J.J. Increase in hepatic microsomal ethanol oxidation by a single dose of ethanol. J. Pharmacol. Exp. Ther. 1982, 221, 275–281. [Google Scholar] [PubMed]
- Teschke, R.; Hauptmeier, K.H.; Frenzel, H. Effect of an acute dose of ethanol on the hepatotoxicity due to carbon tetrachloride. Liver Int. 1983, 3, 100–109. [Google Scholar] [CrossRef]
- Docherty, J.F.; Burgess, E. The action of carbon tetrachloride on the liver. Br. Med. J. 1922, 2, 907–908. [Google Scholar] [CrossRef] [PubMed]
- McGuire, L.W. Carbon tetrachloride poisoning. J. Am. Med. Assoc. 1932, 99, 988–989. [Google Scholar] [CrossRef]
- Lehnherr, E.R. Acute carbon tetrachloride poisoning: Report of a case. Arch. Int. Med. 1935, 56, 98–104. [Google Scholar] [CrossRef]
- Moon, H.D. The pathology of fatal carbon tetrachloride poisoning with special reference to the histogenesis of the hepatic and renal lesions. Am. J. Pathol. 1950, 26, 1041–1057. [Google Scholar] [PubMed]
- Jennings, R.B. Fatal fulminant acute carbon tetrachloride poisoning. Arch. Pathol. 1955, 59, 269–284. [Google Scholar]
- Guild, W.R.; Young, J.V.; Merrill, J.P. Anuria due to carbon tetrachloride intoxication. Ann. Intern. Med. 1958, 48, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- New, P.S.; Lubash, G.D.; Scherr, L.; Rubin, A.L. Acute renal failure: Associated with carbon tetrachloride intoxication. J. Am. Med. Assoc. 1962, 181, 197–200. [Google Scholar] [CrossRef]
- Fischl, J.; Labi, M. Gas chromatic determination of carbon tetrachloride in a case of accidental poisoning. Isr. J. Med. Sci. 1966, 2, 84–85. [Google Scholar] [PubMed]
- Dume, T.; Herms, W.; Schröder, E.; Wetzels, E. Klinik und Therapie der Tetrachlorkohlenstoffvergiftung. Dtsch. Med. Wochenschr. 1969, 94, 1646–1651. [Google Scholar] [CrossRef] [PubMed]
- Kennaugh, R.C. Carbon tetrachloride overdose. A case report. S. Afr. Med. J. 1975, 49, 635–636. [Google Scholar] [PubMed]
- Hadi, S.F.; Mikatti, N.E. Acute carbon tetrachloride poisoning. Intensive Care Med. 1981, 7, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, K.; Ukida, M.; Bode, C. Aminoacid concentration in plasma of patients with liver necrosis after carbon tetrachloride poisoning (three cases). Dtsch. Med. Wochenschr. 1982, 107, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Truss, C.D.; Killenberg, P.G. Treatment of carbon tetrachloride poisoning with hyperbaric oxygen. Gastroenterology 1982, 82, 767–769. [Google Scholar] [PubMed]
- Fogel, R.P.; Davidman, M.; Poleski, M.H.; Spanier, A.H. Carbon tetrachloride poisoning treated with hemodialysis and total parenteral nutrition. Can. Med. Assoc. Med. J. 1983, 128, 560–561. [Google Scholar]
- Ruprah, M.; Mant, T.G.K.; Flanagan, R.J. Acute carbontetrachloride poisoning in 19 patients: Implications for diagnosis and treatment. Lancet 1985, 325, 1027–1029. [Google Scholar] [CrossRef]
- Hoshino, T.; Komatsu, M.; Ono, T.; Funaoka, M.; Kato, J.; Ishii, T.; Kuramitsu, T.; Miura, K.; Masamune, O.; Hojo, H. A case of acute carbon tetrachloride poisoning. Acta Hepatol. 1994, 35, 882–886. [Google Scholar] [CrossRef][Green Version]
- Mydlík, M.; Derzsiová, K.; Frank, K. Renal replacement therapy in acute poisonings—One center experience. Prz. Lek. 2013, 70, 381–385. [Google Scholar] [PubMed]
- Teschke, R. Intoxications by aliphatic halogenated hydrocarbons: Hepatotoxic risks for patients and clinical issues including role of CO2-induced hyperventilation as therapy option. J. Clin. Exp. Toxicol. 2018, 2, 20–24. [Google Scholar]
- Draeger. Draeger Gas Detection Devise. Available online: https://www.gasdetectionwarehouse.com/draeger-gas-detector-tubes-10-per-box-halogenated-hydrocarbons/ (accessed on 8 March 2018).
