Chemistry of Ammonothermal Synthesis


Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
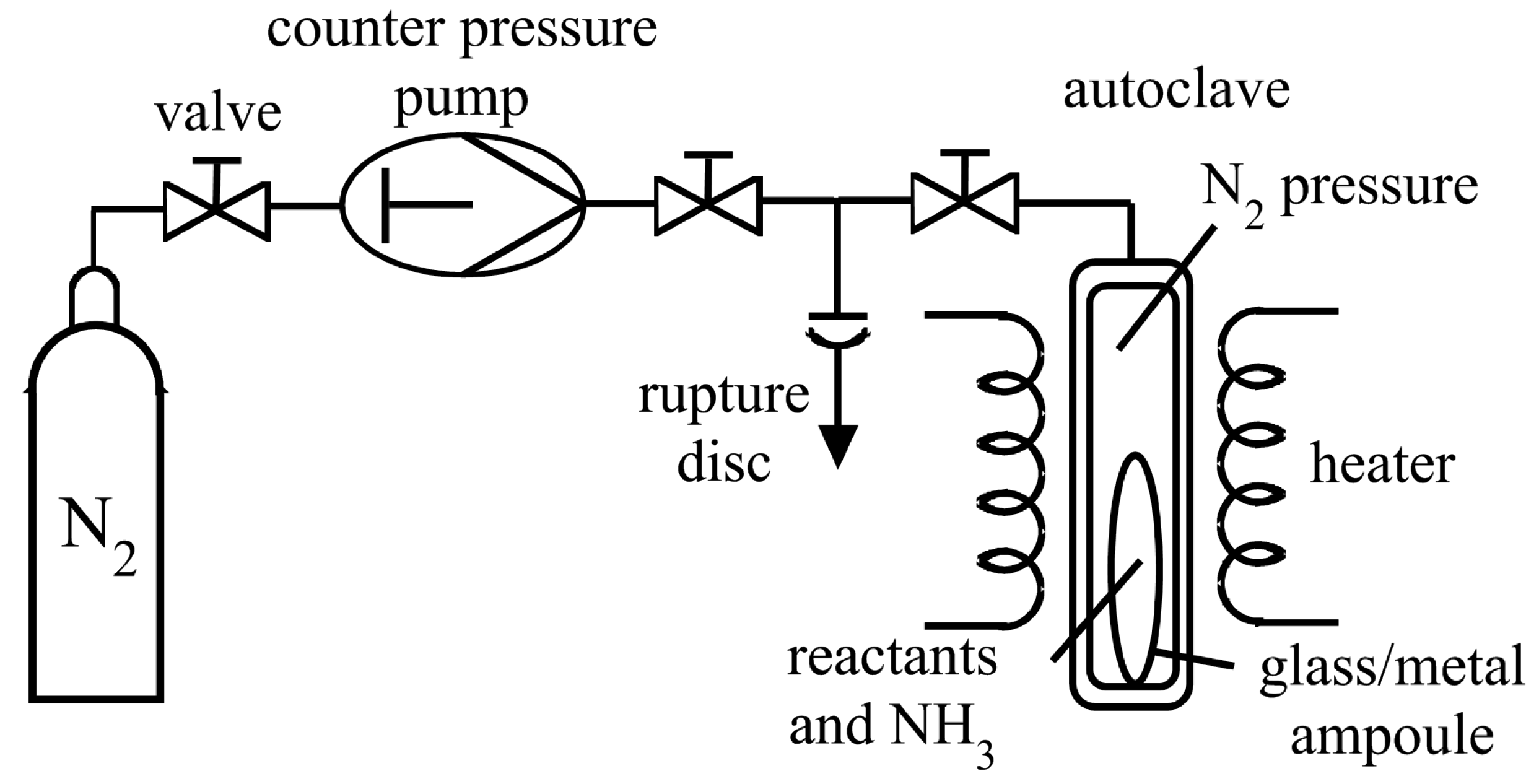{kind=link}
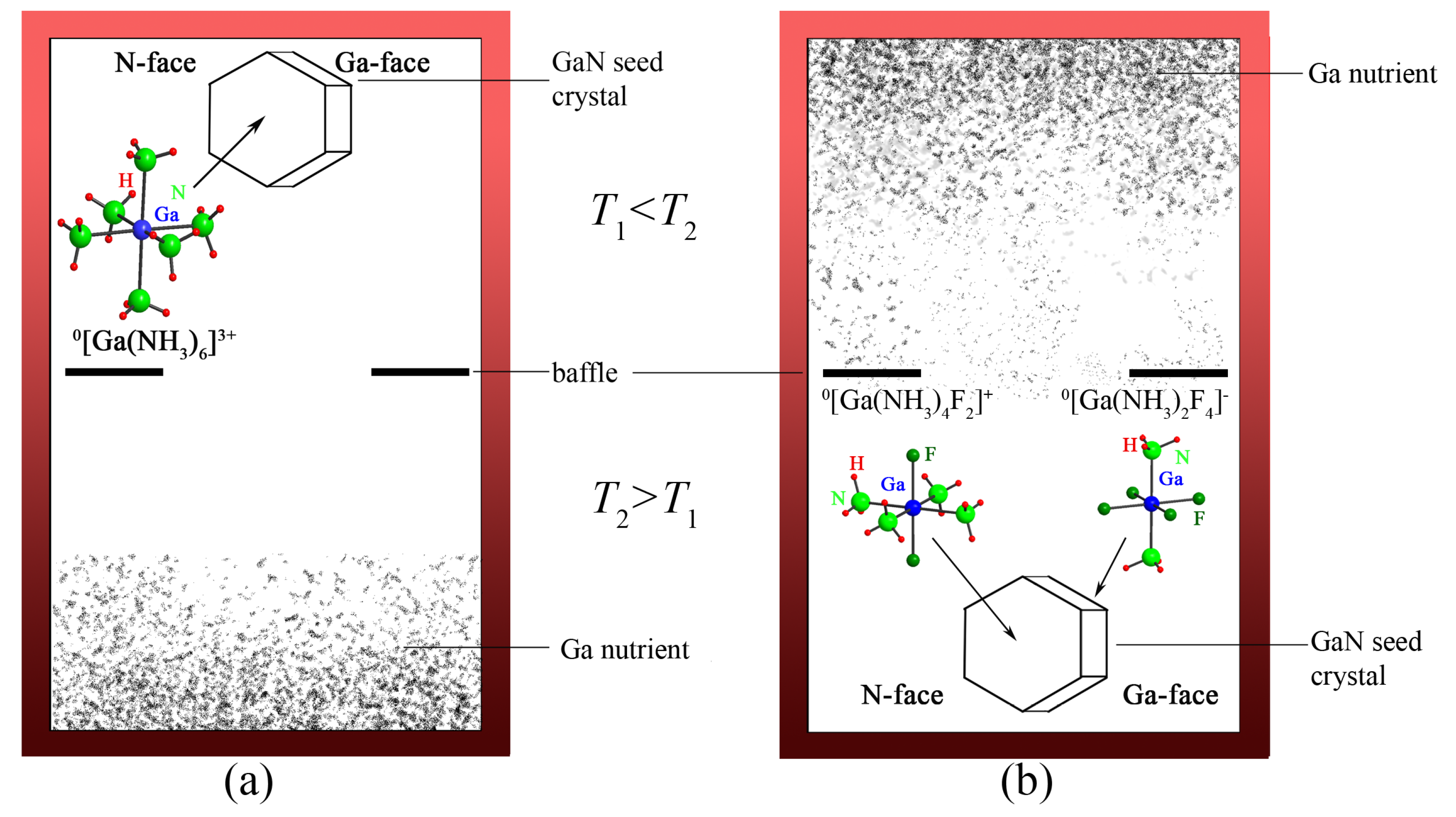{kind=link}
| Solvent | Examples for products | References | |
|---|---|---|---|
| (i) | H2O | ZnO, α-quartz, α-Al2O3, GaPO4 | [12,13,14,18] |
| H2O + isopropyl alcohol | BaTiO3 | [17] | |
| H2O + C2H3Cl3 | Diamond | [19] | |
| NH3 | GaN, AlN, Cu3N, Cs2S2, NaOH | [10,11,20,21,22] | |
| HCl, HBr, HI | BiSCl, BiTeBr, SbSeI | [23] | |
| Ethanol | LiMnO | [15] | |
| Benzyl alcohol | LaCa/Sr/BaMnO | [16] | |
| (ii) | Ethylendiamine | Cu7Te4, CuInSe2 | [8,24] |
| Diethylamine | CuInSe2 | [8] | |
| THF | β-MnS | [25] | |
| (iii) | C6H6 | Se, c/h-BN, γ-MnS | [25,26] |
| Xylene | InAs | [9] | |
| Toluene | CuCr2Se4 | [27] | |
| CO2 | Poly vinyl chloride | [28] | |
| Br2 | SbSBr | [23] |
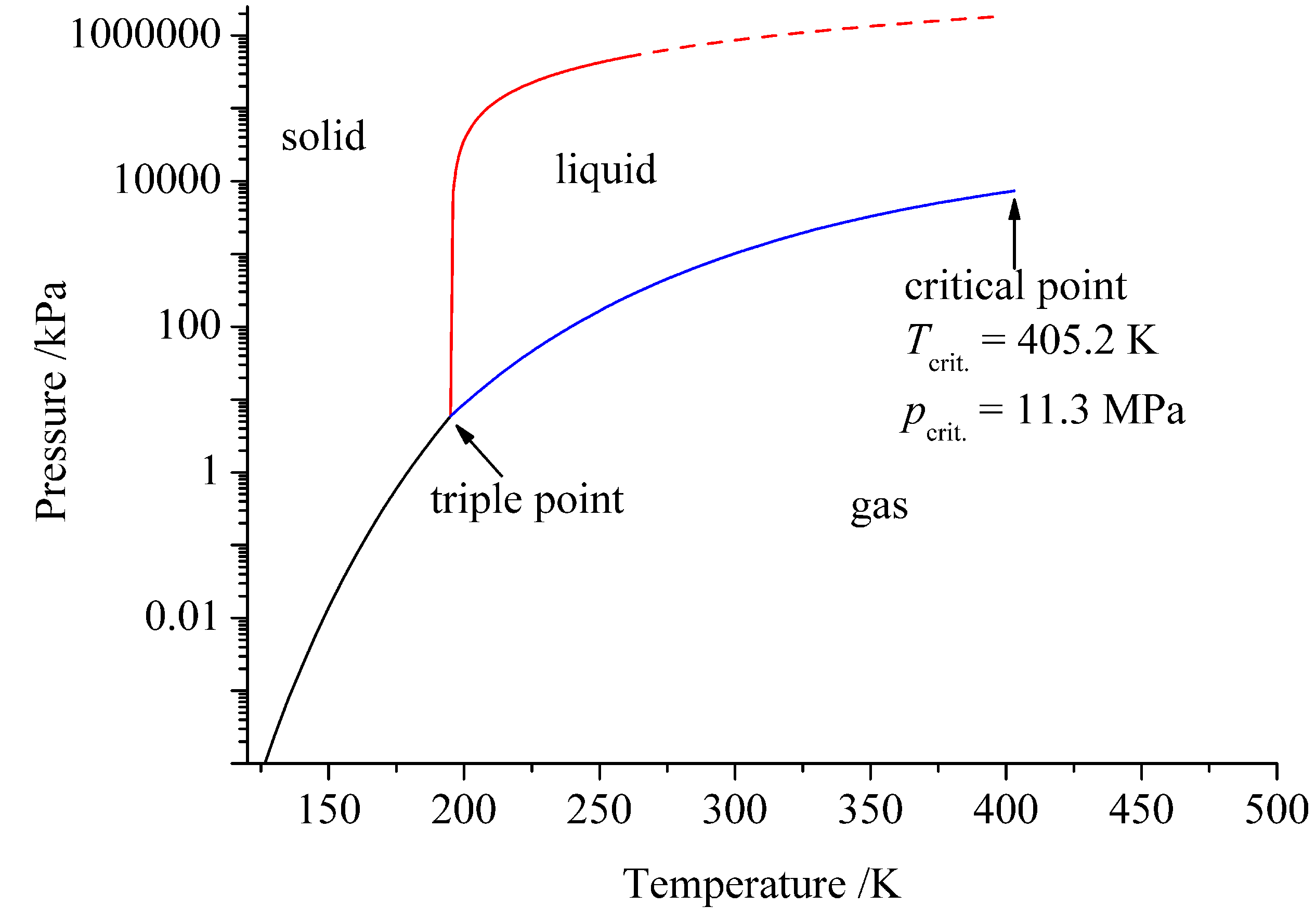
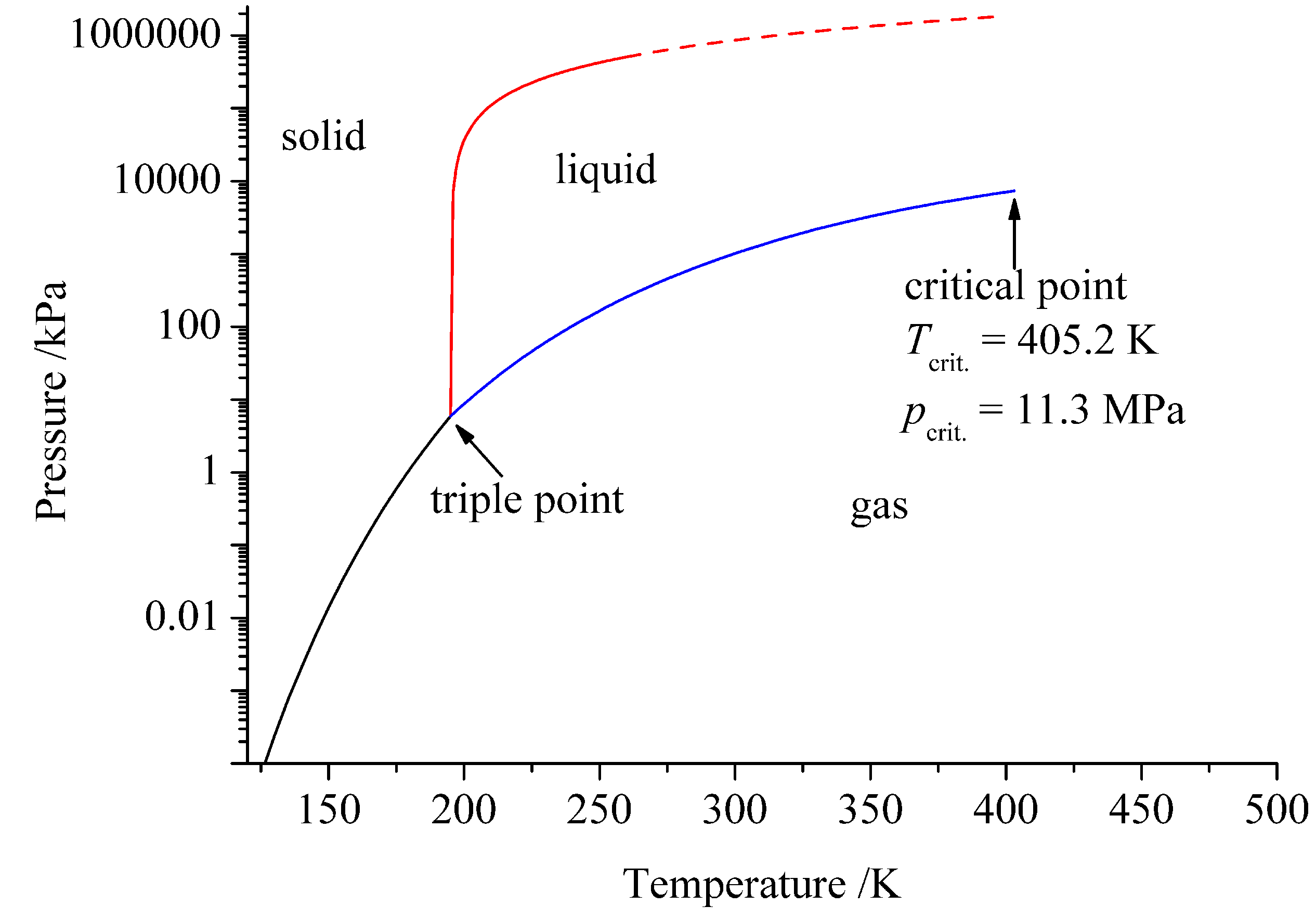
2. Ammonia as Solvent
| Water | Ammonia | |
|---|---|---|
| T/K | 647.65 | 405.2 |
| p/MPa | 22.1 | 11.3 |
| 78.3 (298 K) | 16.9 (298 K) | |
| Autoprotolysis | 2H2O ⇄ H3O+ + OH− | 2NH3 ⇄ NH + NH |
| Ionic product | (298 K) | (239 K) |
| pk | 15.7 | 4.75 |
| Proton affinity/eV | −7.9 | −9.2 |
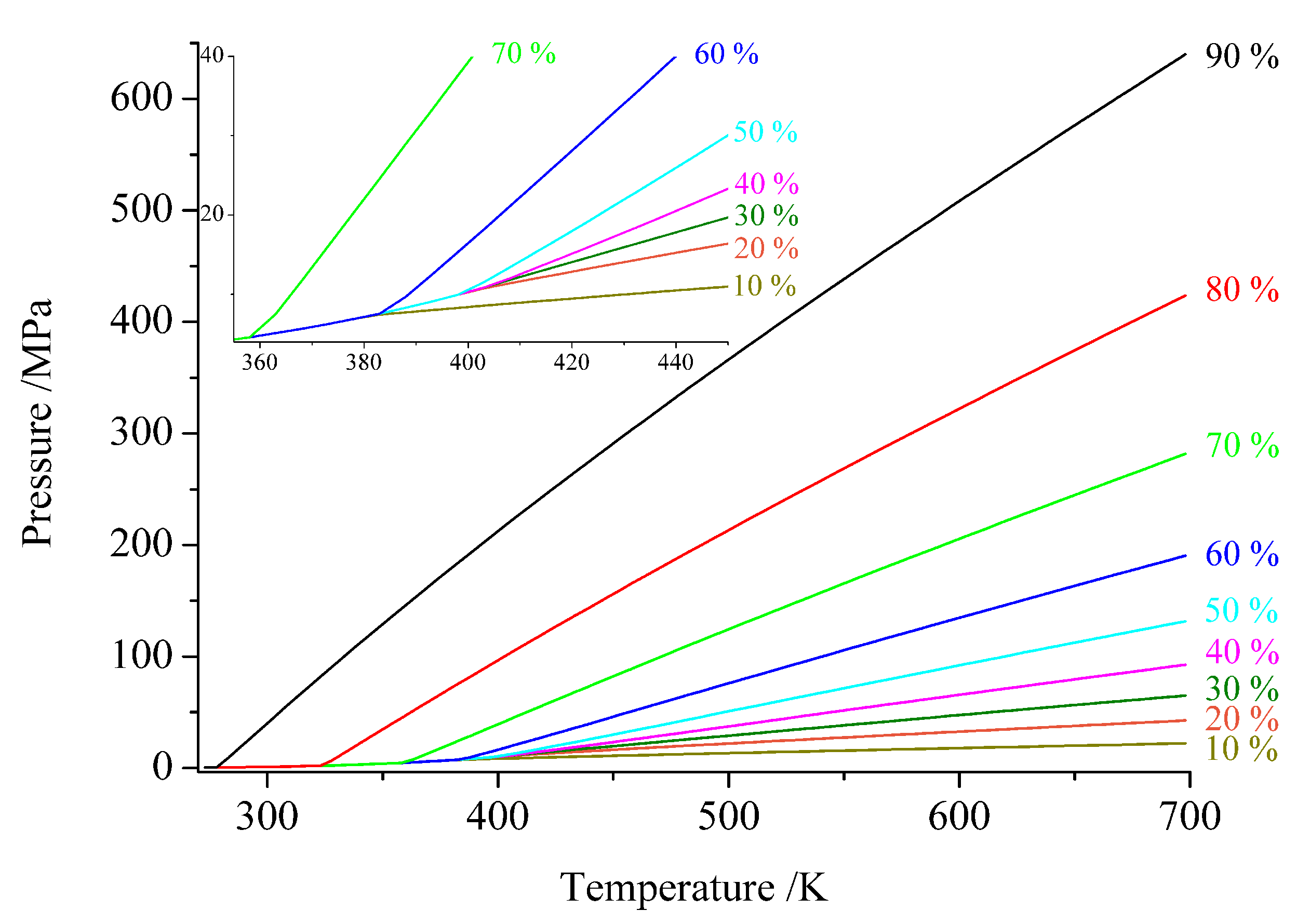
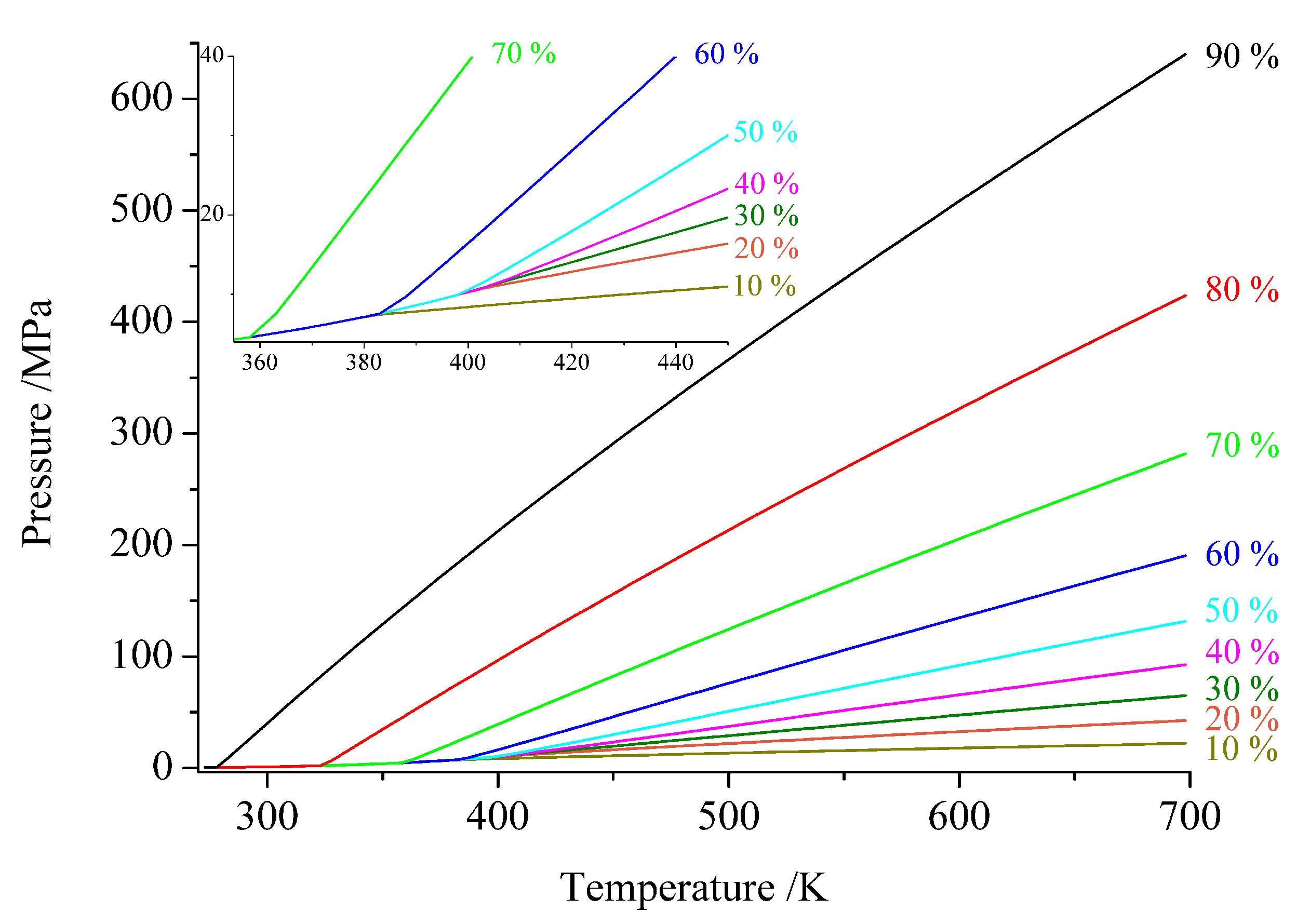
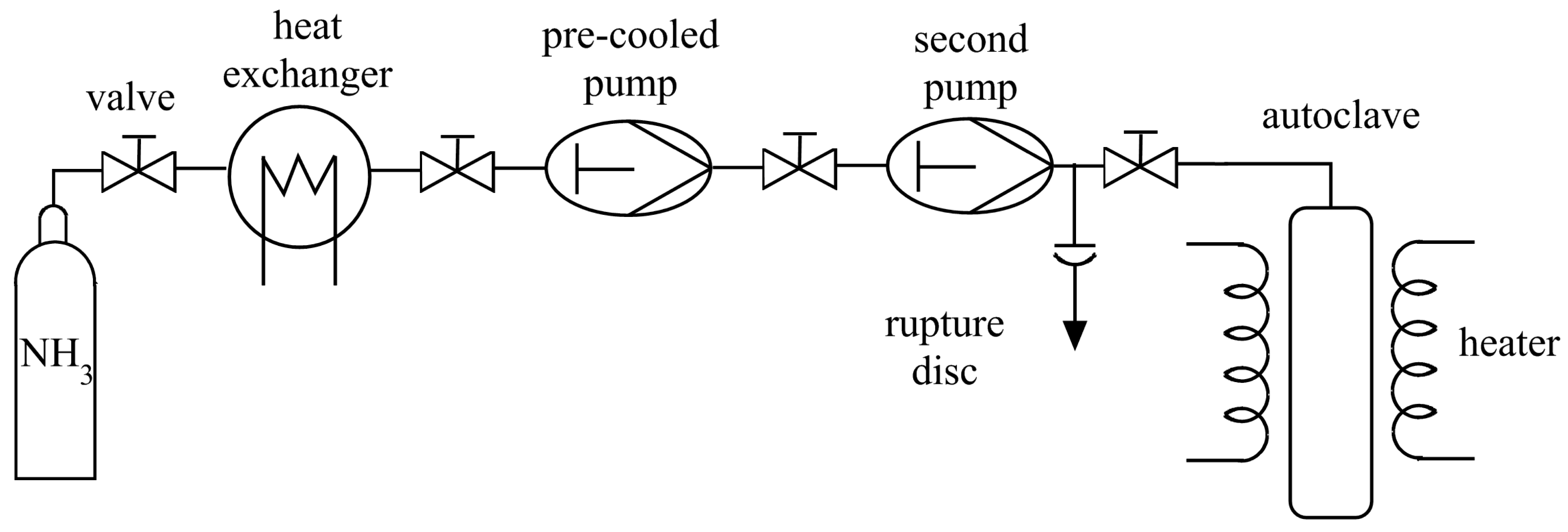
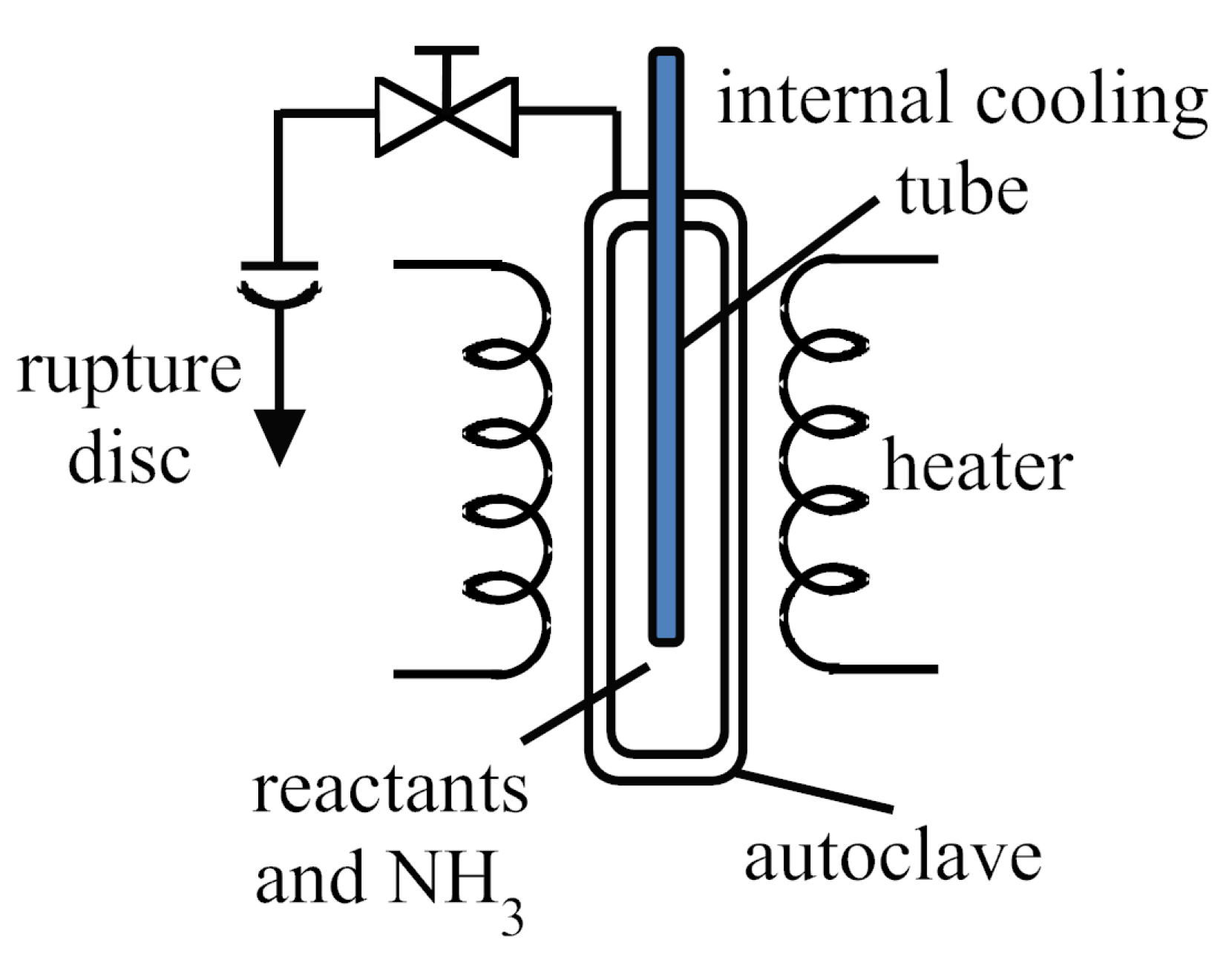
3. Technical Details for Ammonothermal Reactions
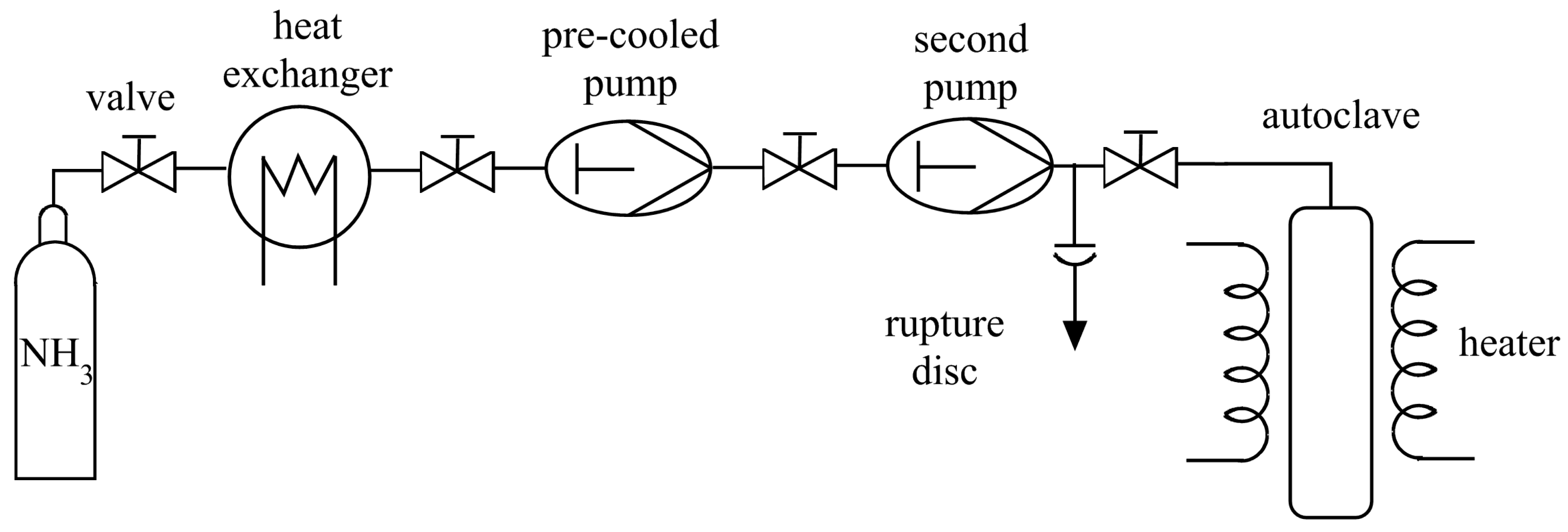

3.1. Reaction Vessels

4. Crystallization Process
4.1. Thermodynamical Parameters
4.2. Chemical Parameters
4.2.1. Ammonobasic Systems
4.2.2. Ammonoacidic Systems
4.2.3. Intermediate Species controlling Solubility and Growth Rates
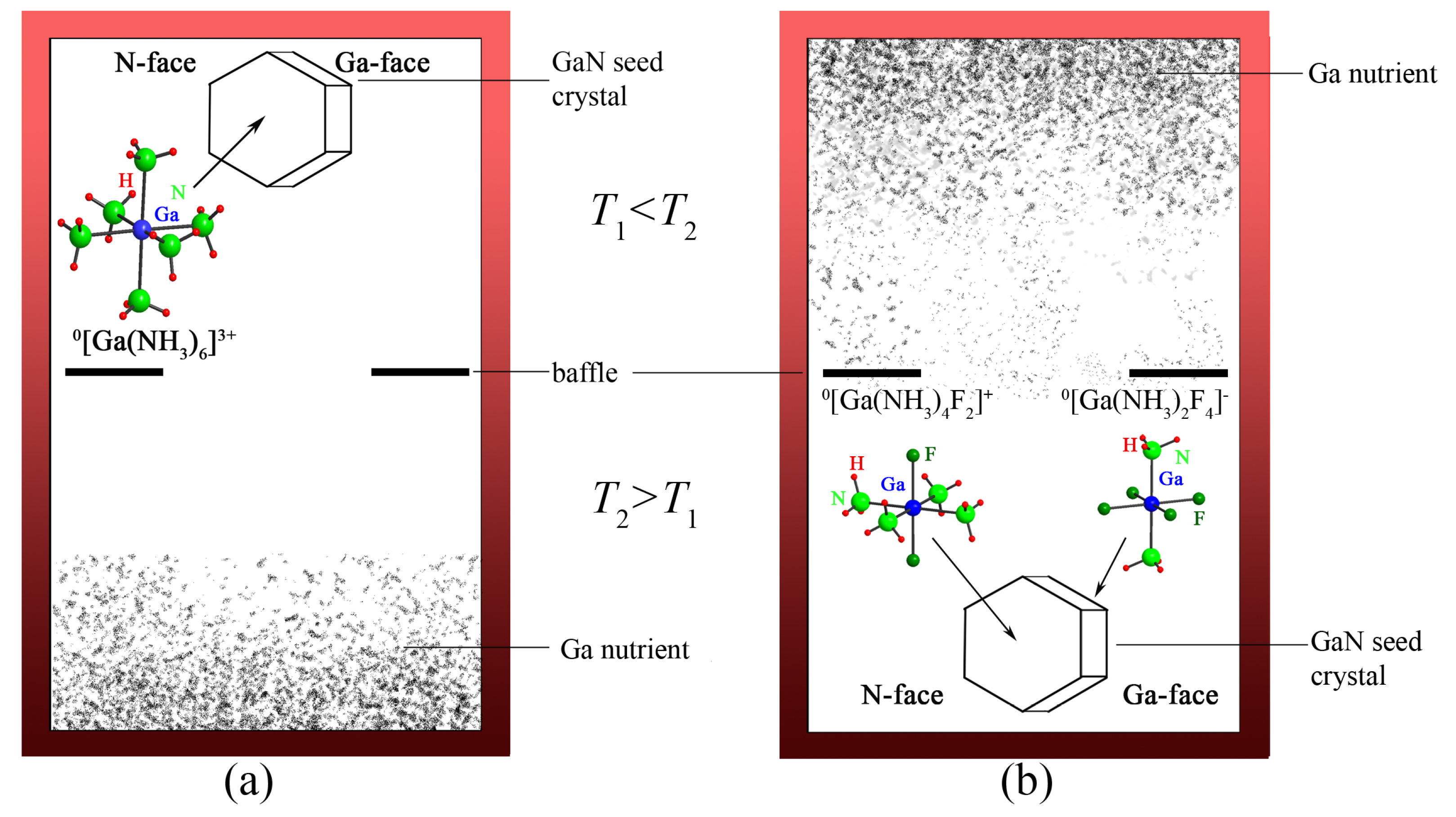
5. Compounds from Ammonothermal Synthesis
5.1. Ammoniates of Metal Halides
| Compound | Reactants + mineralizer | T/K | p/MPa | t/d | References |
|---|---|---|---|---|---|
| Al(NH3)2F3 | AlN + NH4F | 673 | – | 3 | [83] |
| [Al(NH3)5Cl]Cl2 | AlCl3 | 603 | – | 3–6 | [84] |
| [Al(NH3)5Br]Br2 | AlBr3 | 623 | – | 3–6 | [84] |
| [Al(NH3)5I]I2 | AlI3 | 673 | – | 3–6 | [84] |
| [Al(NH3)6]I3 · NH3 | Al + NH4I | 393 | 9 | 1 | [89] |
| Ga(NH3)3F3 | Ga + NH4F | 753 | 238 | 3 | [66] |
| [Ga(NH3)5Cl]Cl2 | Ga + NH4Cl | 853 | 95 | 1 | [66] |
| [Ga(NH3)6]Br3 · NH3 | GaBr3 | 197–373 | ≤6 | – | [66] |
| [Ga(NH3)6]I3 · NH3 | GaI3 | 197–373 | ≤6 | – | [66] |
| [Mn(NH3)6]I2 | Mn + I2 | 673–873 | 600 | ≤7 | [90] |
| [Fe(NH3)6]I2 | Fe + I2 | 673–873 | 600 | ≤7 | [90] |
| Cs3La(NH2)6 · NH3 | Cs + La | 490–570 | 400–600 | 31–103 | [100] |
| Cs4La(NH2)7 · NH3 | Cs + La | 490–570 | 400–600 | 31–103 | [100] |
| BaAl2(NH2)8 · 2NH3 | Al + BaAl2 | 823 | 245 | 29 | [101] |
| InF2(NH2) · NH3 | InN + NH4F | 673 | 220 | 1 | [83] |
5.2. Binary Amides and Deuteroamides
5.2.1. Alkali Metal Amides
| Compound | Reactants + mineralizer | T/K | p/MPa | t/d | Sample | References |
|---|---|---|---|---|---|---|
| LiND2 | Li | 473 | 304 | – | m.c. | [110] |
| NaNH2 | Na | 393 | ≤10 | 14 | s.c. | [111] |
| NaND2 | Na | 423–473 | 405 | 8 | m.c. | [111] |
| KND2 | K | 320 | ≤10 | 4 | m.c. | [112] |
| CsNH2 | Cs | 423 | 180 | 2 | m.c. | [113] |
| CsND2 | Cs | 423 | 180 | 2 | m.c. | [113] |
| Be(NH2)2 | Be | 633 | 253 | 5 | s.c. | [2] |
| Be(NH2)2 | Be + NaN3 | 643 | 355 | 20 | s.c. | [114] |
| Mg(NH2)2 | Mg | 613–653 | 10 | 2 | m.c. | [115] |
| Mg(NH2)2 | Mg + NaN3 | 523 | 253 | 2–4 | s.c. | [1,2] |
| Mg(NH2)2 | Mg3N2 | 633–648 | 1 | ≤7 | m.c. | [115] |
| Mg(NH2)2 | Mg + NaNH2 | 623–653 | 212–345 | 2–4 | s.c. | [115] |
| Ca(NH2)2 | Ca | 370 | 6 | 14 | s.c. | [116] |
| Sr(NH2)2 | Sr + K | 625 | 550 | 7 | s.c. | [117] |
| Sr(ND2)2 | Sr | 625 | 550 | 9 | m.c. | [117] |
| Ba(NH2)2 | Ba | 533 | 324 | 3 | s.c. | [1] |
| Ba(NH2)2 | Ba | 398 | ≤20 | 120 | s.c. | [118] |
| Mn(NH2)2 | Mn + Na2[Mn(NH2)4] | 393 | 10 | 10 | s.c. | [119] |
| Zn(NH2)2 | Zn + Na2[Zn(NH2)4]·0·5NH3 | 523 | 380 | 60 | s.c. | [119] |
| La(NH2)3 | La + KNH2 | 623 | 405 | 6 | s.c. | [120] |
| Sm(NH2)3 | Sm | 403–493 | 200–500 | – | – | [121] |
| Eu(NH2)2 | Eu + K | 523–673 | 500–557 | 7–9 | s.c. | [122] |
| Eu(NH2)2 | Eu | 323 | ≥0.9 | 3 | m.c. | [123] |
| Yb(NH2)3 | Yb | 453 | 507 | 32 | m.c. | [123] |
5.2.2. Alkaline-Earth Metal Amides
5.2.3. Lanthanum Amide, Samarium Amide, Europium Amide and Ytterbium Amide
5.2.4. Transition Metal, Group III and Group IV Metal Amides
5.3. Ternary Amides
| Compound | CN A(1), A(1)/A(2) by NH | Reactants | T/K | p/MPa | t/d | References |
|---|---|---|---|---|---|---|
| K2Li(NH2)3 | 6, 4 | K + Li 2:1 | 333 | 70 | 60 | [145,146] |
| KLi(NH2)2 | a, 4 | K + Li 1:1 | 333–473 | 70–210 | 4 | [145] |
