Abstract
As the overall turnover-limiting step (TOLS) in the homogeneous conversion of N2O, the oxygen-atom transfer (OAT) from an N2O to an Ru-H complex to generate an N2 and Ru-OH complex has been comprehensively investigated by density functional theory (DFT) computations. Theoretical results show that the proton transfer from Ru-H to the terminal N of endo N2O is most favorable pathway, and the generation of N2 via OAT is accomplished by a three-step mechanism [N2O-insertion into the Ru-H bond (TS-1-2, 24.1 kcal mol−1), change of geometry of the formed (Z)-O-bound oxyldiazene intermediate (TS-2-3, 5.5 kcal mol−1), and generation of N2 from the proton transfer (TS-3-4, 26.6 kcal mol−1)]. The Gibbs free energy of activation (ΔG‡) of 29.0 kcal mol−1 for the overall turnover-limiting step (TOLS) is determined. With the participation of potentially existing traces of water in the THF solvent serving as a proton shuttle, the Gibbs free energy of activation in the generation of N2 (TS-3-4-OH2) decreases to 15.1 kcal mol−1 from 26.6 kcal mol−1 (TS-3-4). To explore the structure–activity relationship in the conversion of N2O to N2, the catalytic activities of a series of Ru-H complexes (C1–C10) are investigated. The excellent linear relationships (R2 > 0.91) between the computed hydricities (ΔGH−) and ΔG‡ of TS-3-4, between the computed hydricities (ΔGH−) and the ΔG‡ of TOLS, were obtained. The utilization of hydricity as a potential parameter to predict the activity is consistent with other reports, and the current results suggest a more electron-donating ligand could lead to a more active Ru-H catalyst.
1. Introduction
As a dominant ozone-depleting emission [1,2,3] and a greenhouse gas with about 300 times the global warming potential than that of CO2 (based on the 100-year timescale) [4,5,6], nitrous oxide (N2O) has been brought to the frontier of climate and environmental protection [7,8,9]. Efforts that aim to terminate environmentally detrimental N2O have been examined in the last few decades [10,11]. Beyond the commonly studied decomposition of N2O catalyzed by metal oxides (MOs) [12,13,14,15], a variety of possible conversions of N2O have also been investigated, including O-atom insertion into metal-H and metal-C bonds [16,17,18,19,20,21], cleavage of N-O and N-N bonds [22,23,24,25], and reactions with organic substrates [11,26,27,28]. Among the reactions, the exothermic/exergonic conversion of N2O mediated by CO generating N2 and less harmful CO2 (N2O + CO → N2 + CO2, = −86.3 kcal mol−1) [29,30] is considered as a practicable and cost-effective method to simultaneously address the environmental dilemma created by the emission of N2O and CO [30,31,32,33].
Milstein and co-workers showed that a dicarbonyl PNN-Ru-H pincer complex (I, Scheme 1, PNN = 6-((di-tert-butylphosphino)methylene)-6H-[2,2′-bipyridin]-1-ide) could serve as an efficient catalyst in the homogeneous conversion of N2O and CO [31]. The dicarbonyl PNN-Ru-H complex (I, Scheme 1) catalyzed the conversion of N2O and CO to generate N2 and CO2 with a turnover number (TON) of up to 579 referenced to N2 and 561 referenced to CO2 after heating for 22 h at 70 °C.
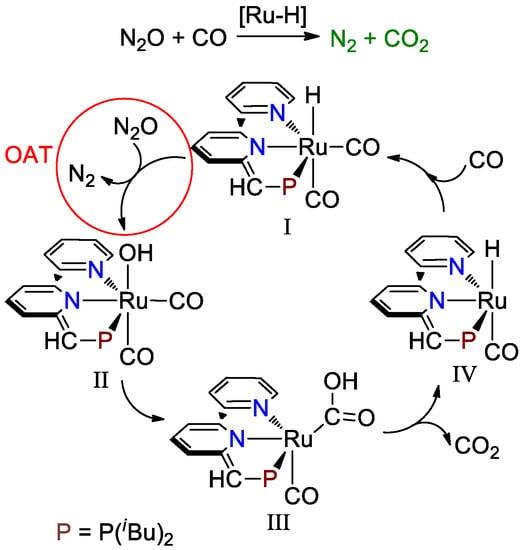
Scheme 1.
Proposed pathway for the conversion of N2O and CO by PNN-Ru-H complex.
The overall mechanism of the conversion of N2O and CO by the Ru-H species includes: (1) the generation of N2 and a dicarbonyl Ru-OH complex (II, Scheme 1) via oxygen-atom transfer (OAT), (2) the formation of a dicarbonyl Ru-COOH complex (III, Scheme 1) via the intramolecular nucleophilic attack of OH on the nearby CO group, (3) the release of CO2 and the formation of a monocarbonyl Ru-H complex (IV, Scheme 1) via decarboxylation, and (4) the regeneration of the dicarbonyl PNN-Ru-H active species (I, Scheme 1) via the nucleophilic attack of a free CO. The generation of N2 via oxygen-atom transfer was proposed as the turnover-limiting step (TOLS), which was supported by the observation of a relatively fast formation of CO2 via an intramolecular reaction between the Ru-OH species and CO [31]. This experimentally established turnover-limiting step is also verified by computational studies [34,35,36].
It has come to our attention that the oxygen-atom transfer (OAT) between N2O and the Ru-H complex has not been fully investigated yet. Three categories of reactions between the N2O and Ru-H complex that need to be considered are: (1) the proton transfer from Ru-H to the terminal N of N2O (I, Chart 1), (2) the proton transfer from Ru-H to the terminal O of N2O (II, Chart 1), and (3) the hydride transfer from Ru-H to the central N of N2O (III, Chart 1). The generation of N2 via proton transfer and hydride transfer from Ru-H to N2O must be thoroughly evaluated and compared. Various transition states of proton transfer from Ru-H to N2O could be proposed, as induced by different geometries of N2O adduct (endo vs. exo isomer). The effect of the geometries of N2O (endo vs. exo isomer) in the oxygen-atom transfer (OAT) from N2O to Ru-H to generate N2 and Ru-OH must also be appropriately addressed.
