
Neonatal Hemolytic Jaundice: Causes, Diagnostic Approach, and Management

 ,
,  ,
, 
Abstract
1. Introduction
2. Immune Causes of Unconjugated Hyperbilirubinemia
2.1. ABO Blood Group Incompatibility
2.2. Rh Incompatibility
2.3. Other RBC Antigen Incompatibility
Hemolytic Disease of the Fetus and Newborn (HDFN)
3. Non-Immune Causes of Unconjugated Hyperbilirubinemia
3.1. RBC Enzymopathies
3.1.1. Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency
3.1.2. Pyruvate Kinase (PK) Deficiency
3.1.3. Other RBC Enzymopathies
3.2. RBC Membrane Defects
3.2.1. Hereditary Spherocytosis (HS)
3.2.2. Hereditary Elliptocytosis and Hereditary Pyropoikilocytosis
3.2.3. Rarer RBC Membrane Disorders
3.3. Hemoglobinopathies
3.4. Congenital Dyserythropoietic Anemia
3.5. Sepsis
3.6. Other Causes
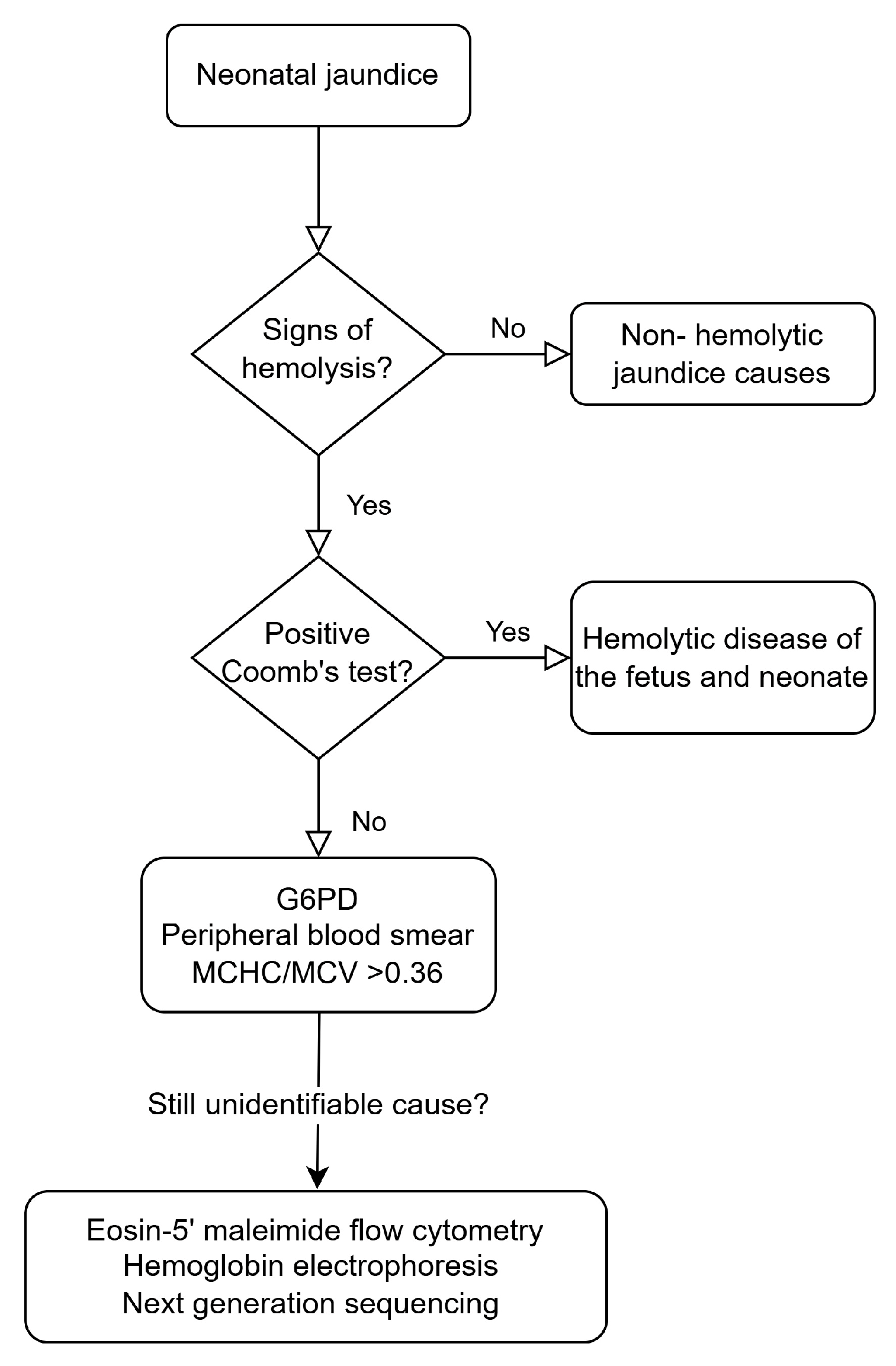
4. Diagnostic Approach
5. Management
6. Conclusions
Funding
Conflicts of Interest
References
- Mitra, S.; Rennie, J. Neonatal jaundice: Aetiology, diagnosis and treatment. Br. J. Hosp. Med. 2017, 78, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.W.R.; Wong, R.J.; Stevenson, D.K. Molecular Physiology and Pathophysiology of Bilirubin Handling by the Blood, Liver, Intestine, and Brain in the Newborn. Physiol. Rev. 2020, 100, 1291–1346. [Google Scholar] [CrossRef] [PubMed]
- Ansong-Assoku, B.; Shah, S.D.; Adnan, M.; Ankola, P.A. Neonatal Jaundice. In StatPearls; Treasure Island (FL) Ineligible Companies: Tampa, FL, USA, 2024. [Google Scholar]
- Pillai, A.; Pandita, A.; Osiovich, H.; Manhas, D. Pathogenesis and Management of Indirect Hyperbilirubinemia in Preterm Neonates Less Than 35 Weeks: Moving Toward a Standardized Approach. NeoReviews 2020, 21, e298–e307. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.B.; Calkins, K.L. Neonatal Indirect Hyperbilirubinemia. NeoReviews 2020, 21, e749–e760. [Google Scholar] [CrossRef]
- Ullah, S.; Rahman, K.; Hedayati, M. Hyperbilirubinemia in Neonates: Types, Causes, Clinical Examinations, Preventive Measures and Treatments: A Narrative Review Article. Iran. J. Public Health 2016, 45, 558–568. [Google Scholar]
- Bhutani, V.K.; Wong, R. Bilirubin-induced neurologic dysfunction (BIND). Semin. Fetal Neonatal Med. 2015, 20, 1. [Google Scholar] [CrossRef]
- Brites, D.; Silva, R.F.M. Bilirubin neurotoxicity: A narrative review on long lasting, insidious, and dangerous effects. Pediatr. Med. 2021, 4, 1–24. [Google Scholar] [CrossRef]