| KLi3(NH2)4 | 8, 4 | K ≤ Li | 333–473 | 70–210 | 4 | [145] |
| KLi7(NH2)8 | 8, 4 | K ≤ Li | 333–473 | 70–210 | 4 | [145] |
| K2[Mg(NH2)4] | 7, 4 | K + Mg | 423 | 200 | 3 | [147] |
| Rb2[Mg(NH2)4] | 7, 4 | Rb + Mg | 423 | 200 | 3 | [147] |
| Cs[Mg(NH2)4] | 9/11, 4 | Cs + Mg | 415 | 200 | 2 | [148] |
| NaCa(NH2)3 | 6, 6 | Na + Ca 5:1–1:2 | 740–773 | 500 | 60 | [149,150] |
| KCa(NH2)3 | 6, 6 | K + Ca 1:1 | 573 | 500 | 20 | [151] |
| RbCa(NH2)3 | 8, 6 | Rb + Ca 1:2–3:1 | 573 | 500 | 17 | [152] |
| CsCa(NH2)3 | 8, 6 | Cs + Ca | 573–773 | 500–600 | 10–35 | [153] |
| KSr(NH2)3 | 6, 6 | K + Sr 1:1 | 570 | 500 | 7 | [149] |
| RbSr(NH2)3 | 6, 6 | Rb + Sr 3:1–1:1 | 540–573 | 800 | 8 | [150] |
| CsSr(NH2)3 | 8, 6 | Cs + Sr | 573–673 | 500–600 | 14–55 | [153] |
| KBa(NH2)3 | 6, 6 | K + Ba 3:1–1:1 | 540–573 | 500 | 7 | [150] |
| RbBa(NH2)3 | 6, 6 | Rb + Ba 3:1–1:1 | 540–573 | 500 | 7 | [150] |
| CsBa(NH2)3 | 8, 6 | Cs + Ba | 473 | 500 | 11 | [154] |
| Na2Sr3(NH2)8 | 6, 6 | Na + Sr 1:2 | 570 | 500 | 4 | [149] |
| Compound | CN A(1), R by NH | Reactants | T/K | p/MPa | t/d | References |
|---|---|---|---|---|---|---|
| Na3[La(NH2)6] | 6, 6 | Na + La 1:1 | 523 | 507 | 30 | [131] |
| KLa2(NH2)7 | 6, 8 | K + La 1:2 | 623 | 507 | 6 | [155] |
| K3[La(NH2)6] | 6, 6 | K + La | 473 | 405 | – | [156] |
| RbLa2(NH2)7 | a | [3] | ||||
| Rb3[La(NH2)6] | 6, 6 | Rb + La 3:1 | 573 | 400–450 | 6–12 | [157] |
| CsLa2(NH2)7 | 9, 8 | Cs + La | 470–570 | 400–600 | 3–100 | [3,158] |
| Na3[Ce(NH2)6] | a | [3] | ||||
| KCe2(NH2)7 | 6, 8 | K + Ce 1:1 | 455 | 400–500 | 5–10 | [76] |
| K3[Ce(NH2)6] | 6, 6 | K + Ce 3:1 | 455 | 400–500 | 5–10 | [76] |
| Cs3Ce2(NH2)9 | 12, 6 | Cs + Ce | 490 | 600 | 21 | [159] |
| Cs3Ce2(NH2)9 | 12, 6 | Cs + Ce | 490 | 200 | 21 | [159] |
| Na3[Nd(NH2)6] | 6, 6 | a | [3] | |||
| KNd2(NH2)7 | 6, 8 | a | [3] | |||
| K3[Nd(NH2)6] | 6, 6 | a | [3] | |||
| Rb3[Nd(NH2)6] | 6, 6 | Rb + Nd 3:1 | 573 | 400 | 7 | [157] |
| Cs3Nd2(NH2)9] | 12, 6 | Cs + Nd | 430–530 | 300 | 7–100 | [159] |
| Na3[Sm(NH2)6] | 6, 6 | a | [3] | |||
| KSm2(NH2)7 | 6, 8 | K + Sm 1:2 | 403–493 | 200–500 | – | [121] |
| K3[Sm(NH2)]6 | 6, 6 | K + Sm 3:1 | 403–493 | 200–500 | – | [121] |
| Cs3Sm2(NH2)9 | 12, 6 | Cs + Sm | 470 | 600 | 60 | [159] |
| KEu(NH2)3 | 6, 6 | K + Eu 1:1 | 573 | 500 | 3 | [122] |
| RbEu(NH2)3 | 6, 6 | Rb + Eu 1:1-2:1 | 540–73 | 500 | 28 | [150] |
| Rb3[Eu(NH2)6] | 10, 6 | Rb + Eu 10:1 | 423 | 500 | 40 | [160] |
| CsEu(NH2)3 | 6, 6 | Cs + Eu | 573 | 500–600 | 9–14 | [153] |
| K3[Eu(NH2)6] | 6, 6 | K + Eu 12:1 | 573 | 500 | 3 | [122,149] |
| Na2Eu3(NH2)8 | 6, 6 | Na + Eu 1:1 | 570 | 500 | 8 | [149] |
| NaGd(NH2)4 | 4, 6 | NaNH2 + Gd 1:1 | 493 | 507 | 20 | [161] |
| Na3[Gd(NH2)6] | 6, 6 | NaNH2 + Gd 3:1 | 573 | 304 | 51 | [3,161] |
| K3[Gd(NH2)6] | 6, 6 | a | [3] | |||
| Cs3Gd2(NH2)9 | 12, 6 | Cs + Gd | 440 | 600 | 160 | [162] |
| NaY(NH2)4 | 6, 6 | Na + Y 1:1 | 523 | 507 | 7 | [163] |
| KY(NH2)4 | 6, 6 | K + Y 1:4–6 | 485–505 | 600 | 22 | [164] |
| RbY(NH2)4 | 11, 6 | Rb + Y 1:4–6 | 485–505 | 600 | 22 | [164] |
| Na3[Y(NH2)6] | 6, 6 | Na + Y 3:1 | 523 | 507 | 7 | [163] |
| Cs3Y2(NH2)9 | 12, 6 | Cs + Y | 490 | 600 | 70 | [162] |
| Rb3[Y(NH2)6] | 10, 6 | Rb + Y 3:1 | 473 | 500 | 14 | [157,160] |
| K3[Y(NH2)6] | 10, 6 | K + Y 3:1 | 473 | 500 | 14 | [157,160] |
| NaYb(NH2)4 | 4, 6 | Na + Yb 1:1 | 413–463 | 507 | 14 | [163] |
| Na3[Yb(NH2)6] | 6, 6 | Na + Yb 3:1 | 453 | 608 | 8 | [165] |
| KYb(NH2)7 | a | [3,121] | ||||
| K3[Yb(NH2)6] | 10, 6 | K + Yb 3:1 | 473 | 500 | 14 | [157,160] |
| Rb3[Yb(NH2)6] | 8, 6 | Rb + Yb 3:1 | 473 | 500 | 14 | [157,160] |
| Cs3Yb2(NH2)9 | 12, 6 | Cs + Yb | 450 | 600 | 160 | [162] |
| Compound | CN A(1), B/M by NH | Reactants | T/K | p/MPa | t/d | References |
|---|---|---|---|---|---|---|
| Li[Ga(NH2)4] | 4, 4 | Ga + LiNH2 | 673 | 250 | 3 | [67] |
| Li[Ga(NH2)4] | 4, 4 | Ga + LiNH2 | 673 | 250 | 3 | [67] |
| Na2[Ga(NH2)4]NH2 | 4, 4 | Ga + NaNH2 | 853 | 130 | 2 | [67,82] |
| Na[Ga(NH2)4] | 4, 4 | Na + Ga | 853 | 130 | 2 | [67,81] |
| Rb[Al(NH2)4] | 12, 4 | Rb + Al | 393–473 | 80–120 | 20 | [166] |
| Cs[Al(NH2)4] | 12, 4 | Cs + Al | 423–473 | 120–600 | 15 | [166] |
| Na2[Mn(NH2)4] | 4/6, 4 | Na + Mn | 373 | 10 | – | [167] |
| K2[Zn(NH2)4] | 7, 4 | Zn + KNH2 | 720 | 249 | 2 | [168] |
5.4. Imides, Nitride Imides, Nitride Amides and Amide Azides
| Compound | Reactants mineralizer | T/K | p/MPa | t/d | References |
|---|---|---|---|---|---|
| MgNH | Mg3N2 | 773 | ≥5 | 7 | [115] |
| Th(NH)2 | Th + Li/Na/K | 573 | 608 | 29 | [33] |
| Th2N2NH | Th | 823 | 507 | 2 | [1] |
| ThN(NH2) | ThNJ + A(1)NH2 | 573 | 405 | 2 | [33] |
| Th3N2(NH)3 | ThNJ + A(1)NH2 | 623 | 608 | 27 | [33] |