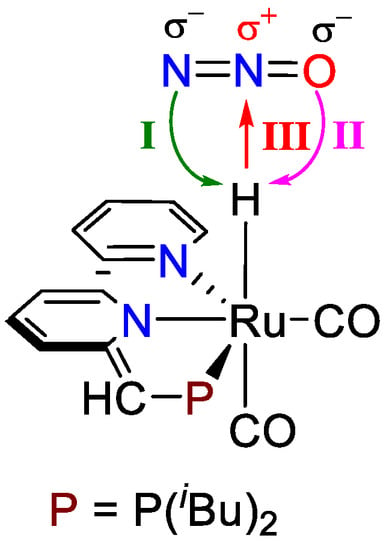
Chart 1.
Proton transfer (I and II) and hydride transfer (III) between Ru-H and N2O.
In this contribution, the detailed reaction mechanism for the oxygen-atom transfer (OAT) from N2O to Ru-H in the conversion of N2O has been comprehensively investigated using density functional theory (DFT) computations. With the detailed reaction mechanism for the conversion of N2O to N2 in hand (Figure 1), the turnover-limiting step for the conversion of N2O and CO by a series of Ru-H complexes (Chart 2) with different electron-donating and electron-withdrawing groups have been investigated to explore the structure–activity relationship, and the results provided here are the continuous efforts for the homogeneous conversion of N2O by the transition metal complex.
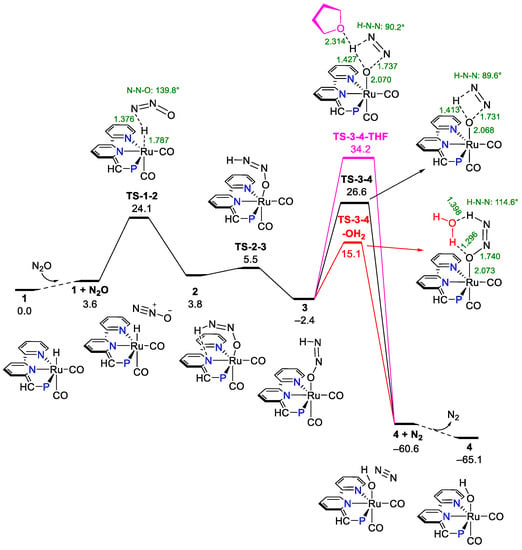
Figure 1.
Free energy diagram for N2 generation from proton transfer to the terminal N of endo N2O. Selected atom distances are given in Å, selected bond angles are given in degrees, and ΔG°/ΔG‡ are in kcal mol−1.
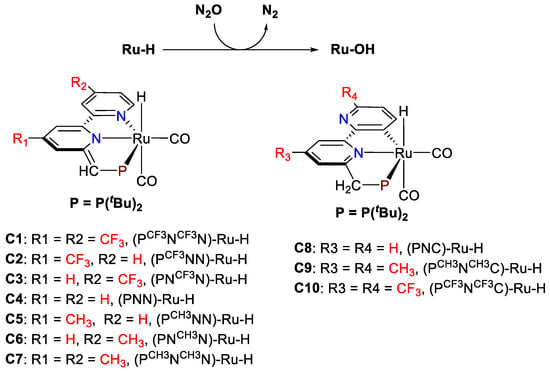
Chart 2.
Studied Ru-H complexes for the conversion of N2O to N2.
2. Computational Methods
Full gas-phase geometry optimizations were performed using the method of B3LYP [37,38,39,40] with Grimme’s D3 [41] dispersion with Becke–Johnson damping [D(3BJ)] [42] and basis set 1 (BS1) (B3LYP-D3(BJ)/BS1) through Gaussian 16 [43]. To account for the solvent effect of tetrahydrofuran (THF), B3LYP-D3(BJ)/BS2 single-point computations using the SMD [44] solvation model with parameters consistent with tetrahydrofuran (THF) as the solvent were performed on the B3LYP-D3(BJ)/BS1 optimized geometries [SMD(THF)-B3LYP-D3(BJ)/BS2//B3LYP-D3(BJ)/BS1]. For comparison, optimizations using B3LYP-D3(BJ)/BS2 with the SMD [44] solvation model in THF were also performed [SMD(THF)-B3LYP-D3(BJ)/BS2, see SI]. In basis set 1 (BS1), the modified LANL2DZ [45,46] basis set and LANL2DZ effective core potentials (ECP) were used for Ru, the LANL2DZ(d,p) [45,47] basis set and LANL2DZ ECP were used for P, and the 6-31G(d’) [48,49,50] basis sets were used for all other atoms (C, N, O, and H). In basis set 2 (BS2), the Ahlrichs Def2-TZVP [51,52] basis sets and related ECP were used for Ru, and TZVP [53] basis sets were used for all other atoms (C, N, O, P, F, and H). The Gaussian 16 default ultrafine integration grid, 2-electron integral accuracy of 10−12, and SCF convergence criterion of 10−8 were used for all computations, and vibrational frequency computations were performed to verify the nature of all stationary points. All located transition states were obtained with only one imaginary frequency, and minima without any imaginary frequencies were obtained. The default rigid-rotor-harmonic-oscillator (RRHO) approximation was used to calculate the vibrational contribution to entropy. The intrinsic reaction coordinate (IRC) computations from the located transition states were performed, and both directions of the reaction path following the transition state were computed (see SI for the IRC plots) [54,55]. Gibbs free energies of activation (ΔG‡) and free energies of reaction (ΔG°) were determined with standard conditions of 1 atm and 298.15 K, which are relative to the Ru-H complex (in kcal mol−1). The Gibbs free energies from the SMD(THF)-B3LYP-D3(BJ)/BS2//B3LYP-D3(BJ)/BS1 computations are presented in the main text. The Gibbs free energies from the SMD(THF)-B3LYP-D3(BJ)/BS2 computations are presented in the supporting information. Gibbs free energies for the overall turnover-limiting steps (TOLS) were determined based on the energetic span/transition state theory. The accuracy and reliability of the computational method [SMD(THF)-B3LYP-D3(BJ)/BS2//B3LYP-D3(BJ)/BS1] was verified. Good agreement between the SMD(THF)-B3LYP-D3(BJ)/BS2//B3LYP-D3(BJ)/BS1 computations and the SMD(THF)-B3LYP-D3(BJ)/BS2 computations was obtained, and the mean absolute deviation (MAD) was 0.5 (Table S1) and the coefficient linear regression (R2) was 0.9967 (Figure S1) [56,57,58].