- Kemper, A.R.; Newman, T.B.; Slaughter, J.L.; Maisels, M.J.; Watchko, J.F.; Downs, S.M.; Grout, R.W.; Bundy, D.G.; Stark, A.R.; Bogen, D.L.; et al. Clinical Practice Guideline Revision: Management of Hyperbilirubinemia in the Newborn Infant 35 or More Weeks of Gestation. Pediatrics 2022, 150, e2022058859. [Google Scholar] [CrossRef]
- Christensen, R.D.; Yaish, H.M. Hemolytic Disorders Causing Severe Neonatal Hyperbilirubinemia. Clin. Perinatol. 2015, 42, 515–527. [Google Scholar] [CrossRef]
- Branch, D.R. Anti-A and anti-B: What are they and where do they come from? Transfusion 2015, 55 (Suppl. S2), S74–S79. [Google Scholar] [CrossRef]
- Matteocci, A.; De Rosa, A.; Buffone, E.; Pierelli, L. Retrospective analysis of HDFN due to ABO incompatibility in a single institution over 6 years. Transfus. Med. 2019, 29, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Akanmu, A.S.; Oyedeji, O.A.; Adeyemo, T.A.; Ogbenna, A.A. Estimating the Risk of ABO Hemolytic Disease of the Newborn in Lagos. J. Blood Transfus. 2015, 2015, 560738. [Google Scholar] [CrossRef] [PubMed]
- Alshammari, S.; Alqashami, A.; Alhumud, S.; Aladadh, M.; Alsaif, S.; Ali, K. Neonatal ABO Incompatibility, Influence of Blood Group, and Coomb’s Test on Outcome. J. Clin. Neonatol. 2022, 11, 212–218. [Google Scholar] [CrossRef]
- de Winter, D.P.; Kaminski, A.; Tjoa, M.L.; Oepkes, D. Hemolytic disease of the fetus and newborn: Systematic literature review of the antenatal landscape. BMC Pregnancy Childbirth 2023, 23, 12. [Google Scholar] [CrossRef]
- Routray, S.S.; Mishra, D.; Kanungo, G.N.; Behera, R. Hemolytic Disease of Newborn due to ABO Incompatibility between B Blood Group Mother and A Blood Group Neonate. J. Lab. Physicians 2023, 15, 146–148. [Google Scholar] [CrossRef]
- Metcalf, R.A.; Khan, J.; Andrews, J.; Mayock, D.; Billimoria, Z.; Pagano, M.B. Severe ABO Hemolytic Disease of the Newborn Requiring Exchange Transfusion. J. Pediatr. Hematol. Oncol. 2019, 41, 632–634. [Google Scholar] [CrossRef]
- Rosenkrans, D.; Zubair, M.; Doyal, A. Rh Blood Group System. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2025. [Google Scholar]
- Sarwar, A.; Citla Sridhar, D. Rh Hemolytic Disease. In StatPearls; Treasure Island (FL) Ineligible Companies: Tampa, FL, USA, 2025. [Google Scholar]
- Myle, A.K.; Al-Khattabi, G.H. Hemolytic Disease of the Newborn: A Review of Current Trends and Prospects. Pediatr. Health Med. Ther. 2021, 12, 491–498. [Google Scholar] [CrossRef]