| Si2N2NH | Si + KNH2 | 873 | 600 | 5 | [173] |
| ZrN(NH2) | ZrNI + KN3 | 633 | 507 | 10 | [75] |
| Cs2(NH2)N3 | Cs + Y | 463–493 | 500–600 | 21–26 | [174] |
5.5. Metal Hydrides and Nitride Hydrides
5.6. Nitrides
| Compound | Reactants + Mineralizer | T/K | p/MPa | t/d | Sample | References |
|---|---|---|---|---|---|---|
| c-Be3N2 | Be | 673 | 20.3 | 7 | m.c. | [2] |
| YN | Y + NH4I | 623 | 61 | 7 | m.c. | [163,174] |
| EuN | Eu + K 40:1 | 673 | 500 | 7 | s.c. | [122] |
| LaN | K3[La(NH2)6] + KNH2 | 650 | 500 | 10 | s.c. | [3] |
| LaN | La + Na | 523–773 | 300–507 | 10 | s.c. | [3,131] |
| CeN | Ce + Cs | 490 | 200 | 12 | – | [159] |
| SmN | Sm + K | 433–453 | 500 | 10–30 | – | [121] |
| GdN | Gd + NH4I | 523 | 507 | 23 | – | [161] |
| GaN | Ga + LiNH2/K | 823 | 500 | ≥7 | s.c. | [77] |
| GaN | GaN + NH4I | ≤1123 | ≤150 | – | s.c. | [62] |
| GaN | GaN + KNH2 + KI | 673 | 240 | 7 | s.c. | [180] |
| c-GaN | [Ga(NH)] + NH4I | 753 | – | 2–3 | s.c. | [181] |
| GaN:Mn | Ga + Mn + KNH2 | 723–823 | 400–500 | 3–10 | s.c. | [182] |
| GaN:Cr | Ga + CrBr3 + KNH2 | 723–823 | 400–500 | 3–10 | s.c. | [182] |
| GaN:Fe | Ga + Fe + LiNH2 | 723–773 | 400–500 | 3–10 | s.c. | [182] |
| AlN | Al + K | 723–873 | 200 | 1–18 | s.c | [78] |
| AlN | Al + NH4Cl | 723 | – | 2 | s.c | [11] |
| InN | In + KNH2 | 723 | – | – | m.c. | [43] |
| FeNiN | Fe + FeI2 | 733–853 | 600–800 | – | s.c. | [183,184,185] |
| -Mn6N | MnI2 + NaNH2 | 673–723 | 600 | 60–120 | s.c. | [167] |
| η-Mn3N2 | Mn + I2 or MnI2 + NaNH2 | 673–873 | 600 | 30 | s.c. | [167,186] |
| η-Mn3N2 | Mn + K/Rb | 673–873 | ≤600 | 35 | s.c. | [186] |
| ϵ-Mn4N | Mn + GaN A(1)NH2 | 723–823 | 400–500 | 3–10 | m.c. | [182] |
| Ni3N | [Ni(NH3)6]Cl2 + NaNH2 | 523 | 200 | 7 | s.c. | [187] |
| Cu3N | [Cu(NH3)4]NO3 | 623–853 | ≥600 | – | s.c. | [20] |
| CuPdN | [Cu(NH3)4]NO3 + Cu | 723 | 600 | 7 | s.c. | [188] |
| 0.020/0.989 | + [Pd(NH3)4](NO3)2 |
5.6.1. Alkaline-Earth Metal Nitrides
5.6.2. Group III and IV Nitrides
5.6.3. Rare-Earth Metal Nitrides
5.6.4. Transition Metal Nitrides
5.7. Non-Nitrogen Compounds
| Compound | Reactants + mineralizer | T/K | p/MPa | t/d | References |
|---|---|---|---|---|---|
| NaOH | NaOH | 523–473 | ≤600 | 10 | [22] |
| KOH | K(H2O)OH + KNH2 | ≤423 | ≤600 | ∼10 | [22] |
| RbOH | Rb(H2O)OH + RbNH2 | ≤365 | ≤600 | ∼10 | [22] |
| CsOH | Cs(H2O)OH + CsNH2 | 460 | 300 | 5 | [227] |
| CaOH | CaO + NH4I | 723 | 69–207 | 1 | [57,229] |
| K(H2O)OH | K(H2O)OH | 390–410 | 500 | 7–10 | [228] |
| Rb(H2O)OH | Rb(H2O)OH | 400–420 | 180 | 6 | [228] |
| Cs(H2O)OH | Cs(H2O)OH | 450 | 250 | 8 | [227] |
| LiHS | LiNH2 + H2S | 300–370 | – | – | [3,230] |
| KHS | KNH2 + H2S | 393 | ≤30 | 7 | [3,231] |
| CaS | CaS + NH4I | 573–673 | 69–207 | 1 | [57,229] |
| SrS | SrS + NH4I | 573–673 | 69–207 | 1 | [57,229] |
| CdS | CdS + NH4I | 573–673 | 69–207 | 1 | [57,229] |
| CuS | CuI + CaS + NH4I | 643 | 69–207 | 1 | [229] |
| Cu7S4 | CuI + CaS + NH4I | 643 | 69–207 | 1 | [229] |
| CaCu2S2 | CuI + CaS + NH4I | 573–673 | 69–207 | 1 | [57,229] |
| NH4Cu4S3 | CuI + CaS + NH4I | 573–673 | 69–207 | 1 | [229] |
| Na2S2 | a | [3,232] | |||
| K2S2 | a | [3,232] | |||
| Rb2S2 | a | [3,232] | |||
| Cs2S2 | Cs + Se | 573 | 200–300 | – | [21] |
| Na2S3 · NH3 | Na + S | 300–320 | 200 | – | [233] |
| K2S3 | K + S | 423 | 50 | – | [234] |
| Rb2S3 | Rb + S | 670 | 300 | – | [235] |
| Cs2S3 | Cs + S | 370 | 50 | – | [235] |
| K2S5 | a | [3,232] | |||
| Rb2S5 | Rb + S | 450 | 5 | – | [236] |
| Cs2S5 | Cs + S | 323–373 | 10–200 | – | [237] |
| Cs2Se | Cs + Se | 573 | 200 | – | [3,238] |
| Na2Se2 | a | [3,232] | |||
| K2Se2 | a | [3,232] | |||
| Rb2Se2 | a | [3,232] | |||
| Cs2Se2NH3 | a | [3,232] | |||
| K2Se3 | K + Se | 423 | 50 | – | [234] |
| Rb2Se3 | Rb + Se | 600 | 100 | – | [235] |
| Cs2Se3 | Cs + Se | 570 | 300 | – | [235] |
| Rb2Se5 | Rb + S | 450 | 500 | – | [236] |
| K2Te3 | a | [3,232] | |||
| Rb2Te3 | Rb + Te | 500 | 100 | – | [239] |
| Cs2Te3 | Cs + Te | 500 | 100 | – | [239] |
| Rb2Te5 | Rb + Te | 473 | 200 | 7 | [56] |
| Cs2Te5 | Cs + Te | 473 | 200 | 90 | [240] |
| K2[CN2] | Ga + K | 853 | 90 | – | [177] |
6. Conclusions
Acknowledgments
Conflicts of Interest
Nomenclature:
| A | Alkali or alkaline-earth metal, A(1) = alkali metal, A(2) = alkaline-earth metal |
| B | Further metal, might be main group metal, transition or rare-earth metal |
| M | Transition metal, excluding Sc, Y, La |
| R | Rare-earth metal, Sc, Y, La-Lu |
| X | Halide |
| E | Other main group element |
References
- Juza, R.; Jacobs, H.; Gerke, H. Ammonothermalsynthese von Metallamiden und Metallnitriden. Ber. Bunsenges. Phys. Chem. 1966, 70, 1103–1105. [Google Scholar]