3. Results and Discussion
To fully understand the oxygen-atom transfer (OAT) from N2O to an Ru-H complex (Scheme 1), the proposed pathway for the proton transfer from Ru-H to the terminal N of endo N2O is presented in the following Section 3.1 (Figure 1), and other higher energetic pathways including: (1) the proton transfer from Ru-H to the terminal O of N2O (Scheme S2) and (2) hydride transfer from Ru-H to the central N of N2O (Figures S6 and S7) are presented in the supporting information.
3.1. Proton Transfer from Ru-H to the Terminal N of Endo N2O
The release of N2 from an oxygen-atom transfer from N2O via the pathway of the proton transfer from Ru-H to the terminal N of endo N2O involves two important (Z)-O-bound oxyldiazene intermediates (structures 2 and 3, Figure 1). Structure 2, which has an intramolecular hydrogen bond between the oxyldiazene substrate and the anionic pyridinyl unit, is generated by the insertion of N2O into the Ru-H bond of the Ru-H complex (structure 1) via TS-1-2 at 24.1 kcal mol−1 (Figure 1). It should be noted that the bent geometry of the O atom in the six-membered ring caused by the intramolecular hydrogen bond in (Z)-O-bound oxyldiazene intermediate 2 prevents the direct proton transfer from the terminal N-H to the O atom. Intermediate 2 must go through a necessary change of geometry to form its structural isomer 3 in order to accomplish the proton transfer. There is a facile geometry isomerization from intermediate (Z)-O-bound oxyldiazene intermediate 2 to 3 via TS-2-3 (5.5 kcal mol−1, Figure 1). The O-bound oxyldiazene intermediate 3 is 6.4 kcal mol−1 lower in energy than 2 (–2.4 vs. 3.8 kcal mol−1), which is partially due to the breaking of the six-membered ring formed from the intramolecular hydrogen bond. In structure 2, the orientation of the lone pair of electrons is unfavorable for the proton transfer to the oxygen from the terminal N-H. However, from structure 3, the proton transfer from the terminal N-H to the O may occur. The geometry of (Z)-O-bound oxyldiazene intermediate 3 is consistent with reported Ru/Rh intermediates [59,60,61], but is dissimilar to the recent results of Xie and co-workers [34]. From the O-bound diazene intermediate 3, the cyclic four-membered ring transition state for the proton transfer (TS-3-4, 26.6 kcal mol−1, Figure 1) forms the Ru-OH complex (structure 4, Figure 1) and molecular N2. The generation of N2 from the proton transfer (TS-3-4) is the TOLS for the conversion of N2O to N2. The Gibbs free energy of activation for the overall turnover-limiting steps (TOLS) based on the energetic span/transition state theory is determined as 29.0 kcal mol−1 (3 to TS-3-4, Figure 1) [62,63]. The generation of separated N2 and the Ru-OH complex (structure 4) from the Ru-H complex and N2O is favorable by –65.1 kcal mol−1 (Figure 1). The effect of potentially existing traces of water in the THF solvent in the homogeneous conversion of N2O was also considered. Anticipated lower Gibbs free energy of activation in the generation of N2 (15.1 kcal mol−1, TS-3-4-OH2, Figure 1) with the participation of potentially existing H2O serving as a proton shuttle compared to the non-assisted generation of N2 (26.6 kcal mol−1, TS-3-4, Figure 1) was obtained. No such assistance of solvent THF was found (34.2 kcal mol−1, TS-3-4-THF, Figure 1). The result of the H2O-assisted generation of N2 is consistent with Poater’s results on the hydrogenation of N2O by the PNP-Ru-dihydride pincer complex [35,36].
Another structural isomer that created a pathway for the N2 generation from the N2O oxygen-atom transfer involves an intermediate without an intramolecular hydrogen bond, and is presented in Figure S6. Structures 2b, 3b, and 4b in Figure S6 are the structural isomers of structures 2, 3, and 4 in Figure 1 with a different orientation of diazene substrate and the OH group. For this alternative pathway, slightly higher ΔG‡ are found (26.5 kcal mol−1 for TS-1b-2b vs. 24.1 kcal mol−1 for TS-1-2, 27.5 kcal mol−1 for TS-3b-4b vs. 26.6 kcal mol−1 for TS-3-4). The effect on the ΔG‡ for the generation of N2 from the anionic pyridinyl is relatively small. The possible proton transfer from Ru-H to the terminal O of N2O was also investigated (Scheme S2), and significantly higher ΔG‡ were obtained (40.6 kcal mol−1 for TS-1-2d and 38.6 kcal mol−1 for TS-1b-2c). Compared to the formation of the Ru-O bond in Figure 1, the relatively small electronegativity of terminal N of N2O made it difficult to form Ru-N bonds (2c and 2d in Scheme S2), which caused the higher ΔG‡. Even higher ΔG‡ were also obtained for the hydride transfer from Ru-H to the central N of N2O (42.7 kcal mol−1 for TS-1-5 and 45.2 kcal mol−1 for TS-1b-5b, Figures S6 and S7).