- Tyndall, C.; Cuzzilla, R.; Kane, S.C. The rhesus incompatible pregnancy and its consequences for affected fetuses and neonates. Transfus. Apher. Sci. 2020, 59, 102948. [Google Scholar] [CrossRef]
- Yu, D.; Ling, L.E.; Krumme, A.A.; Tjoa, M.L.; Moise, K.J., Jr. Live birth prevalence of hemolytic disease of the fetus and newborn in the United States from 1996 to 2010. AJOG Glob. Rep. 2023, 3, 100203. [Google Scholar] [CrossRef]
- Zakerihamidi, M.; Moradi, A.; Boskabadi, H. Comparison of severity and prognosis of jaundice due to Rh incompatibility and G6PD deficiency. Transfus. Apher. Sci. 2023, 62, 103714. [Google Scholar] [CrossRef]
- Ozcan, M.; Sevinc, S.; Erkan, V.B.; Yurdugul, Y.; Sarici, S.U. Hyperbilirubinemia due to minor blood group (anti-E) incompatibility in a newborn: A case report. Turk. Arch. Pediatr. 2017, 52, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Tugcu, A.U.; Ince, D.A.; Turan, O.; Belen, B.; Olcay, L.; Ecevit, A. Hemolytic anemia caused by non-D minor blood incompatibilities in a newborn. Pan Afr. Med. J. 2019, 33, 262. [Google Scholar] [CrossRef]
- Agrawal, A.; Hussain, K.S.; Kumar, A. Minor blood group incompatibility due to blood groups other than Rh(D) leading to hemolytic disease of fetus and newborn: A need for routine antibody screening during pregnancy. Intractable Rare Dis. Res. 2020, 9, 43–47. [Google Scholar] [CrossRef]
- Thornton, N.M.; Grimsley, S.P. Clinical significance of antibodies to antigens in the ABO, MNS, P1PK, Rh, Lutheran, Kell, Lewis, Duffy, Kidd, Diego, Yt, and Xg blood group systems. Immunohematology 2019, 35, 95–101. [Google Scholar] [CrossRef]
- Odabasi, I.O.; Uslu, S.; Bas, E.K.; Bulbul, A.; Unal, E.T.; Acar, D.B.; Tellioglu, A.; Ileri, M.F. Hemolytic Anemia Due To Anti-c Incompatibility in The Newborn Period: A Case Report. Med. Bull. Sisli Etfal Hosp. 2020, 54, 502–504. [Google Scholar] [CrossRef]
- Aykut, S.; Demir, S.C.; Evruke, I.C.; Sucu, M.; Uzay, F.I.; Avan, M.; Bayer, O.K.; Yalcin, E. Approach to Pregnancy Affected by Kell Alloimmunization. Case Rep. Hematol. 2024, 2024, 1929147. [Google Scholar] [CrossRef]
- Khafagy, S.; Khaled, A.; Ibrahim, M. Minor Blood Group Incompatibility in Patients with Neonatal Jaundice: A Single Centre Study in Egypt. QJM Int. J. Med. 2024, 117, hcae175.749. [Google Scholar] [CrossRef]