- Juza, R.; Jacobs, H. Ammonothermal synthesis of magnesium and beryllium amides. Angew. Chem. Int. Ed. 1966, 5, 247. [Google Scholar] [CrossRef]
- Jacobs, H.; Schmidt, D. High-pressure ammonolysis in solid-state chemistry. Curr. Top. Mater. Sci. 1982, 8, 387–427. [Google Scholar]
- Byrappa, K.; Yoshimura, M. Handbook of Hydrothermal Technology a Technology for Crystal Growth and Materials Processing; Noyes Publications/Wiliam Andrew Publishing, LLC: Park Ridge, NY, USA, 2001. [Google Scholar]
- Largeteau, A.; Darracq, S.; Goglio, G.; Demazeau, G. Solvothermal crystal growth of functional materials. High Press. Res. 2008, 28, 503–508. [Google Scholar] [CrossRef]
- Rabenau, A. The role of hydrothermal synthesis in preparative chemistry. Angew. Chem. Int. Ed. 1985, 24, 1026–1040. [Google Scholar] [CrossRef]
- Demazeau, G. Solvothermal reactions: An original route for the synthesis of novel materials. J. Mater. Sci. 2007, 43, 2104–2114. [Google Scholar] [CrossRef]
- Li, B.B.; Xie, Y.; Huang, J.; Qian, Y. Synthesis by a solvothermal route and characterization of CuInSe2 nanowhiskers and nanoparticles. Adv. Mater. 1999, 11, 1456–1459. [Google Scholar] [CrossRef]
- Li, Y.D.; Duan, X.F.; Qian, Y.T.; Yang, L.; Ji, M.R.; Li, C.W. Solvothermal co-reduction route to the nanocrystalline III–V semiconductor InAs. J. Am. Chem. Soc. 1997, 119, 7869–7870. [Google Scholar] [CrossRef]
- Dwiliński, R.; Wysmolek, A.; Baranowski, J.; Kamińska, M.; Doradziński, R.; Jacobs, H. GaN synthesis by ammonothermal method. Acta Phys. Pol. A 1995, 88, 833–836. [Google Scholar]
- Lan, Y.C.; Chen, X.L.; Cao, Y.G.; Xu, Y.P.; Xun, L.; Xu, T.; Liang, J.K. Low-temperature synthesis and photoluminescence of AlN. J. Cryst. Growth 1999, 207, 247–250. [Google Scholar] [CrossRef]
- Nicholas, N.J.; Franks, G.V.; Ducker, W.A. The mechanism for hydrothermal growth of zinc oxide. CrystEngComm 2012, 14, 1232–1240. [Google Scholar] [CrossRef]
- Nacken, R. Hydrothermale Mineralsynthese zur Züchtung von Quarzkristallen. Chem. Ztg. 1950, 74, 745–749. [Google Scholar]
- Gleichmann, H.; Richert, H.; Hergt, R.; Barz, R.U.; Grassl, M. Hydrothermal liquid phase epitaxy of gallium orthophosphate on quartz crystal substrates. Cryst. Res. Technol. 2001, 36, 1181–1188. [Google Scholar] [CrossRef]
- Li, W.J.; Shi, E.W.; Chen, Z.Z.; Zhen, Y.Q.; Yin, Z.W. Solvothermal synthesis of superfine Li1−xMn2O4-σ powders. J. Solid State Chem. 2002, 163, 132–136. [Google Scholar] [CrossRef]
- Vázquez-Vázquez, C.; López-Quintela, A.M. Solvothermal synthesis and characterisation of La1−xAxMnO3 nanoparticles. J. Solid State Chem. 2006, 179, 3229–3237. [Google Scholar] [CrossRef]
- Bocquet, J.F.; Chhor, K.; Pommier, C. Barium titanate powders synthesis from solvothermal reaction and supercritical treatment. Mater. Chem. Phys. 1999, 57, 273–280. [Google Scholar] [CrossRef]
- Suchanek, W. Hydrothermal synthesis of alpha alumina (α-Al2O3) powders: Study of the processing variables and growth mechanisms. J. Am. Ceram. Soc. 2010, 93, 399–412. [Google Scholar] [CrossRef]
- Korablov, S.; Yokosawa, K.; Korablov, D.; Tohji, K.; Yamasaki, N. Hydrothermal formation of diamond from chlorinated organic compounds. Mater. Lett. 2006, 60, 3041–3044. [Google Scholar] [CrossRef]
- Zachwieja, U.; Jacobs, H. Ammonothermalsynthese von Kupfernitrid, Cu3N. J. Less-Common Met. 1990, 161, 175–184. [Google Scholar] [CrossRef]
- Böttcher, P. Zur Synthese und Struktur von Cs2S2. J. Less-Common Met. 1979, 63, 99–103. [Google Scholar] [CrossRef]
- Jacobs, H.; Kockelkorn, J.; Tacke, T. Hydroxide des Natriums, Kaliums und Rubidiums: Einkristallzüchtung und Röntgenographische Strukturbestimmung an der bei Raumtemperatur stabilen Modifikation. Z. Anorg. Allg. Chem. 1985, 531, 119–124. [Google Scholar] [CrossRef]
- Popolitov, V.; Litvin, B.; Lobachev, A. Hydrothermal crystallization of semiconducting compounds of group AV BVI CVII (AV:Sb, Bi; BVI:S, Se, Te; CVII:I, Br, Cl). Phys. Status Solidi A 1970, 3, K1–K4. [Google Scholar] [CrossRef]
- Li, B.; Xie, Y.; Huang, J.; Su, H.; Qian, Y. A Solvothermal route to nanocrystalline Cu7Te4 at low temperature. J. Solid State Chem. 1999, 146, 47–50. [Google Scholar] [CrossRef]
- Lu, J.; Qi, P.; Peng, Y.; Meng, Z.; Yang, Z. Metastable MnS crystallites through solvothermal synthesis. Chem. Mater. 2001, 13, 2169–2172. [Google Scholar] [CrossRef]
- Hao, X.; Yu, M.; Cui, D.; Xu, X.; Wang, Q.; Jiang, M. The effect of temperature on the synthesis of BN nanocrystals. J. Cryst. Growth 2002, 241, 124–128. [Google Scholar] [CrossRef]
- Ramesha, K.; Seshadri, R. Solvothermal preparation of ferromagnetic sub-micron spinel CuCr2Se4 particles. Solid State Sci. 2004, 6, 841–845. [Google Scholar] [CrossRef]