The above discussed mechanistic studies of the homogeneous oxygen-atom transfer (OAT) from N2O to the Ru-H complex to generate an N2 and Ru-OH complex clearly showed that the pathway of the proton transfer from Ru-H to the terminal N of endo N2O is most favorable (Figure 1). It is accomplished by three asynchronous steps including N2O insertion into the Ru-H bond (TS-1-2), change of geometry of the formed O-bound diazene intermediate (TS-2-3), and the generation of N2 from the proton transfer (TS-3-4). The last step (TS-3-4) forming the molecular N2 and Ru-OH complex is the overall turnover-limiting step (TOLS) in the proposed three-step mechanism.
3.2. Hydricity as A Parameter to Predict the Activity
The concept of hydricity has been previously utilized to interpret the structure–activity relationships in transition-metal hydride species involved homogeneous catalysis [64,65,66,67,68,69,70]. The hydrogenation of CO2 to formate catalyzed by molecular Co-H complexes presented an excellent linear relationship between the logarithm of the catalytic turnover frequency and the hydricity of Co-H complexes (R2 = 0.9956), and significantly improved activity for Co-H complexes with relatively stronger hydride-donating ability were observed [66]. The model using the relationship between hydricities and the one-electron reduction potential of the transition-metal complexes is also used to study the reactivity of transition-metal hydride complexes in the CO2 reduction [68]. The hydricity (ΔGH−) of each Ru-H complex was calculated using the equation presented in Scheme 2 [64,65].
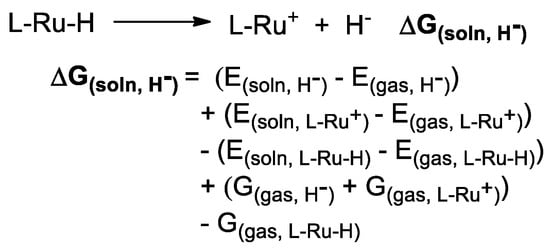
Scheme 2.
Equation used to calculate the hydricity (ΔGH−) of Ru-H complex.
To explore the structure–activity relationship in the conversion of N2O to N2, the catalytic pathways of a series of Ru-H complexes (C1–C7, Chart 2) were investigated. The parent PNN-Ru-H complex was modified by the introduced electron-donating (CH3) and electron-withdrawing groups (CF3) on the para positions of these two pyridinyl fragments. For comparison, the structural isomers of the Ru-H complex with the PNC ligand [PNC = 6′-((di-tert-butylphosphino)methyl)-(2,2′-bipyridin)-3-ide] were also modeled (C8–C10, Chart 2). It is noted that the Ru-H complex with the PNC ligand [PNC = 6′-((di-tert-butylphosphino)methyl)-(2,2′-bipyridin)-3-ide] was significantly less active than the PNN-Ru-H complex in the homogeneous conversion of N2O and CO [31].
The most favorable intramolecular hydrogen-bond-involved pathway (Figure 1) for the conversion of N2O to N2 catalyzed by Ru-H complexes (C1–C10, Chart 2) is studied. The ΔG‡ for TS-1-2 (formation of O-bound diazene intermediate from insertion of N2O into the Ru-H bond) and TS-3-4 (generation of N2 from proton transfer) are summarized in Table 1. Catalysts with an electron-withdrawing group (CF3) generally produce higher ΔG‡ for TS-1-2 and TS-3-4 compared to catalysts with an electron-donating group (CH3). The ΔG‡ of TS-1-2 for C1 [(PCF3NCF3N)-Ru-H], C4 [(PNN)-Ru-H], and C7 [(PCH3NCH3N)-Ru-H] are 24.9, 24.1, and 23.6 kcal mol−1, respectively (Table 1). The ΔG‡ of TS-3-4 for C1 [(PCF3NCF3N)-Ru-H], C4 [(PNN)-Ru-H], and C7 [(PCH3NCH3N)-Ru-H] are 27.3, 26.6, and 26.0 kcal mol−1, respectively (Table 1). It is noted the C8 [(PNC)-Ru-H] has a higher ΔG‡ for the turnover-limiting step (TOLS, TS-3-4) compared to C4 [(PNN)-Ru-H] (28.4 vs. 26.6 kcal mol−1), which is consistent with the experimental reports [31].

Table 1.
Computed hydricities, Ru-H stretching frequencies, and Gibbs free energies of activation, ΔG‡.
Computed Ru-H harmonic stretching frequencies (νRu-H, gas-phase B3LYP-GD3BJ/BS1 computations) and computed hydricities (SMD(THF)-B3LYP-GD3BJ/BS2//B3LYP-GD3BJ/BS1) are also summarized in Table 1. Computational results show that introducing the CF3 electron-withdrawing group strengthens the Ru-H bond (higher Ru-H stretching frequencies) compared to the CH3 electron-donating group (1950.9 cm−1 for C1 [(PCF3NCF3N)-Ru-H], 1943.3 for C4 [(PNN)-Ru-H], and 1941.9 for C7 [(PCH3NCH3N)-Ru-H]. These results are consistent with the computed hydricities, and a stronger Ru-H bond has a poorer hydride-donating ability (the more positive hydricity value). The computed hydricities are 54.7 kcal mol−1 for C1 [(PCF3NCF3N)-Ru-H], 50.9 for C4 [(PNN)-Ru-H], and 49.1 for C7 [(PCH3NCH3N)-Ru-H] (Table 1). The computed hydricities, together with the Ru-H stretching frequencies, demonstrate that the hydride-donating ability for the Ru-H complexes with the CF3 electron-withdrawing group is poorer compared to the complexes with the CH3 electron-donating group.