- Committee on Practice Bulletins—Obstetrics. ACOG Practice Bulletin No. 192: Management of Alloimmunization During Pregnancy. Obstet. Gynecol. 2018, 131, e82–e90. [Google Scholar] [CrossRef]
- Norton, M.E.; Chauhan, S.P.; Dashe, J.S. Society for Maternal-Fetal Medicine (SMFM) Clinical Guideline #7: Nonimmune hydrops fetalis. Am. J. Obstet. Gynecol. 2015, 212, 127–139. [Google Scholar] [CrossRef]
- Hendrickson, J.E.; Delaney, M. Hemolytic Disease of the Fetus and Newborn: Modern Practice and Future Investigations. Transfus. Med. Rev. 2016, 30, 159–164. [Google Scholar] [CrossRef]
- Nakayama, A.; Oshiro, M.; Yamada, Y.; Hattori, T.; Wakano, Y.; Hayashi, S.; Kokubo, M.; Takemoto, K.; Honda, S.; Ieda, K.; et al. Prognostic factors of hydrops fetalis with pleural effusion. Pediatr. Int. Off. J. Jpn. Pediatr. Soc. 2017, 59, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Hall, V.; Vadakekut, E.S.; Avulakunta, I.D. Hemolytic Disease of the Fetus and Newborn. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2025. [Google Scholar]
- Dziegiel, M.H.; Krog, G.R.; Hansen, A.T.; Olsen, M.; Lausen, B.; Norgaard, L.N.; Bergholt, T.; Rieneck, K.; Clausen, F.B. Laboratory Monitoring of Mother, Fetus, and Newborn in Hemolytic Disease of Fetus and Newborn. Transfus. Med. Hemotherapy 2021, 48, 306–315. [Google Scholar] [CrossRef]
- Luzzatto, L.; Ally, M.; Notaro, R. Glucose-6-phosphate dehydrogenase deficiency. Blood 2020, 136, 1225–1240. [Google Scholar] [CrossRef]
- Lee, H.Y.; Ithnin, A.; Azma, R.Z.; Othman, A.; Salvador, A.; Cheah, F.C. Glucose-6-Phosphate Dehydrogenase Deficiency and Neonatal Hyperbilirubinemia: Insights on Pathophysiology, Diagnosis, and Gene Variants in Disease Heterogeneity. Front. Pediatr. 2022, 10, 875877. [Google Scholar] [CrossRef] [PubMed]
- Orrico, F.; Laurance, S.; Lopez, A.C.; Lefevre, S.D.; Thomson, L.; Moller, M.N.; Ostuni, M.A. Oxidative Stress in Healthy and Pathological Red Blood Cells. Biomolecules 2023, 13, 1262. [Google Scholar] [CrossRef]
- Nannelli, C.; Bosman, A.; Cunningham, J.; Dugue, P.A.; Luzzatto, L. Genetic variants causing G6PD deficiency: Clinical and biochemical data support new WHO classification. Br. J. Haematol. 2023, 202, 1024–1032. [Google Scholar] [CrossRef]
- Luzzatto, L.; Bancone, G.; Dugué, P.A.; Jiang, W.; Minucci, A.; Nannelli, C.; Pfeffer, D.; Prchal, J.; Sirdah, M.; Sodeinde, O.; et al. New WHO classification of genetic variants causing G6PD deficiency. Bull. World Health Organ. 2024, 102, 615–617. [Google Scholar] [CrossRef]