- Kendall, J.L.; Canelas, D.A.; Young, J.L.; deSimone, J.M. Polymerizations in supercritical carbon dioxide. Chem. Rev. 1999, 99, 543–564. [Google Scholar] [CrossRef] [PubMed]
- Bunsen, R. Über die Spannkraft einiger condensirten Gase. Ann. Phys. 1839, 46, 97–103. [Google Scholar] [CrossRef]
- Bunsen, R. Bemerkungen zu einigen Einwürfen gegen mehrere Ansichten über die chemisch-geologischen Erscheinungen in Island. Liebigs Ann. 1848, 65, 70–85. [Google Scholar] [CrossRef]
- Schafhäutl, A. Die neuesten geologischen Hypothesen und ihr Verhältnis zur Naturwissenschaft überhaupt. Gelehrte Anzeigen München 1845, 20, 577. [Google Scholar]
- De Sénarmont, M. Sur la formation des minéraux par voie humide dans les gites métallifères concrétionnés. Ann. Chim. Phys. 1851, 32, 129–175. [Google Scholar]
- Blunck, H.; Juza, R. Verbindungen des Thoriums mit Stickstoff und Wasserstoff. Z. Anorg. Allg. Chem. 1974, 410, 9–20. [Google Scholar] [CrossRef]
- Akasaki, I. Nitride semiconductors–Impact on the future world. J. Cryst. Growth 2002, 237–239, 905–911. [Google Scholar] [CrossRef]
- Zhu, D.; Wallis, D.; Humphreys, C. Prospects of III-nitride optoelectronics grown on Si. Rep. Progr. Phys. 2013, 76, 106501. [Google Scholar] [CrossRef] [PubMed]
- Ehrentraut, D.; Meissner, E.; Bockowski, M. Technology of Gallium Nitride Crystal Growth; Springer Materials Sciences: Berlin Heidelberg, Germany, 2010. [Google Scholar]
- Denis, A.; Goglio, G.; Demazeau, G. Gallium nitride bulk crystal growth processes: A review. Mater. Sci. Eng. R. 2006, R50, 167–194. [Google Scholar] [CrossRef]
- Dwiliński, R.; Doradziński, R.; Garczyński, J.; Sierzputowski, L.; Puchalski, A.; Kanbara, Y.; Yagi, K.; Minakuchi, H.; Hayashi, H. Bulk ammonothermal GaN. J. Cryst. Growth 2009, 311, 3015–3018. [Google Scholar] [CrossRef]
- Ehrentraut, D.; Pakalapati, R.T.; Kamber, D.S.; Jiang, W.; Pocius, D.W.; Downey, B.C.; Mclaurin, M.; D’Evelyn, P.M. High quality, low cost ammonothermal bulk GaN substrates. Jpn. J. Appl. Phys. 2013, 52, 1–4. [Google Scholar] [CrossRef]
- Kim, E. Next Generation LED-SORAA. In Proceedings of Strategies in Light: Santa Clara Convention, Silicon Valley, CA, USA, 2 August 2012.
- Letts, E. Development of GaN wafers for solid-state lighting via the ammonothermal method. In Proceedings of 8th International Workshop on Bulk Nitride Semiconductors, Seeon, Germany, 1 October 2013.
- D’Evelyn, P.M.; Park, D.S.; LeBoeuf, S.F.; Rowland, L.B.; Narang, K.J.; Hong, H.; Arthur, S.D.; Sandvik, M. Gallium nitride crystals and wafers and method of making. US Patent 7,786,503 B2, 31 August 2010. [Google Scholar]
- Wang, B.; Callahan, M. Ammonothermal synthesis of III-nitride crystals. Cryst. Growth Des. 2006, 6, 1227–1246. [Google Scholar] [CrossRef]
- Ehrentraut, D.; Sato, H.; Kagamitani, Y.; Sato, H.; Yoshikawa, A.; Fukuda, T. Solvothermal growth of ZnO. Progr. Cryst. Growth Charact. Mater. 2006, 52, 280–335. [Google Scholar] [CrossRef]
- Tomida, D.; Kagamitani, Y.; Bao, Q.; Hazu, K.; Sawayama, H.; Chichibu, S.F.; Yokoyama, C.; Fukuda, T.; Ishiguro, T. Enhanced growth rate for ammonothermal gallium nitride crystal growth using ammonium iodide mineralizer. J. Cryst. Growth 2012, 353, 59–62. [Google Scholar] [CrossRef]
- Bao, Q.; Saito, M.; Hazu, K.; Furusawa, K.; Kagamitani, Y.; Kayano, R.; Tomida, D.; Qiao, K.; Ishiguro, T.; Yokoyama, C.; et al. Ammonothermal crystal growth of GaN using an NH4F mineralizer. Cryst. Growth Des. 2013, 13, 4158–4161. [Google Scholar] [CrossRef]
- Holleman, A.; Wiberg, N.; Wiberg, E. Lehrbuch der Anorganischen Chemie, 102nd ed.; de Gruyter: Berlin, Germany, 2007. [Google Scholar]
- Franklin, E.C.; Fernelius, W.C. The Nitrogen System of Compounds; Reinhold Publishing Corporation: New York, NY, USA, 1935. [Google Scholar]
- Bronn, J. Über den Zustand der metallischen Lösungen. Ann. Phys. 1905, 16, 166–171. [Google Scholar] [CrossRef]
- Kraus, C.A. Solutions of metals in non-metallic solvents. VI. The conductance of the alkali metals in liquid ammonia. J. Am. Chem. Soc. 1921, 43, 749–770. [Google Scholar]
- Haar, L.; Gallagher, J. Thermodynamic properties of ammonia. J. Phys. Chem. Ref. Data 1978, 7, 635–792. [Google Scholar] [CrossRef]
- Xiang, H.W. Vapor pressures, critical parameters, boiling points, and triple points of ammonia and trideuteroammonia. J. Phys. Chem. Ref. Data 2004, 33, 1005. [Google Scholar] [CrossRef]
- Lemmon, E.W.; McLinden, M.O.; Friend, D.G. Thermophysical properties of fluid systems, NIST standard reference data. Available online: http://webbook.nist.gov/chemistry/fluid/ (accessed on 09 December 2013).