- Teschke, R. Forced ventilation as therapy for acute intoxications due to halogenated aliphatic hydrocarbons. Experience in 60 patients. Therapiewoche 1987, 37, 339–344. [Google Scholar]
- Pebay-Peyroula, F.; Nicaise, A.M. Élimination pulmonaire des toxiques-mesure-applications toxicologiques. J. Eur. Toxicol. 1970, 5, 300–308. [Google Scholar]
- Kindler, U.; Goeckenjahn, G.; Barthels, F.; Grabensee, B. Hyperventilationstherapie bei Patienten mit Vergiftungen durch Halogenkohlenwasserstoffewasserstoffe. Intensivmedizin 1977, 14, 362–367. [Google Scholar]
- Lemburg, P.; Sprock, I.; Bretschneider, A.; Storm, W.; Göbel, U. A new concept of therapy in accidental intoxications with halogenated hydrocarbons. Vet. Hum. Toxicol. 1979, 21, S37–S40. [Google Scholar]
- Teschke, R.; Jehle, J.; Altrogge, A. Leberenzymveränderungen bei Vergiftungen durch halogenierte Kohlenwasserstoffe unter Hyperventilationstherapie. In Pädiatrische Intensivmedizin II; Lemburg, P., Ed.; Thieme Stuttgart: Stuttgart, Germany, 1981; pp. 39–48. [Google Scholar]
- Pothmann, R.; Lemburg, P.; Sprock, I.; Göbel, U. Hyperventilationstherapie bei oraler Vergiftung mit halogenierten Kohlenwasserstoffen. In Pädiatrische Intensivmedizin II; Lemburg, P., Ed.; Thieme Stuttgart: Stuttgart, Germany, 1981; pp. 229–233. [Google Scholar]
- Teschke, R.; Goldermann, L.; Vierke, W.; Hanrath, R.D.; Gellert, J. Forcierte Ventilation. In Aktuelle Intensivmedizin 1, Diagnose, Verlaufskontrolle und Therapie Schwerer Exogener Vergiftungen; Deutsch, E., Kleinberger, G., Ritz, R., Schuster, H.P., Eds.; Schattauer: Stuttgart, Germany, 1984; pp. 117–131. [Google Scholar]
- Gellert, J.; Goldermann, L.; Teschke, R. Effect of CO2-induced hyperventilation on carbon tetrachloride (CCl4) levels following acute CCl4 poisoning. Intensive Care Med. 1983, 9, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, H.; Heidenreich, T.; Gellert, J.; Teschke, R. Protective effect of CO2-induced hyperventilation on the hepatotoxicity elicited by carbon tetrachloride. Liver Int. 1982, 2, 376–384. [Google Scholar] [CrossRef]
- Gellert, J.; Frenzel, H.; Heidenreich, T.; Goldermann, L.; Vierke, W.; Teschke, R. Effektivität der CO2-induzierten Hyperventilationstherapie durch halogenierte Kohlenwasserstoffe. Intensivmedizin 1982, 19, 293–297. [Google Scholar]
- Morgan, A.; Black, A.; Belcher, D.R. The excretion in breath of some aliphatic halogenated hydrocarbons following administration by inhalation. Ann. Occup. Hyg. 1970, 14, 219–233. [Google Scholar]
- McLean, A.E.M. Drugs, diet and liver injury. In Drugs and the Liver; Gerok, W., Sickinger, K., Eds.; Schattauer: Stuttgart, Germany, 1975; pp. 143–148. [Google Scholar]
- Zangar, R.C.; Benson, J.M.; Burnett, V.L.; Springer, D.L. Cytochrome P450 2E1 is the primary enzyme responsible for low-dose carbon tetrachloride metabolism in human liver microsomes. Chem. Biol. Interact. 2000, 15, 233–243. [Google Scholar] [CrossRef]
- Park, S.M.; Park, E.J.; Ko, G.; Kim, J.; Sohn, D.H. A study for regulation of ethanol-inducible P450 (CYP2E1) on CCl4-induced hepatic damage. Arch. Pharm. Res. 1995, 18, 179. [Google Scholar] [CrossRef]
- Sohn, D.H.; Yun, Y.P.; Park, S.S.; Veech, R.L.; Song, B.J. Post-translational reduction of cytochrome P450 2E1 by CCl4, its substrate. Biochem. Biophys. Res. Commun. 1991, 179, 449–454. [Google Scholar] [CrossRef]
- Manno, M.; De Matteis, F.; King, L.J. The mechanism of suicidal reductive inactivation of microsomal cytochrome P450 by carbon tetrachloride. Biochem. Pharmacol. 1988, 38, 1001–1007. [Google Scholar]
- Poyer, J.L.; Floyd, R.A.; McCay, P.B.; Janzen, E.G.; Davis, E.R. Spin-trapping of the trichloromethyl radical produced during enzyme NADPH oxidation in the presence of carbon tetrachloride or bromotrichloromethane. Biochem. Biophys. Acta 1978, 539, 402–409. [Google Scholar] [CrossRef]