In order to quantitatively explore the structure–activity relationship in the conversion of N2O to N2, the relationship between the computed ΔG‡ and Ru-H stretching frequencies (Figure 2), and the relationship between the computed ΔG‡ and computed hydricities (Figure 3) were fitted. Good linear relationships between computed ΔG‡ and Ru-H stretching frequencies (R2 = 0.9658 for C1–C7, and R2 = 0.8578 for C8–C10, Figure 2) were obtained, and excellent correlations exist between ΔG‡ of TS-3-4 and the computed hydricities (R2 = 0.9158 for C1–C7, and R2 = 0.9765 for C8–C10, Figure 3). Excellent linear fittings between the ΔG‡ of TOLS and the computed hydricities (R2 = 0.9381 for C1–C7, and R2 = 0.9272 for C8–C10, Figure 4) were also obtained. The structure–activity relationship using hydricity to predict the activity is consistent with the results from studies on the molecular transition-metal hydride involved CO2 hydrogenation, CO2 reduction, and H2 evolution [66,67,68,71,72]. This result suggests that a more active Ru-H catalyst with a higher turnover frequency for the conversion of N2O to N2 would come from introducing a more electron-donating ligand.
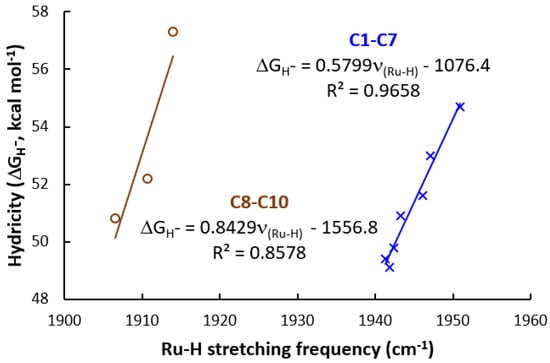
Figure 2.
Linear fitting between the computed hydricities (ΔGH−) and Ru-H stretching frequencies.
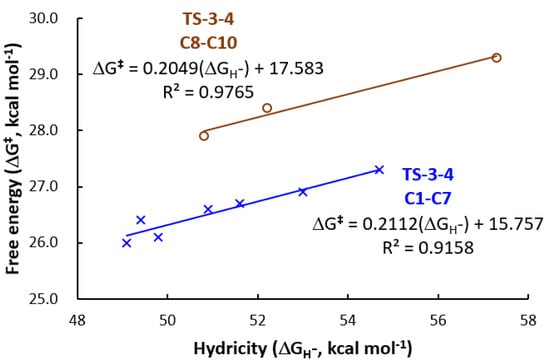
Figure 3.
Linear fitting between the Gibbs free energies of activation (ΔG‡) of TS-3-4 and the computed hydricities (ΔGH−).
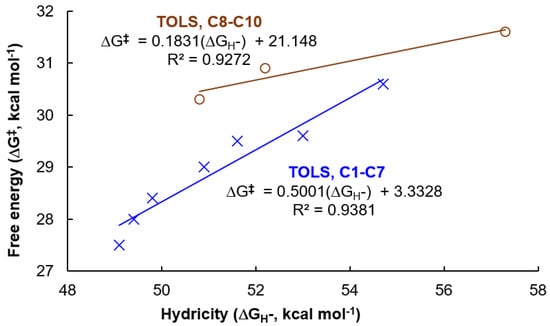
Figure 4.
Linear fitting between the computed hydricities (ΔGH−) and the Gibbs free energy of activation (ΔG‡) of TOLS.
4. Conclusions
A comprehensive theoretical investigation of the reaction between N2O to an Ru-H complex using DFT computations was performed. The proton transfer from Ru-H to the terminal N of endo N2O (Figure 1) was shown as the most favorable pathway, which includes N2O insertion into the Ru-H bond (TS-1-2, 24.1 kcal mol−1), change of geometry of the formed (Z)-O-bound oxyldiazene intermediate (TS-2-3, 5.5 kcal mol−1), and the formation of an Ru-OH complex and generation of N2 from a proton transfer step (TS-3-4, 26.6 kcal mol−1). Significantly low Gibbs free energy of activation in the generation of N2 (15.1 kcal mol−1, TS-3-4-OH2) with the participation of potentially existing traces of H2O in the THF solvent serving as a proton shuttle was observed. The excellent linear relationships between the computed hydricities (ΔGH−) and the Gibbs free energies of activation (ΔG‡) of TS-3-4, between the computed hydricities (ΔGH−) and the Gibbs free energy of activation (ΔG‡) of TOLS (R2 > 0.91), suggest that hydricity could be utilized as a potential parameter to predict the catalytic activities, and the design of more active Ru-H catalysts could benefit from ligand modification with more electron-donating groups.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics10060069/s1, Table S1. Comparisons of the Gibbs free energies; Figure S1. Linear relationship; Scheme S1. Computed APT charges of N2O; Figure S2. IRC plots; Figure S3. IRC plots; Figure S4. IRC plots; Figure S5. IRC plots; Figure S6. Free energy diagram for an alternative higher-energy pathway; Scheme S2. Free energy diagram for proton transfer; Figure S7. Free energy diagram for N2 generation from hydride transfer; Figure S8. Free energy diagram for a higher-energy hydride transfer; Figure S9. Linear fitting; Table S2. DFT computed energies; Table S3. SMD(THF)-B3LYP-GD3BJ/BS2 computed energies; Table S4. Cartesian coordinates.
Author Contributions
G.L.: conceptualization, investigation, formal analysis, methodology, writing—reviewing and editing, and funding acquisition; M.Z.: investigation and formal analysis; C.E.W.: conceptualization, formal analysis, writing—reviewing and editing, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the start-up funds from Xidian University (1018/10251210050), and also partially supported by the Mississippi State University Office of Research and Economic Development and the United States National Science Foundation (OIA-1539035).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank the high-performance computing platform of Xidian University (XDHCPP) and the Mississippi Center for Supercomputing Research (MCSR) for computing support. We are grateful for the financial support from the Academy of Advanced Interdisciplinary Research and the start-up funds from Xidian University (1018/10251210050). This study was also partially supported by the Mississippi State University Office of Research and Economic Development and the United States National Science Foundation (OIA-1539035).