- Shatenshtein, A.I. A study of acid catalysis in liquid ammonia. J. Am. Chem. Soc. 1937, 59, 432–435. [Google Scholar] [CrossRef]
- Rondinini, S.; Longhi, P.; Mussini, P.R.; Mussini, T. Autoprotolysis constants in nonaqueous solvents and aqueous organic solvent mixtures. Pure Appl. Chem. 1987, 59, 1693–1702. [Google Scholar] [CrossRef]
- Böttcher, P.; Kretschmann, U. Darstellung und Kristallstruktur von Dirubidiumpentatellurid, Rb2Te5. J. Less-Common Met. 1983, 95, 81–91. [Google Scholar] [CrossRef]
- Purdy, A.P. Ammonothermal crystal growth of sulfide materials. Chem. Mater. 1998, 10, 692–694. [Google Scholar] [CrossRef]
- Glasser, L. Equations of state and phase diagrams of ammonia. J. Chem. Educ. 2009, 86, 1457–1458. [Google Scholar] [CrossRef]
- Alt, N.S.A.; Schlücker, E. Hochdruck-Sichtzelle für Untersuchungen des ammonothermalen Prozesses. Chem. Ing. Tech. 2011, 83, 280–285. [Google Scholar] [CrossRef]
- Alt, N.S.A.; Meissner, E.; Schlücker, E. Development of a novel in situ monitoring technology for ammonothermal reactors. J. Cryst. Growth 2012, 350, 2–4. [Google Scholar] [CrossRef]
- Hüttig, G.F. Apparat zur gleichzeitigen Druck- und Raummessung von Gasen (Tensi-eudiometer). Z. Anorg. Allg. Chem. 1920, 114, 161–173. [Google Scholar] [CrossRef]
- Yoshida, K.; Aoki, K.; Fukuda, T. High-temperature acidic ammonothermal method for GaN crystal growth. J. Cryst. Growth 2013, in press. [Google Scholar] [CrossRef]
- Callahan, M.J.; Chen, Q.S.C. Hydrothermal and Ammonothermal Growth of ZnO and GaN. In Handbook of Crystal Growth, 1st ed.; Dhanaraj, G., Byrappa, K., Prasad, V., Dudley, M., Eds.; Springer: Berlin/ Heidelberg, Germany, 2010; pp. 655–689. [Google Scholar]
- Binnewies, M.; Glaum, R.; Schmidt, M.; Schmidt, P. Chemical Vapor Transport Reactions; Walter de Gruyter: Berlin, Germany & Boston, MA, USA, 2012. [Google Scholar]
- Wang, B.; Callahan, M. Transport growth of GaN crystals by the ammonothermal technique using various nutrients. J. Cryst. Growth 2006, 291, 455–460. [Google Scholar] [CrossRef]
- Zhang, S.; Hintze, F.; Schnick, W.; Niewa, R. Intermediates in ammonothermal GaN crystal growth under ammonoacidic conditions. Eur. J. Inorg. Chem. 2013, 2013, 5387–5399. [Google Scholar] [CrossRef]
- Zhang, S.; Alt, N.S.A.; Schlücker, E.; Niewa, R. Novel alkali metal amidogallates as intermediates in ammonothermal GaN crystal growth. J. Cryst. Growth 2013, in press. [Google Scholar] [CrossRef]
- Alt, N.S.A.; Meissner, E.; Schlücker, E.; Frey, L. In situ monitoring technologies for ammonothermal reactors. Phys. Status Solidi C 2012, 9, 436–439. [Google Scholar] [CrossRef]
- Erlekampf, J.; Seebeck, J.; Savva, P.; Meissner, E.; Friedrich, J.; Alt, N.; Schlücker, E.; Frey, L. Numerical time-dependent 3D simulation of flow pattern and heat distribution in an ammonothermal system with various baffle shapes. J. Cryst. Growth 2014, in press. [Google Scholar] [CrossRef]
- Vogelsang, K.; Schröter, W.; Hoffmann, R.; Jacobs, H. Ein Beitrag zum Problem der Porenbildung. Härterei Techn. Mitt. 2002, 57, 42–48. [Google Scholar]
- Cundy, C.S.; Cox, P.a. The hydrothermal synthesis of zeolites: Precursors, intermediates and reaction mechanism. Microporous Mesoporous Mater. 2005, 82, 1–78. [Google Scholar] [CrossRef]
- Moolenaar, R.J.; Evans, J.C.; McKeever, L.D. The structure of the aluminate ion in solutions at high pH. J. Phys. Chem. 1970, 74, 3629–3636. [Google Scholar] [CrossRef]
- Juza, R. Amides of the alkali and the alkaline earth metals. Angew. Chem. Int. Ed. 1964, 3, 471–481. [Google Scholar] [CrossRef]
- Juza, R. Über die Amide der 1. und 2. Gruppe des periodischen Systems. Z. Anorg. Allg. Chem. 1937, 231, 121–135. [Google Scholar] [CrossRef]
- Juza, R. Über ein Nitridamid des Zirkoniums. Z. Anorg. Allg. Chem. 1974, 406, 145–152. [Google Scholar]
- Jacobs, H.; Kablitz, D. Untersuchung des Systems Kalium/Cer/Ammoniak. Z. Anorg. Allg. Chem. 1979, 454, 35–42. [Google Scholar] [CrossRef]
- Dwiliński, R.; Baranowski, J.; Kamińska, M. On GaN crystallization by ammonothermal method. Acta Phys. Pol. A 1996, 90, 763–766. [Google Scholar]
- Peters, D. Ammonothermal synthesis of aluminum nitride. J. Cryst. Growth 1990, 104, 411–418. [Google Scholar] [CrossRef]
- Lan, Y.; Chen, X.L.; Xu, Y.; Cao, Y.; Huang, F. Syntheses and structure of nanocrystalline gallium nitride obtained from ammonothermal method using lithium metal as mineralizator. Mater. Res. Bull. 2000, 35, 2325–2330. [Google Scholar] [CrossRef]
- Guarino, R.; Rouxel, J. L’amidogallate de potassium KGa(NH2)4 et l’imidogallate KGa(NH)2. L’obtention de l’amidure de gallium Ga(NH2)3. Bull. Soc. Chim. Fr. 1969, 7, 2284–2287. [Google Scholar]
- Jacobs, H.; Nöcker, B. Neubestimmung von Struktur und Eigenschaften isotyper Natriumtetraamidometallate des Aluminiums und Galliums. Z. Anorg. Allg. Chem. 1993, 619, 381–386. [Google Scholar] [CrossRef]
- Molinié, P.; Brec, R.; Rouxel, J. Le pentaamidogallate de sodium: Na2Ga(NH2)5. C. R. Hebd. Seances Acad. Sci. C 1972, 274, 1388–1391. [Google Scholar]
- Ketchum, D.; Schimek, G.; Pennington, W.; Kolis, J. Synthesis of new group III fluoride–ammonia adducts in supercritical ammonia: Structures of AlF3(NH3)2 and InF2(NH2)(NH3). Inorg. Chim. Act. 1999, 294, 200–206. [Google Scholar] [CrossRef]
- Jacobs, H.; Schröder, F.O. Penta-ammoniates of aluminium halide: The crystal structures of AlX3·5NH3 with X = Cl , Br , I. Z. Anorg. Allg. Chem. 2002, 628, 951–955. [Google Scholar] [CrossRef]
- Johnson, W.C.; Parsons, J.B. Nitrogen compounds of gallium. J. Phys. Chem. 1932, 36, 2588–2594. [Google Scholar] [CrossRef]
- Wang, B.; Callahan, M.; Rakes, K.; Bouthillette, L.; Wang, S.Q.; Bliss, D.; Kolis, J. Ammonothermal growth of GaN crystals in alkaline solutions. J. Cryst. Growth 2006, 287, 376–380. [Google Scholar] [CrossRef]
- Ehrentraut, D.; Kagamitani, Y.; Yokoyama, C.; Fukuda, T. Physico-chemical features of the acid ammonothermal growth of GaN. J. Cryst. Growth 2008, 310, 891–895. [Google Scholar] [CrossRef]
- Purdy, A.P. Ammonothermal synthesis of cubic gallium nitride. Chem. Mater. 1999, 11, 1648–1651. [Google Scholar] [CrossRef]
- Peters, D.; Bock, J.; Jacobs, H. Hexaaminaluminiumiodidammoniakat-[Al(NH3)6]I3NH3-Darstellung und Kristallstruktur. J. Less-Common Met. 1989, 154, 243–250. [Google Scholar] [CrossRef]
- Jacobs, H.; Bock, J.; Stüve, C. Röntgenographische Strukturbestimmung und IR-spektroskopische Untersuchungen an Hexaamindiiodiden, [M(NH3)6]I2, von Eisen und Mangan. J. Less-Common Met. 1987, 134, 207–214. [Google Scholar] [CrossRef]
- Leineweber, A.; Friedriszik, M.W.; Jacobs, H. Preparation and crystal structures of Mg(NH3)2Cl2 , Mg(NH3)2Br2, and Mg(NH3)2I2. J. Solid State Chem. 1999, 234, 229–234. [Google Scholar] [CrossRef]
- Essmann, R. Influence of coordination on N-H...X- hydrogen bonds. Part 1. [Zn(NH3)4]Br2 and [Zn(NH3)4]I2. J. Mol. Struct. 1995, 356, 201–206. [Google Scholar]
- Meyer, G.; Roos, M. Zwei Galliumfluorid-Ammoniakate: Ga(NH3)F3 und Ga(NH3)2F3. Z. Anorg. Allg. Chem. 1999, 625, 1129–1134. [Google Scholar]
- Bremm, S.; Meyer, G. Reactivity of ammonium halides: Action of ammonium chloride and bromide on iron and iron(III) chloride and bromide. Z. Anorg. Allg. Chem. 2003, 629, 1875–1880. [Google Scholar] [CrossRef]
- Bremm, S.; Meyer, G.; Möller, A.; Amann, P.; Sobotka, B. Einwirkung von Ammoniumhalogeniden auf Eisen und Eisenhalogenide. Z. Anorg. Allg. Chem. 2002, 628, 2190. [Google Scholar] [CrossRef]
- Widenmeyer, M.; Hansen, T.C.; Meissner, E.; Niewa, R. Formation and decomposition of iron nitrides observed by in situ powder neutron diffraction and thermal analysis. Z. Anorg. Allg. Chem. 2014, in press. [Google Scholar] [CrossRef]
- Hambley, T.W.; Lay, P.A. Comparisons of π-bonding and hydrogen bonding in isomorphous compounds: [M(NH3)4Cl]Cl2 (M = Cr, Co, Rh, Ir, Ru, Os). Inorg. Chem. 1986, 25, 4553–4558. [Google Scholar] [CrossRef]
- Podberezskaya, N.; Yudanova, T.; Magarill, S.; Ipatova, E.; Romanenko, G.; Pervukhina, N.; Borisov, S. Structures of crystals of inorganic coordination compounds with complex ions [MA5X] and [MX5A] containing neutral (A) and acid (X) ligands, at very high packing densities. J. Struct. Chem. 1992, 32, 894–904. [Google Scholar] [CrossRef]
- Weishaupt, M.; Bezler, H.; Strähle, J. Darstellung und Kristallstruktur von (NH4)2[V(NH3)Cl5]. Die Kristallchemie der Salze (NH4)2[V(NH3)Cl5], [Rh(NH3)Cl]Cl2 und M2VXCl5 mit M = K, NH4, Rb, Cs und X = Cl, O. Z. Anorg. Allg. Chem. 1978, 440, 52–64. [Google Scholar]
- Jacobs, H.; Schmidt, D.; Schmitz, D. Struktur und Eigenschaften der Caesiumamidolanthanatmonoammoniakate Cs3La(NH2)6·NH3 und Cs4La(NH2)7·NH3. J. Less-Common Met. 1981, 81, 121–133. [Google Scholar] [CrossRef]
- Pust, P.; Schmiechen, S.; Hintze, F.; Schnick, W. Ammonothermal synthesis and crystal structure of BaAl2(NH2)8·2NH3. Z. Anorg. Allg. Chem. 2013, 639, 1185–1187. [Google Scholar] [CrossRef]
- Joannis, A. Sur l’amidure de sodium et sur un chlorure de disodammonium. C. R. Hebd. Seances Acad. Sci. C 1891, 112, 392–394. [Google Scholar]
- Mammano, N.; Sienko, M. Low-temperature X-ray study of the compound tetraaminelithium. J. Am. Chem. Soc. 1968, 90, 6322–6324. [Google Scholar] [CrossRef]
- Kirschke, E.J.; Jolly, W.L. The reversibility of the reaction of alkali metals with liquid ammonia. Inorg. Chem. 1967, 6, 855–862. [Google Scholar] [CrossRef]
- Ruff, O.; Geisel, E. Über die Natur der sogenannten Metallammoniumverbindungen. Ber. Dtsch. Chem. Ges. 1906, 39, 828–843. [Google Scholar] [CrossRef]
- Juza, R.; Fasold, K.; Haeberle, C. Untersuchungen über die Amide der Alkalimetalle. Z. Anorg. Allg. Chem. 1937, 234, 75–85. [Google Scholar] [CrossRef]
- Juza, R.; Opp, K. Die Kristallstruktur des Lithiumamides. Z. Anorg. Allg. Chem. 1951, 266, 313–324. [Google Scholar] [CrossRef]
- Juza, R.; Weber, H.H.; Opp, K. Kristallstruktur des Natriumamids. Z. Anorg. Allg. Chem. 1956, 284, 73–82. [Google Scholar] [CrossRef]
- Jacobs, H.; Juza, R. Neubestimmung der Kristallstruktur des Lithiumamids. Z. Anorg. Allg. Chem. 1972, 391, 271–279. [Google Scholar] [CrossRef]
- Nagib, M.; Jacobs, H. Neutronenbeugung am Lithiumdeuteroamid. Atomkernenergie 1973, 21, 275–278. [Google Scholar]
- Nagib, M.; Kistrup, H.; Jacobs, H. Neutronenbeugung am Natriumdeuteroamid. Atomkernenergie 1975, 26, 87–90. [Google Scholar]
- Bohger, P.; Zeiske, T.; Jacobs, H. Neutronenbeugung an der Tieftemperaturmodifikation von Rubidiumdeuteroamid. Z. Anorg. Allg. Chem. 1998, 624, 364–366. [Google Scholar] [CrossRef]
- Nagib, M.; von Osten, E.; Jacobs, H. Röntgen- und Neutronenbeugung und Bestimmung der Wärmekapazität an Caesiumamid–CsNH2–und Caesiumdeuteroamid–CsND2–bei Temperaturen von 348 bis 33 K. Atomkernenergie 1983, 43, 47–54. [Google Scholar]
- Jacobs, H.; Juza, R. Darstellung und Eigenschaften von Berylliumamid und -imid. Z. Anorg. Allg. Chem. 1969, 370, 248–253. [Google Scholar] [CrossRef]
- Jacobs, H.; Juza, R. Darstellung und Eigenschaften von Magnesiumamid und -imid. Z. Anorg. Allg. Chem. 1969, 370, 254–261. [Google Scholar] [CrossRef]
- Senker, J.; Jacobs, H.; Müller, M. Reorientational dynamics of amide ions in isotypic phases of strontium and calcium amide. 1. Neutron diffraction experiments. J. Phys. Chem. B 1998, 102, 931–940. [Google Scholar]
- Nagib, M.; Jacobs, H.; Kistrup, H. Neutronenbeugung am Strontiumdeuteroamid, Sr(ND2)2, bei Temperaturen von 31 bis 570 K. Atomkernenergie 1979, 33, 38–42. [Google Scholar]