- Ibrahim, Z.S.; Ishizuka, M.; Soliman, M.; ElBohi, K.; Sobhy, W.; Muzandu, K.; Elkattawy, A.M.; Sakamoto, K.Q.; Fujita, S. Protection by Nigella sativa against carbon tetrachloride-induced downregulation of hepatic cytochrome P450 isozymes in rats. Jpn. J. Vet. Res. 2008, 56, 119–128. [Google Scholar] [PubMed]
- McCay, P.B.; Lai, E.K.; Poyer, J.L.; DuBose, C.M.; Janzen, E.G. Oxygen and carbon-centered free radical formation during carbon tetrachloride metabolism. J. Biol. Chem. 1984, 259, 2135–2143. [Google Scholar] [PubMed]
- Tierney, D.J.; Haas, A.; Koop, D.R. Degradation of cytochrome P450 2E1: Selective loss after labilization of the enzyme. Arch. Biochem. Biophys. 1992, 293, 9–16. [Google Scholar] [CrossRef]
- Li, X.X.; Zheng, Q.C.; Wang, Y.; Zhang, H.X. Theoretical insights into the reductive metabolism of CCl4 by cytochrome P450 enzymes and the CCl4-dependent suicidal inactivation of P450. Dalton Trans. 2014, 43, 14833–14840. [Google Scholar] [CrossRef] [PubMed]
- Pischik, E.; Kauppinen, R. An update of clinical management of acute intermittent porphyria. Appl. Clin. Genet. 2015, 8, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Tschudy, D.P.; Welland, F.H.; Collins, A. The effect of carbohydrate feeding on the induction of delta-aminolevulinic acid synthetase. Metabolism 1964, 13, 396–406. [Google Scholar] [CrossRef]
- Puy, H.; Deybach, J.C. Haem biosynthesis and excretion of porphyrins. In Textbook of Hepatology, 3rd ed.; Rodés, J., Benhamou, J.-P., Blei, A., Reichen, J., Rizzetto, M., Eds.; Wiley: Hoboken, NJ, USA, 2007; pp. 207–214. Available online: http://www.gastrohep.com/ebooks/rodes/Rodes_2_3_10.pdf (accessed on 8 March 2018).
- Levin, W.; Jacobson, M.; Kutzman, R. Incorporation of radioactive-delta-aminolevulinic acid into microsomal cytochrome P450. Arch. Biochem. Biophys. 1972, 148, 262–269. [Google Scholar] [CrossRef]
- Homann, J.; Rotter, S.; Schneider, S.; Röttger, P.; Kratz, F.; Kroker, R.; Kamenisch, W.; Paul, F.; Matthes, K.J. Influence of cimetidine on CCl4-induced liver injury and survival in rats. Biochem. Pharmacol. 1985, 34, 415–416. [Google Scholar] [CrossRef]
- Das, J.; Ghosh, J.; Manna, P.; Sil, P.C. Taurine protects acetaminophen-induced oxidative damage in mice kidney through APAP urinary excretion and CYP2E1 inactivation. Toxicology 2010, 269, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Vierke, W.; Gellert, J. Effect of ethanol on carbon tetrachloride levels and hepatotoxicity after acute carbon tetrachloride poisoning. Arch. Toxicol. 1984, 56, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R. Alcoholic steatohepatitis (ASH) and acute alcoholic hepatitis (AH): Cascade of events, clinical features, and pharmacotherapy options. Exp. Opin. Pharmacother. 2018, in press. [Google Scholar]
- Lauschke, V.M.; Ingelman-Sundberg, M. The importance of patient-specific factors for hepatic drug response and toxicity. Int. J. Mol. Sci. 2016, 17, 1714. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, W.; Clària, J.; Arroyo, V.; Rodés, J. Review article: Carbon tetrachloride induced cirrhosis in rats: A useful tool for investigating the pathogenesis of ascites in chronic liver disease. J. Gastroenterol. Hepatol. 1992, 7, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Delire, B.; Stärkel, P.; Leclercq, I. Animal models for fibrotic liver diseases: What we have, what we need, and what is under development. J. Clin. Transl. Hepatol. 2015, 3, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Proctor, E.; Chatamra, K. High yield micronodular cirrhosis in the rat. Gastroenterology 1982, 83, 1183–1190. [Google Scholar] [PubMed]
- Leung, T.M.; Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Nehoda, H.; Wieser, C.; Koller, J.; Konigsrainer, K.A.; Battista, H.J.; Vogel, W.; Margreiter, R. Recurrent liver failure with severe rhabdomyolysis after liver transplantation for carbon tetrachloride intoxication. Hepatogastroenterology 1998, 45, 191–195. [Google Scholar] [PubMed]





















© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).