Conflicts of Interest
The authors declare no conflict of interest.
References and Note
- Ravishankara, A.R.; Daniel, J.S.; Portmann, R.W. Nitrous Oxide (N2O): The Dominant Ozone-Depleting Substance Emitted in the 21st Century. Science 2009, 326, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Montzka, S.A.; Dlugokencky, E.J.; Butler, J.H. Non-CO2 greenhouse gases and climate change. Nature 2011, 476, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Chipperfield, M. Nitrous oxide delays ozone recovery. Nat. Geosci. 2009, 2, 742–743. [Google Scholar] [CrossRef]
- Griffis, T.J.; Chen, Z.; Baker, J.M.; Wood, J.D.; Millet, D.B.; Lee, X.; Venterea, R.T.; Turner, P.A. Nitrous oxide emissions are enhanced in a warmer and wetter world. Proc. Natl. Acad. Sci. USA 2017, 114, 12081–12085. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Lee, J.W.; Chandran, K.; Kim, S.; Khanal, S.K. Nitrous Oxide (N2O) Emission from Aquaculture: A Review. Environ. Sci. Technol. 2012, 46, 6470–6480. [Google Scholar] [CrossRef] [PubMed]
- IPCC. Climate Change 2013: The Physical Science Basis: Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2013. [Google Scholar]
- Yang, G.; Peng, Y.; Marushchak, M.E.; Chen, Y.; Wang, G.; Li, F.; Zhang, D.; Wang, J.; Yu, J.; Liu, L.; et al. Magnitude and Pathways of Increased Nitrous Oxide Emissions from Uplands Following Permafrost Thaw. Environ. Sci. Technol. 2018, 52, 9162–9169. [Google Scholar] [CrossRef]
- Richard, S.S.; Anne, R.D.; Luke, D.O.; Darryn, W.W. Impact of future nitrous oxide and carbon dioxide emissions on the stratospheric ozone layer. Environ. Res. Lett. 2015, 10, 034011. [Google Scholar] [CrossRef]
- Kanter, D.; Mauzerall, D.L.; Ravishankara, A.R.; Daniel, J.S.; Portmann, R.W.; Grabiel, P.M.; Moomaw, W.R.; Galloway, J.N. A post-Kyoto partner: Considering the stratospheric ozone regime as a tool to manage nitrous oxide. Proc. Natl. Acad. Sci. USA 2013, 110, 4451–4457. [Google Scholar] [CrossRef]
- Tolman, W.B. Binding and Activation of N2O at Transition-Metal Centers: Recent Mechanistic Insights. Angew. Chem. Int. Ed. 2010, 49, 1018–1024. [Google Scholar] [CrossRef]
- Severin, K. Synthetic chemistry with nitrous oxide. Chem. Soc. Rev. 2015, 44, 6375–6386. [Google Scholar] [CrossRef]
- You, Y.; Chang, H.; Ma, L.; Guo, L.; Qin, X.; Li, J.; Li, J. Enhancement of N2O decomposition performance by N2O pretreatment over Ce-Co-O catalyst. Chem. Eng. J. 2018, 347, 184–192. [Google Scholar] [CrossRef]
- Konsolakis, M. Recent Advances on Nitrous Oxide (N2O) Decomposition over Non-Noble-Metal Oxide Catalysts: Catalytic Performance, Mechanistic Considerations, and Surface Chemistry Aspects. ACS Catal. 2015, 5, 6397–6421. [Google Scholar] [CrossRef]
- Kaczmarczyk, J.; Zasada, F.; Janas, J.; Indyka, P.; Piskorz, W.; Kotarba, A.; Sojka, Z. Thermodynamic Stability, Redox Properties, and Reactivity of Mn3O4, Fe3O4, and Co3O4 Model Catalysts for N2O Decomposition: Resolving the Origins of Steady Turnover. ACS Catal. 2016, 6, 1235–1246. [Google Scholar] [CrossRef]
- Yan, L.; Zhang, X.; Ren, T.; Zhang, H.; Wang, X.; Suo, J. Superior performance of nano-Au supported over Co3O4 catalyst in direct N2O decomposition. Chem. Commun. 2002, 860–861. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Feller, M.; Ben-David, Y.; Milstein, D. Hydrogenation and Hydrosilylation of Nitrous Oxide Homogeneously Catalyzed by a Metal Complex. J. Am. Chem. Soc. 2017, 139, 5720–5723. [Google Scholar] [CrossRef]
- Vaughan, G.A.; Rupert, P.B.; Hillhouse, G.L. Selective O-atom transfer from nitrous oxide to hydride and aryl ligands of bis(pentamethylcyclopentadienyl)hafnium derivatives. J. Am. Chem. Soc. 1987, 109, 5538–5539. [Google Scholar] [CrossRef]
- Matsunaga, P.T.; Hillhouse, G.L.; Rheingold, A.L. Oxygen-atom transfer from nitrous oxide to a nickel metallacycle. Synthesis, structure, and reactions of (2,2’-bipyridine)Ni(OCH2CH2CH2CH2). J. Am. Chem. Soc. 1993, 115, 2075–2077. [Google Scholar] [CrossRef]
- Kaplan, A.W.; Bergman, R.G. Nitrous Oxide Mediated Oxygen Atom Insertion into a Ruthenium−Hydride Bond. Synthesis and Reactivity of the Monomeric Hydroxoruthenium Complex (DMPE)2Ru(H)(OH). Organometallics 1997, 16, 1106–1108. [Google Scholar] [CrossRef]
- Kaplan, A.W.; Bergman, R.G. Nitrous Oxide Mediated Synthesis of Monomeric Hydroxoruthenium Complexes. Reactivity of (DMPE)2Ru(H)(OH) and the Synthesis of a Silica-Bound Ruthenium Complex. Organometallics 1998, 17, 5072–5085. [Google Scholar] [CrossRef]