- Jacobs, H.; Hadenfeldt, C. Die Kristallstruktur von Bariumamid, Ba(NH2)2. Z. Anorg. Allg. Chem. 1975, 418, 132–140. [Google Scholar] [CrossRef]
- Fröhling, B.; Kreiner, G.; Jacobs, H. Synthesis and crystal structure of manganese(II) and zinc amides, Mn(NH2)2 and Zn(NH2)2. Z. Anorg. Allg. Chem. 1999, 625, 211–216. [Google Scholar] [CrossRef]
- Hadenfeldt, C.; Gieger, B.; Jacobs, H. Die Kristallstruktur von Lanthanamid, La(NH2)3. Z. Anorg. Allg. Chem. 1974, 410, 104–112. [Google Scholar]
- Jacobs, H.; Kistrup, H. Über das System Kalium/Samarium/Ammoniak. Z. Anorg. Allg. Chem. 1977, 435, 127–136. [Google Scholar] [CrossRef]
- Jacobs, H.; Fink, U. Untersuchung des Systems Kalium/Europium/Ammoniak. Z. Anorg. Allg. Chem. 1978, 438, 151–159. [Google Scholar] [CrossRef]
- Hadenfeldt, C.; Jacobs, H.; Juza, R. Über die Amide des Europiums und Ytterbiums. Z. Anorg. Allg. Chem. 1970, 379, 144–156. [Google Scholar] [CrossRef]
- Nagib, M.; von Osten, E.; Jacobs, H. Neutronenbeugung an drei Modifikationen des Kaliumdeuteroamids KND2. Atomkernenergie 1977, 29, 41–47. [Google Scholar]
- Nagib, M.; Jacobs, H.; von Osten, E. Neutronenbeugung am Kaliumdeuteroamid KND2 bei 31 K. Atomkernenergie 1977, 29, 303–304. [Google Scholar]
- Dachs, H. Bestimmung der Lage des Wasserstoffs in LiOH durch Neutronenbeugung. Z. Kristallogr. 1956, 112, 60–67. [Google Scholar] [CrossRef]
- Juza, R.; Jacobs, H.; Klose, W. Die Kristallstrukturen der Tieftemperaturmodifikationen von Kalium- und Rubidiumamid. Z. Anorg. Allg. Chem. 1965, 338, 171–178. [Google Scholar] [CrossRef]
- Jacobs, H.; Nagib, M.; Osten, E.V. Einkristallzüchtung und Kristallchemie der Alkali- und Erdalkalimetallamide. Acta Crystallogr. A 1978, 34, 168. [Google Scholar]
- Schenk, P.; Tulhoff, H. Das System Kaliumamid/Ammoniak. Angew. Chem. 1962, 74, 962. [Google Scholar] [CrossRef]
- Juza, R.; Schumacher, H. Zur Kenntnis der Erdalkalimetallamide. Z. Anorg. Allg. Chem. 1963, 324, 278–286. [Google Scholar] [CrossRef]
- Jacobs, H.; Scholze, H. Untersuchung des Systems Na/La/NH3. Z. Anorg. Allg. Chem. 1976, 427, 8–16. [Google Scholar] [CrossRef]
- Juza, R.; Fasold, K.; Kuhn, W. Untersuchungen über Zink- und Cadmiumamid. Z. Anorg. Allg. Chem. 1937, 234, 86–96. [Google Scholar] [CrossRef]
- Fitzgerald, F.F. Reactions in liquid ammonia. Potassium ammonozincate, cuprous nitride and an ammonobasic mercuric bromide. J. Am. Chem. Soc. 1907, 29, 656–665. [Google Scholar]
- Joannis, M. Action du chlorure de bore sur le gaz ammoniac. C. R. Hebd. Seances Acad. Sci. 1902, 135, 1106. [Google Scholar]
- Janik, J.F.; Wells, R.L. Gallium imide, {Ga(NH)3/2}n, a new polymeric precursor for gallium nitride powders. Chem. Mater. 1996, 8, 2708–2711. [Google Scholar] [CrossRef]
- Wiberg, E.; May, A. Über die Umsetzung von Aluminiumwasserstoff mit Ammoniak und Aminen. Z. Naturforsch. 1955, 10b, 229. [Google Scholar]
- Semenenko, K.N.; Bulychev, B.M.; Shevlyagina, E.A. Aluminium hydride. Russ. Chem. Rev. (Engl. Transl.) 1966, 35, 649–658. [Google Scholar] [CrossRef]
- Purdy, A.P. Indium(III) amides and nitrides. Inorg. Chem. 1994, 33, 282–286. [Google Scholar] [CrossRef]
- Vigouroux, E.; Hugot, C. Silicon amide and imide. C. R. Hebd. Seances Acad. Sci. C 1903, 136, 1670–1672. [Google Scholar]
- Glemser, O.; Naumann, P. Über den thermischen Abbau von Siliciumdiimid Si(NH)2. Z. Anorg. Allg. Chem. 1959, 298, 134–141. [Google Scholar] [CrossRef]
- Molinié, P.; Brec, R.; Rouxel, J.; Herpin, P. Structures des amidoaluminates alcalins MAl(NH2)4 (M = Na, K, Cs). Structure de l’amidogallate de sodium NaGa(NH2)4. Acta Crystallogr. B 1973, 29, 925–934. [Google Scholar] [CrossRef]
- Drew, M.; Goulter, J.; Guémas-Brisseau, L.; Palvadeau, P.; Rouxel, J.; Herpin, P. Etude structurale d’amidobéryllates de rubidium et de potassium. Acta Crystallogr. B 1974, 30, 2579–2582. [Google Scholar] [CrossRef]
- Jacobs, H.; Jänichen, K. Lithiumaluminiumamid, LiAl(NH2)4, Darstellung, röntgenographische Untersuchung, Infrarotspektrum und thermische Zersetzung. Z. Anorg. Allg. Chem. 1985, 531, 125–139. [Google Scholar] [CrossRef]
- Tenten, A.; Jacobs, H. Strukturen und thermisches Verhalten von Kaliumtetraamidoaluminat, α- und β-KAl(NH2)4. Z. Kristallogr. 1989, 186, 289–291. [Google Scholar]
- Jacobs, H.; Harbrecht, B. Substitution in layers of cations in lithium amide: Potassium trilithium amide, KLi3(NH2)4, and potassium heptalithium amide, KLi7(NH2)8. Z. Anorg. Allg. Chem. 1984, 518, 87–100. [Google Scholar] [CrossRef]
- Kraus, F.; Korber, N. K2Li(NH2)3 and K2Na(NH2)3–Synthesis and crystal structure of two crystal-chemically isotypic mixed-cationic amides. J. Solid State Chem. 2005, 178, 1241–1246. [Google Scholar] [CrossRef]
- Jacobs, H.; Kockelkorn, J. Darstellung und Eigenschaften der Amidomagnesate des Kaliums und Rubidiums K2[Mg(NH2)4]- und Rb2[Mg(NH2)4]-Verbindungen mit isolierten [Mg(NH2)4]2− Tetraedern. J. Less-Common Met. 1984, 97, 205–214. [Google Scholar] [CrossRef]
- Jacobs, H.; Birkenbeul, J.; Schmitz, D. Strukturverwandschaft des Dicaesiumamidomagnesats, Cs[Mg(NH2)4], zum β-K2SO4-Typ. J. Less-Common Met. 1982, 85, 79–86. [Google Scholar] [CrossRef]
- Jacobs, H.; Fink, U. Über Natrium- und Kaliumamidometallate des Calciums, Strontiums und Europiums. J. Less-Common Met. 1979, 63, 273–286. [Google Scholar] [CrossRef]
- Jacobs, H.; Kockelkorn, J.; Birkenbeul, J. Struktur und Eigenschaften der ternären Metallamide NaCa(NH2)3, KBa(NH2)3, RbBa(NH2)3, Rb(Eu(NH2)3 und RbSr(NH2)3. J. Less-Common Met. 1982, 87, 215–224. [Google Scholar] [CrossRef]
- Jacobs, H.; Fink, U. Darstellung und Kristallstruktur von KCa(NH2)3. Z. Anorg. Allg. Chem. 1977, 435, 137–145. [Google Scholar] [CrossRef]
- Jacobs, H.; Kockelkorn, J. Darstellung und Kristallstruktur des Rubdiumcalciumamids, RbCa(NH2)3. Z. Anorg. Allg. Chem. 1979, 456, 147–154. [Google Scholar] [CrossRef]
- Jacobs, H.; Kockelkorn, J. Über Caesiumamidometallate (CsM(NH2)3) des Calciums, Strontiums und Europiums; Verbindungen mit der Struktur “Hexagonaler Perowskite”. J. Less-Common Met. 1981, 81, 143–154. [Google Scholar] [CrossRef]
- Jacobs, H. Darstellung und Eigenschaften des Caesiumbariumamids, CsBa(NH2)3: Strukturverwandschaft zum NH4CdCl3-Typ. J. Less-Common Met. 1982, 85, 71–78. [Google Scholar] [CrossRef]
- Hadenfeldt, C.; Gieger, B.; Jacobs, H. Darstellung und Kristallstruktur von KLa2(NH2)7. Z. Anorg. Allg. Chem. 1974, 408, 27–36. [Google Scholar] [CrossRef]
- Hadenfeldt, C.; Gieger, B.; Jacobs, H. Darstellung und Kristallstruktur von K3La(NH2)6. Z. Anorg. Allg. Chem. 1974, 403, 319–326. [Google Scholar] [CrossRef]
- Jacobs, H.; Stüve, C. Rubidiumhexaamidolanthanat und -neodymat, Rb3[La(NH2)6] und Rb3[Nd(NH2)6]; Strukturverwandtschaft zu K3[Cr(OH6)] und K4CdCl6. Z. Anorg. Allg. Chem. 1987, 546, 42–47. [Google Scholar] [CrossRef]
- Jacobs, H.; Schmidt, D. Über ein Caesiumheptaamidodilanthanat CsLa2(NH2)7. J. Less-Common Met. 1981, 78, 51–59. [Google Scholar] [CrossRef]
- Jacobs, H.; Schmidt, D. Struktur und Eigenschaften von perowskitartigen Caesiumamidometallaten des Cers, Neodyms und Samariums Cs3Ln2(NH2)9. J. Less-Common Met. 1980, 76, 227–244. [Google Scholar] [CrossRef]
- Jacobs, H.; Kockelkorn, J. Über Kalium- und Rubidiumamidometallate des Europiums, Yttriums und Ytterbiums, K3M(NH2)6 und Rb3M(NH2)6. J. Less-Common Met. 1982, 85, 97–110. [Google Scholar] [CrossRef]
- Linde, G.; Juza, R. Amidometallate von Lanthan und Gadolinium und Umsetzung von Lanthan, Gadolinium und Scandium mit Ammoniak. Z. Anorg. Allg. Chem. 1974, 409, 191–198. [Google Scholar] [CrossRef]
- Jacobs, H.; Peters, D.; Hassiepen, K. Caesiumamidometallate des Gadoliniums, Ytterbiums und Yttriums mit perowskitverwandten Atomanordnungen Cs3M2(NH2)9. J. Less-Common Met. 1986, 118, 31–41. [Google Scholar] [CrossRef]
- Stuhr, A.; Jacobs, H.; Juza, R. Amide des Yttriums. Z. Anorg. Allg. Chem. 1973, 395, 291–300. [Google Scholar] [CrossRef]
- Peters, D.; Jacobs, H. Übergang von dichter Anionenpackung zu perowskitartiger Struktur bei Kalium- und Rubidiumamidyttriat, KY(NH2)4 und RbY(NH2)4. J. Less-Common Met. 1986, 119, 99–113. [Google Scholar]
- Hadenfeldt, C.; Jacobs, H. Darstellung, Eigenschaften und Kristallstruktur von Na3[Yb(NH2)6]. Z. Anorg. Allg. Chem. 1972, 393, 111–125. [Google Scholar] [CrossRef]
- Jacobs, H.; Jänichen, K. Darstellung und Kristallstruktur von Tetraamidoaluminaten des Rubidiums und Caesiums, Rb[Al(NH2)4] und Cs[Al(NH2)4]. J. Less-Common Met. 1990, 159, 315–325. [Google Scholar] [CrossRef]
- Kreiner, G.; Jacobs, H. Magnetische Struktur von η-Mn3N2. J. Alloys Compd. 1992, 183, 345–362. [Google Scholar] [CrossRef]
- Richter, T.M.M.; Zhang, S.; Niewa, R. Ammonothermal synthesis of dimorphic K2[Zn(NH2)4]. Z. Kristallogr. 2013, 228, 351–358. [Google Scholar] [CrossRef]
- Fröhling, B.; Jacobs, H. Positions of the protons in potassium tetraamidozincate, K2Zn(NH2)4. Z. Anorg. Allg. Chem. 1997, 623, 1103–1107. [Google Scholar] [CrossRef]
- Brec, R.; Novak, A.; Rouxel, J. Etude par spectroscopie infrarouge des amidoaluminates de lithium, sodium et potassium. Bull. Soc. Chim. Fr. 1967, 7, 2432–2435. [Google Scholar]
- Drew, M.; Guémas, L.; Chevalier, P.; Palvadeau, P.; Rouxel, J. Etude structurale de l’amidozincate de rubidium Rb2Zn(NH2)4 et de l’amidomanganite de potassium K2Mn(NH2)4. Rev. Chim. Min. 1975, 12, 419–426. [Google Scholar]
- Jacobs, H.; Gieger, B.; Hadenfeldt, C. Über das System Kalium/Lanthan/Ammoniak. J. Less-Common Met. 1979, 64, 91–99. [Google Scholar] [CrossRef]
- Peters, D.; Jacobs, H. Ammonothermalsynthese von kristallinem Siliciumnitridimid, Si2N2NH. J. Less-Common Met. 1989, 146, 241–249. [Google Scholar] [CrossRef]
- Harbrecht, B.; Jacobs, H. Hochdrucksynthese von Caesiumamidazid, Cs2(NH2)N3 aus Caesiummetall und Ammoniak. Z. Anorg. Allg. Chem. 1983, 500, 181–187. [Google Scholar] [CrossRef]
- Höhn, P.; Kniep, R.; Maier, J. Ba9N[N3][TaN4]2 ein Nitridotantalat(V) mit Nitrid- und Azid-Ionen. Angew. Chem. 1993, 105, 1409–1410. [Google Scholar] [CrossRef]
- Clarke, S.J.; DiSalvo, F.J. Crystal structure of nonabarium bis(tetranitridoniobate) nitride azide, Ba9[NbN4]2N[N3]. Z. Kristallogr. NCS 1997, 212, 309–310. [Google Scholar]
- Zhang, S.; Zherebtsov, D.; DiSalvo, F.J.; Niewa, R. Na5[CN2]2[CN], (Li,Na)5[CN2]2[CN], and K2[CN2]: Carbodiimides from high-pressure synthesis. Z. Anorg. Allg. Chem. 2012, 638, 2111–2116. [Google Scholar] [CrossRef]
- Stock, A.; Blix, M. Über das Borimid, B2(NH)3. Ber. Dtsch. Chem. Ges. 1901, 34, 3039–3048. [Google Scholar] [CrossRef]
- Blix, M.; Wirbelauer, W. Über das Siliciumsulfochlorid, SiSCl2, Siliciumimid, Si(NH)2, Siliciumstickstoffimid (Silicam), Si2N3H und den Siliciumstickstoff, Si3N4. Ber. Dtsch. Chem. Ges. 1903, 36, 4220–4228. [Google Scholar] [CrossRef]
- Ketchum, D.; Kolis, J. Crystal growth of gallium nitride in supercritical ammonia. J. Cryst. Growth 2001, 222, 431–434. [Google Scholar] [CrossRef]
- Jouet, R.J.; Purdy, A.P.; Wells, R.L.; Janik, J.F. Preparation of phase pure cubic gallium nitride, c-GaN, by ammonothermal conversion of gallium imide, {Ga(NH)3/2}n. J. Clust. Sci. 2002, 13, 469–486. [Google Scholar] [CrossRef]
- Zajac, M.; Gosk, J.; Grzanka, E.; Stelmakh, S.; Palczewska, M.; Wysmoek, A.; Korona, K.; Kamiska, M.; Twardowski, A. Ammonothermal synthesis of GaN doped with transition metal ions (Mn, Fe, Cr). J. Alloys Compd. 2008, 456, 324–338. [Google Scholar] [CrossRef]
- Jacobs, H.; Rechenbach, D.; Zachwieja, U. Structure determination of γ’-Fe4N and ϵ-Fe3N. J. Alloys Compd. 1995, 227, 10–17. [Google Scholar] [CrossRef]
- Jacobs, H.; Rechenbach, D.; Zachwieja, D. Untersuchungen zur Struktur und zum Zerfall von Eisennitriden- γ’-Fe4N und ϵ-Fe3N. Härterei Techn. Mitt. 1995, 50, 205–213. [Google Scholar]
- Jacobs, H.; Bock, J. Einkristallzüchtung von γ′-Fe4N in überkritischem Ammoniak. J. Less-Common Met. 1987, 134, 215–220. [Google Scholar] [CrossRef]
- Jacobs, H.; Stüve, C. Hochdrucksynthese der η-phase im System Mn-N: Mn3N2. J. Less-Common Met. 1984, 96, 323–329. [Google Scholar] [CrossRef]
- Leineweber, A.; Jacobs, H.; Hull, S. Ordering of nitrogen in nickel nitride Ni3N determined by neutron diffraction. Inorg. Chem. 2001, 40, 5818–5822. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, H.; Zachwieja, U. Kupferpalladiumnitride, Cu3PdxN mit x = 0,020 und 0,989, Perowskite mit “bindender 3d10-4d10-Wechselwirkung”. J. Less-Common Met. 1991, 170, 185–190. [Google Scholar] [CrossRef]
- von Stackelberg, M.; Paulus, R. Untersuchungen über die Kristallstrukturen der Nitride und Phosphide zweiwertiger Metalle. Z. Physik. Chem. B 1933, 22, 305. [Google Scholar]
- Reckeweg, O.; Lind, C.; DiSalvo, F.J. Rietveld refinement of the crystal structure of α-Be3N2 and the experimental determination of optical band gaps for Mg3N2, Ca3N2 and CaMg2N2. Z. Naturforsch. 2003, 58b, 159–162. [Google Scholar] [CrossRef]
- Karau, F.; Schnick, W. Synthese von Cadmiumnitrid Cd3N2 durch thermischen Abbau von Cadmiumazid Cd(N3)2 und Kristallstrukturbestimmung aus Röntgen-Pulverbeugungsdaten. Z. Anorg. Allg. Chem. 2007, 633, 223–226. [Google Scholar] [CrossRef]
- Partin, D.E.; Williams, D.J.; O’Keeffe, M. The crystal structures of Mg3N2 and Zn3N2. J. Solid State Chem. 1997, 132, 56–59. [Google Scholar] [CrossRef]
- Reckeweg, O.; DiSalvo, F.J. About binary and ternary alkaline earth metal nitrides. Z. Anorg. Allg. Chem. 2001, 627, 371–377. [Google Scholar] [CrossRef]
- Soto, G.; Diaz, J.A.; De la Cruz, W.; Contreras, O.; Moreno, M.; Reyes, A. Epitaxial α-Be3N2 thin films grown on Si substrates by reactive laser ablation. Mater. Sci. Eng., B. 2002, 94, 62–65. [Google Scholar] [CrossRef]
- García Núñez, C.; Pau, J.; Hernández, M.; Cervera, M.; Ruiz, E.; Piqueras, J. On the zinc nitride properties and the unintentional incorporation of oxygen. Thin Solid Films 2012, 520, 1924–1929. [Google Scholar] [CrossRef]
- Soto, G.; Díaz, J.A.; Machorro, R.; Reyes-Serrato, A.; de la Cruz, W. Beryllium nitride thin film grown by reactive laser ablation. Mater. Lett. 2002, 52, 29–33. [Google Scholar] [CrossRef]
- Toyoura, K.; Tsujimura, H.; Goto, T.; Hachiya, K.; Hagiwara, R.; Ito, Y. Optical properties of zinc nitride formed by molten salt electrochemical process. Thin Solid Films 2005, 492, 88–92. [Google Scholar] [CrossRef]
- Maruska, H.P.; Tietjen, J.J. The preparation and properties of vapor-deposited single-crystalline GaN. Appl. Phys. Lett. 1969, 15, 327. [Google Scholar] [CrossRef]
- Perry, P.B.; Rutz, R.F. The optical absorption edge of single-crystal AlN prepared by a close-spaced vapor process. Appl. Phys. Lett. 1978, 33, 319–321. [Google Scholar] [CrossRef]
- Wu, J.; Walukiewicz, W.; Shan, W.; Yu, K.; Ager, J.; Haller, E.E.; Lu, H.; Schaff, W.J. Effects of the narrow band gap on the properties of InN. Phys. Rev. B 2002, 66, 201403. [Google Scholar] [CrossRef]
- Xie, R.J.; Bert Hintzen, H.T. Optical properties of (oxy)nitride materials: A review. J. Am. Ceram. Soc. 2013, 96, 665–687. [Google Scholar] [CrossRef]
- Dwiliński, R.; Doradziński, R.; Garczyński, J.; Sierzputowski, L.; Kucharski, R.; Zajac, M.; Rudziński, M.; Kudrawiec, R.; Serafińczuk, J.; Strupiński, W. Recent achievements in AMMONO-bulk method. J. Cryst. Growth 2010, 312, 2499–2502. [Google Scholar] [CrossRef]
- Gogova, D.; Petrov, P.P.; Buegler, M.; Wagner, M.R.; Nenstiel, C.; Callsen, G.; Schmidbauer, M.; Kucharski, R.; Zajac, M.; Dwiliński, R.; et al. Structural and optical investigation of non-polar (1–100) GaN grown by the ammonothermal method. J. Appl. Phys. 2013, 113, 203513. [Google Scholar] [CrossRef]
- Shubina, T.; Ivanov, S.; Jmerik, V.; Solnyshkov, D.; Vekshin, V.; Kopev, P.; Vasson, A.; Leymarie, J.; Kavokin, A.; Amano, H.; et al. Mie resonances, infrared emission, and the band gap of InN. Phys. Rev. Lett. 2004, 92, 117407. [Google Scholar] [CrossRef] [PubMed]
- Komissarova, T.A.; Jmerik, V.N.; Ivanov, S.V.; Paturi, P. Detection of metallic in nanoparticles in InGaN alloys. Appl. Phys. Lett. 2011, 99, 072107. [Google Scholar] [CrossRef]
- Wu, J.; Walukiewicz, W.; Yu, K.M.; Ager, J.W.; Haller, E.E.; Lu, H.; Schaff, W.J. Small band gap bowing in In1−xGaxN alloys. Appl. Phys. Lett. 2002, 80, 4741. [Google Scholar] [CrossRef]
- Ranade, M.R.; Tessier, F.; Navrotsky, A.; Marchand, R. Calorimetric determination of the enthalpy of formation of InN and comparison with AlN and GaN. J. Mater. Res. 2011, 16, 2824–2831. [Google Scholar] [CrossRef]
- Richter, E.; Stoica, T.; Zeimer, U.; Netzel, C.; Weyers, M.; Tränkle, G. Si doping of GaN in hydride vapor-phase epitaxy. J. Electron. Mater. 2013, 42, 820–825. [Google Scholar] [CrossRef]
- Steckl, A.J.; Birkhahn, R. Visible emission from Er-doped GaN grown by solid source molecular beam epitaxy. Appl. Phys. Lett. 1998, 73, 1700–1702. [Google Scholar] [CrossRef]
- Adekore, B.T.; Callahan, M.J.; Bouthillette, L.; Dalmau, R.; Sitar, Z. Synthesis of erbium-doped gallium nitride crystals by the ammonothermal route. J. Cryst. Growth 2007, 308, 71–79. [Google Scholar] [CrossRef]
- Korona, K.P.; Doradziński, R.; Palczewska, M.; Pietras, M.; Kamiska, M.; Kuhl, J. Properties of zinc acceptor and exciton bound to zinc in ammonothermal GaN. Phys. Status Solidi B 2003, 235, 40–43. [Google Scholar] [CrossRef]
- Scotti, N.; Kockelmann, W.; Senker, J. Sn3N4, a Tin(IV) nitride–syntheses and the first crystal structure determination of a binary tin-nitrogen compound. Z. Anorg. Allg. Chem. 1999, 625, 1435–1439. [Google Scholar] [CrossRef]
- Louhadj, A.; Ghezali, M.; Badi, F.; Mehnane, N.; Cherchab, Y.; Amrani, B.; Abid, H.; Sekkal, N. Electronic structure of ScN, YN, LaN and GdN superlattices. Superlatt. Microstruct. 2009, 46, 435–442. [Google Scholar] [CrossRef]
- Srivastava, V.; Rajagopalan, M.; Sanyal, S.P. Theoretical investigation on structural, magnetic and electronic properties of ferromagnetic GdN under pressure. J. Magn. Magn. Mater. 2009, 321, 607–612. [Google Scholar] [CrossRef]
- Li, D.X.; Haga, Y.; Shida, H.; Suzuki, T. Magnetic properties of ferromagnetic GdN. Physica B 1994, 199/200, 631–633. [Google Scholar] [CrossRef]
- Mancera, L.; Rodr, J.A. First principles calculations of the ground state properties and structural phase transformation in YN. J. Phys.: Condens. Matter 2003, 15, 2625–2633. [Google Scholar] [CrossRef]
- Niewa, R. Nitridocompounds of manganese: Manganese nitrides and nitridomanganates. Z. Kristallogr. 2002, 217, 8–23. [Google Scholar] [CrossRef]
- Takei, W.J.; Heikes, R.R.; Shirane, G. Magnetic structure of Mn4N-type compounds. Phys. Rev. B 1962, 125, 1893–1897. [Google Scholar] [CrossRef]
- Borsa, D.M.; Grachev, S.; Presura, C.; Boerma, D.O. Growth and properties of Cu3N films and Cu3N/γ’-Fe4N bilayers. Appl. Phys. Lett. 2002, 80, 1823–1825. [Google Scholar] [CrossRef]
- Sakuma, A. Self-consistent calculations for the electronic structures of iron nitrides, Fe3N, Fe4N and Fe16N2. J. Magn. Magn. Mater. 1991, 102, 127–134. [Google Scholar] [CrossRef]
- Wang, D.Y. Properties of various sputter-deposited CuN thin films. J. Vac. Sci. Technol. A 1998, 16, 2084–2092. [Google Scholar] [CrossRef]
- Desmoulins-Krawiec, S.; Aymonier, C.; Loppinet-Serani, A.; Weill, F.; Gorsse, S.; Etourneau, J.; Cansell, F. Synthesis of nanostructured materials in supercritical ammonia: nitrides, metals and oxides. J. Mater. Chem. 2004, 14, 228–232. [Google Scholar] [CrossRef]
- Hasegawa, M.; Yagi, T. Systematic study of formation and crystal structure of 3d-transition metal nitrides synthesized in a supercritical nitrogen fluid under 10 GPa and 1800 K using diamond anvil cell and YAG laser heating. J. Alloys Compd. 2005, 403, 131–142. [Google Scholar] [CrossRef]
- Zachwieja, U.; Jacobs, H. Kolumnarstrukturen bei Tri- und Diamminnitraten, [M(NH3)3]NO3 und [M(NH3)2]NO3 des einwertigen Kupfers und Silbers. Z. Anorg. Allg. Chem. 1989, 571, 37–50. [Google Scholar] [CrossRef]
- Hahn, U.; Weber, W. Electronic structure and chemical-bonding mechanism of Cu3N, Cu3NPd, and related Cu(I) compounds. Phys. Rev. B: Condens. Matter 1996, 53, 12684–12693. [Google Scholar] [CrossRef]
- Leineweber, A.; Niewa, R.; Jacobs, H.; Kockelmann, W. The manganese nitrides η-Mn3N2 and Ө-Mn6N5+x: Nuclear and magnetic structures. J. Mater. Chem. 2000, 10, 2827–2834. [Google Scholar] [CrossRef]
- Jacobs, H.; Harbrecht, B. Eine neue Darstellungsmethode für Caesiumhydroxid. Z. Naturforsch. 1981, 36b, 270–271. [Google Scholar] [CrossRef]
- Jacobs, H.; Tacke, T.; Kockelkorn, J. Hydroxidmonohydrate des Kaliums und Rubidiums; Verbindungen, deren Atomanordnungen die Schreibweise K(H2O)OH bzw. Rb(H2O)OH nahelegen. Z. Anorg. Allg. Chem. 1984, 516, 67–78. [Google Scholar] [CrossRef]
- Purdy, A.P. Ammonothermal growth of chalcogenide single crystal materials. US Patent 5,902,396, 11 May 1999. [Google Scholar]
- Jacobs, H.; Kirchgässner, R.; Bock, J. Darstellung und Kristallstruktur von Lithiumhydrogensulfid LiHS. Z. Anorg. Allg. Chem. 1989, 569, 111–116. [Google Scholar] [CrossRef]
- Jacobs, H.; Erten, C. Über Kaliumhydrogensulfid, KHS. Z. Anorg. Allg. Chem. 1981, 473, 125–132. [Google Scholar] [CrossRef]
- Böttcher, P. Beiträge zur Kenntnis der Alkalimetallpolychalkogenide. Habilitation Thesis, RWTH Aachen, Germany, 1980. [Google Scholar]
- Böttcher, P. Zur Kenntnis der Verbindung Na2S3. Z. Anorg. Allg. Chem. 1980, 467, 149–157. [Google Scholar] [CrossRef]
- Böttcher, P. Die Kristallstruktur von K2S3 und K2Se3. Z. Anorg. Allg. Chem. 1977, 432, 167–172. [Google Scholar] [CrossRef]
- Böttcher, P. Preparation and crystal structure of the dialkali metal trichalcogenides Rb2S3, Rb2Se3 and Cs2Se3. Z. Anorg. Allg. Chem. 1980, 461, 13–21. [Google Scholar] [CrossRef]
- Böttcher, P. Synthesis and crystal structure of the dirubidiumpentachalcogenides Rb2S5 and Rb2Se5. Z. Kristallogr. 1979, 150, 65–73. [Google Scholar] [CrossRef]
- Böttcher, P.; Kruse, K. Darstellung und Kristallstruktur von Dicaesiumpentasulfid (Cs2S5). J. Less-Common Met. 1982, 83, 115–125. [Google Scholar] [CrossRef]
- Böttcher, P. Zur Kenntnis von Cs2Se. J. Less-Common Met. 1980, 76, 271–277. [Google Scholar] [CrossRef]
- Böttcher, P. Synthesis and crystal structure of Rb2Te3 and Cs2Te3. J. Less-Common Met. 1980, 70, 263–271. [Google Scholar] [CrossRef]
- Böttcher, P. Darstellung und Kristallstruktur von Dicaesiumpentatellurid, Cs2Te5. Z. Anorg. Allg. Chem. 1982, 491, 39–46. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Richter, T.M.M.; Niewa, R. Chemistry of Ammonothermal Synthesis. Inorganics 2014, 2, 29-78. https://doi.org/10.3390/inorganics2010029
Richter TMM, Niewa R. Chemistry of Ammonothermal Synthesis. Inorganics. 2014; 2(1):29-78. https://doi.org/10.3390/inorganics2010029
Chicago/Turabian StyleRichter, Theresia M. M., and Rainer Niewa. 2014. "Chemistry of Ammonothermal Synthesis" Inorganics 2, no. 1: 29-78. https://doi.org/10.3390/inorganics2010029
APA StyleRichter, T. M. M., & Niewa, R. (2014). Chemistry of Ammonothermal Synthesis. Inorganics, 2(1), 29-78. https://doi.org/10.3390/inorganics2010029