- Koo, K.; Hillhouse, G.L. Formation of a Substituted Tetrahydrofuran by Formal [2 + 2 + 1] Coupling of an Oxygen Atom with Two Olefins at a Nickel Center. Organometallics 1998, 17, 2924–2925. [Google Scholar] [CrossRef]
- Tskhovrebov, A.G.; Solari, E.; Scopelliti, R.; Severin, K. Reactions of Grignard Reagents with Nitrous Oxide. Organometallics 2014, 33, 2405–2408. [Google Scholar] [CrossRef]
- Laplaza, C.E.; Odom, A.L.; Davis, W.M.; Cummins, C.C.; Protasiewicz, J.D. Cleavage of the Nitrous Oxide NN Bond by a Tris(amido)molybdenum(III) Complex. J. Am. Chem. Soc. 1995, 117, 4999–5000. [Google Scholar] [CrossRef]
- Ni, C.; Ellis, B.D.; Long, G.J.; Power, P.P. Reactions of Ar’CrCrAr’ with N2O or N3(1-Ad): Complete cleavage of the Cr-Cr quintuple interaction. Chem. Commun. 2009, 2332–2334. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Michel, R.; Koley, D.; Roesky, H.W.; Stalke, D. Reactivity Studies of a Disilene with N2O and Elemental Sulfur. Inorg. Chem. 2011, 50, 10878–10883. [Google Scholar] [CrossRef]
- Poh, S.; Hernandez, R.; Inagaki, M.; Jessop, P.G. Oxidation of Phosphines by Supercritical Nitrous Oxide. Org. Lett. 1999, 1, 583–586. [Google Scholar] [CrossRef]
- Ben-Daniel, R.; Neumann, R. Activation of Nitrous Oxide and Selective Oxidation of Alcohols and Alkylarenes Catalyzed by the [PV2Mo10O40]5− Polyoxometalate Ion. Angew. Chem. Int. Ed. 2003, 42, 92–95. [Google Scholar] [CrossRef]
- Tskhovrebov, A.G.; Naested, L.C.E.; Solari, E.; Scopelliti, R.; Severin, K. Synthesis of Azoimidazolium Dyes with Nitrous Oxide. Angew. Chem. Int. Ed. 2015, 54, 1289–1292. [Google Scholar] [CrossRef]
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics, 83rd ed.; CRC Press LLC: Boca Raton, FL, USA, 2002. [Google Scholar]
- Lee, J.-D.; Fang, W.-P.; Li, C.-S.; Cheng, C.-H. Catalytic reduction of nitrous oxide by carbon monoxide in the presence of rhodium carbonyl and hydroxide. Evidence for an electron-transfer and an oxygen-transfer mechanism. J. Chem. Soc. Dalton Trans. 1991, 1923–1927. [Google Scholar] [CrossRef]
- Zeng, R.; Feller, M.; Diskin-Posner, Y.; Shimon, L.J.W.; Ben-David, Y.; Milstein, D. CO Oxidation by N2O Homogeneously Catalyzed by Ruthenium Hydride Pincer Complexes Indicating a New Mechanism. J. Am. Chem. Soc. 2018, 140, 7061–7064. [Google Scholar] [CrossRef]
- Pacultová, K.; Obalová, L.; Kovanda, F.; Jirátová, K. Catalytic reduction of nitrous oxide with carbon monoxide over calcined Co–Mn–Al hydrotalcite. Catal. Today 2008, 137, 385–389. [Google Scholar] [CrossRef]
- Ketrat, S.; Maihom, T.; Wannakao, S.; Probst, M.; Nokbin, S.; Limtrakul, J. Coordinatively Unsaturated Metal–Organic Frameworks M3(btc)2 (M = Cr, Fe, Co, Ni, Cu, and Zn) Catalyzing the Oxidation of CO by N2O: Insight from DFT Calculations. Inorg. Chem. 2017, 56, 14005–14012. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Zhang, Y.; Xiang, C.; Li, Y.; Fan, T.; Lei, Q.; Fang, W. Non-innocent PNN ligand is important for CO oxidation by N2O catalyzed by a (PNN)Ru–H pincer complex: Insights from DFT calculations. Dalton Trans. 2018, 47, 15324–15330. [Google Scholar] [CrossRef] [PubMed]
- Luque-Urrutia, J.A.; Poater, A. The Fundamental Noninnocent Role of Water for the Hydrogenation of Nitrous Oxide by PNP Pincer Ru-based Catalysts. Inorg. Chem. 2017, 56, 14383–14387. [Google Scholar] [CrossRef] [PubMed]
- Escayola, S.; Solà, M.; Poater, A. Mechanism of the Facile Nitrous Oxide Fixation by Homogeneous Ruthenium Hydride Pincer Catalysts. Inorg. Chem. 2020, 59, 9374–9383. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results Obtained With The Correlation Energy Density Functionals Of Becke And Lee, Yang And Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Couty, M.; Hall, M.B. Basis sets for transition metals: Optimized outer p functions. J. Comput. Chem. 1996, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Check, C.E.; Faust, T.O.; Bailey, J.M.; Wright, B.J.; Gilbert, T.M.; Sunderlin, L.S. Addition of polarization and diffuse functions to the LANL2DZ basis set for p-block elements. J. Phys. Chem. A 2001, 105, 8111–8116. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular-Orbital Studies of Organic-Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Influence of Polarization Functions on Molecular-Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- In Gaussian software, the 6-31G(d’) basis set has the exponent of d polarization functions for C, N, O taken from the 6-311G(d) basis sets, instead of the original arbitrarily assigned exponent of 0.8 used in the 6-31G(d) basis sets. For H, the 6-31G(d’) keyword in Gaussian software utilizes the 6-31G(d) basis sets.
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Liang, G.; Hollis, T.K.; Webster, C.E. Computational Analysis of the Intramolecular Oxidative Amination of an Alkene Catalyzed by the Extreme π-loading N-Heterocyclic Carbene Pincer Tantalum(V) Bis(imido) Complex. Organometallics 2018, 37, 1671–1681. [Google Scholar] [CrossRef]
- Zhang, M.; Liang, G. Understanding the Sigmatropic Shifts of Cyclopenta-2,4-dien-1-yltrimethylsilane in its Diels-Alder Addition. Org. Biomol. Chem. 2021, 19, 1732–1737. [Google Scholar] [CrossRef] [PubMed]
- Witte, J.; Mardirossian, N.; Neaton, J.B.; Head-Gordon, M. Assessing DFT-D3 Damping Functions Across Widely Used Density Functionals: Can We Do Better? J. Chem. Theory Comput. 2017, 13, 2043–2052. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; An, K.; Wang, Y.; Wu, Y.-D.; Zhang, X.; Yu, Z.-X.; He, W. A Combined Computational and Experimental Study of Rh-Catalyzed C–H Silylation with Silacyclobutanes: Insights Leading to a More Efficient Catalyst System. J. Am. Chem. Soc. 2021, 143, 3571–3582. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Wang, F. Performing Molecular Dynamics Simulations and Computing Hydration Free Energies on the B3LYP-D3(BJ) Potential Energy Surface with Adaptive Force Matching: A Benchmark Study with Seven Alcohols and One Amine. ACS Phys. Chem. Au 2021, 1, 14–24. [Google Scholar] [CrossRef]
- Xie, H.; Li, Y.; Huang, L.; Nong, F.; Ren, G.; Fan, T.; Lei, Q.; Fang, W. Dehydrogenation of benzyl alcohol with N2O as the hydrogen acceptor catalyzed by the rhodium(I) carbene complex: Insights from quantum chemistry calculations. Dalton Trans. 2016, 45, 16485–16491. [Google Scholar] [CrossRef]
- Yu, H.; Jia, G.; Lin, Z. Theoretical Studies on O-Insertion Reactions of Nitrous Oxide with Ruthenium Hydride Complexes. Organometallics 2008, 27, 3825–3833. [Google Scholar] [CrossRef]
- Yao, L.; Li, Y.; Huang, L.; Guo, K.; Ren, G.; Wu, Z.; Lei, Q.; Fang, W.; Xie, H. A DFT study on the mechanisms of hydrogenation and hydrosilylation of nitrous oxide catalyzed by a ruthenium PNP pincer complex. Comput. Theor. Chem. 2018, 1128, 48–55. [Google Scholar] [CrossRef]
- Murdoch, J.R. What is the rate-limiting step of a multistep reaction? J. Chem. Educ. 1981, 58, 32. [Google Scholar] [CrossRef]
- Kozuch, S.; Shaik, S. How to Conceptualize Catalytic Cycles? The Energetic Span Model. Acc. Chem. Res. 2011, 44, 101–110. [Google Scholar] [CrossRef]
- Wiedner, E.S.; Chambers, M.B.; Pitman, C.L.; Bullock, R.M.; Miller, A.J.M.; Appel, A.M. Thermodynamic hydricity of transition metal hydrides. Chem. Rev. 2016, 116, 8655–8692. [Google Scholar] [CrossRef]
- Ilic, S.; Alherz, A.; Musgrave, C.B.; Glusac, K.D. Thermodynamic and kinetic hydricities of metal-free hydrides. Chem. Soc. Rev. 2018, 47, 2809–2836. [Google Scholar] [CrossRef] [PubMed]
- Jeletic, M.S.; Hulley, E.B.; Helm, M.L.; Mock, M.T.; Appel, A.M.; Wiedner, E.S.; Linehan, J.C. Understanding the Relationship Between Kinetics and Thermodynamics in CO2 Hydrogenation Catalysis. ACS Catal. 2017, 7, 6008–6017. [Google Scholar] [CrossRef]
- Ostericher, A.L.; Waldie, K.M.; Kubiak, C.P. Utilization of Thermodynamic Scaling Relationships in Hydricity To Develop Nickel Hydrogen Evolution Reaction Electrocatalysts with Weak Acids and Low Overpotentials. ACS Catal. 2018, 8, 9596–9603. [Google Scholar] [CrossRef]
- Waldie, K.M.; Ostericher, A.L.; Reineke, M.H.; Sasayama, A.F.; Kubiak, C.P. Hydricity of transition-metal hydrides: Thermodynamic considerations for CO2 reduction. ACS Catal. 2018, 8, 1313–1324. [Google Scholar] [CrossRef]
- Goodfellow, A.S.; Bühl, M. Hydricity of 3D Transition Metal Complexes from Density Functional Theory: A Benchmarking Study. Molecules 2021, 26, 4072. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Liu, Z.; Tang, L.; Li, J.; Yang, Z. Trans Influence of Boryl Ligands in CO2 Hydrogenation on Ruthenium Complexes: Theoretical Prediction of Highly Active Catalysts for CO2 Reduction. Catalysts 2021, 11, 1356. [Google Scholar] [CrossRef]
- Mondal, B.; Neese, F.; Ye, S. Toward Rational Design of 3D Transition Metal Catalysts for CO2 Hydrogenation Based on Insights into Hydricity-Controlled Rate-Determining Steps. Inorg. Chem. 2016, 55, 5438–5444. [Google Scholar] [CrossRef]
- Muckerman, J.T.; Achord, P.; Creutz, C.; Polyansky, D.E.; Fujita, E. Calculation of thermodynamic hydricities and the design of hydride donors for CO2 reduction. Proc. Natl. Acad. Sci. USA 2012, 109, 15657–15662. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).