- Zhou, X.; Qiang, Z.; Zhang, S.; Zhou, Y.; Xiao, Q.; Tan, G. Evaluating the relationship between Clinical G6PD enzyme activity and gene variants. PeerJ 2024, 12, e16554. [Google Scholar] [CrossRef]
- Cunningham, A.D.; Hwang, S.; Mochly-Rosen, D. Glucose-6-Phosphate Dehydrogenase Deficiency and the Need for a Novel Treatment to Prevent Kernicterus. Clin. Perinatol. 2016, 43, 341–354. [Google Scholar] [CrossRef]
- Al-Bedaywi, R.R.R.; Salameh, K.M.K.; Abedin, S.; Viswanathan, B.; Khedr, A.A.; Habboub, L.H.M. Glucose-6-phosphate dehydrogenase deficiency and neonatal indirect hyperbilirubinemia: A retrospective cohort study among 40,305 consecutively born babies. J. Perinatol. 2024, 44, 1035–1041. [Google Scholar] [CrossRef]
- Luzzatto, L. Diagnosis and clinical management of enzymopathies. Hematol. Am. Soc. Hematol. Educ. Program 2021, 2021, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, H.; Abdeldaiem, N. Relationship between Mothers’ Ingestion of Fava Beans and Occurrence of Favism Attack among Their Breastfed Infants. Egypt. J. Health Care 2022, 13, 1995–2010. [Google Scholar] [CrossRef]
- Beretta, A.; Manuelli, M.; Cena, H. Favism: Clinical Features at Different Ages. Nutrients 2023, 15, 343. [Google Scholar] [CrossRef] [PubMed]
- Grace, R.F.; Glader, B. Red Blood Cell Enzyme Disorders. Pediatr. Clin. N. Am. 2018, 65, 579–595. [Google Scholar] [CrossRef]
- Secrest, M.H.; Storm, M.; Carrington, C.; Casso, D.; Gilroy, K.; Pladson, L.; Boscoe, A.N. Prevalence of pyruvate kinase deficiency: A systematic literature review. Eur. J. Haematol. 2020, 105, 173–184. [Google Scholar] [CrossRef]
- Grace, R.F.; Zanella, A.; Neufeld, E.J.; Morton, D.H.; Eber, S.; Yaish, H.; Glader, B. Erythrocyte pyruvate kinase deficiency: 2015 status report. Am. J. Hematol. 2015, 90, 825–830. [Google Scholar] [CrossRef]
- Chonat, S.; Eber, S.W.; Holzhauer, S.; Kollmar, N.; Morton, D.H.; Glader, B.; Neufeld, E.J.; Yaish, H.M.; Rothman, J.A.; Sharma, M.; et al. Pyruvate kinase deficiency in children. Pediatr. Blood Cancer 2021, 68, e29148. [Google Scholar] [CrossRef]
- Grace, R.F.; Bianchi, P.; van Beers, E.J.; Eber, S.W.; Glader, B.; Yaish, H.M.; Despotovic, J.M.; Rothman, J.A.; Sharma, M.; McNaull, M.M.; et al. Clinical spectrum of pyruvate kinase deficiency: Data from the Pyruvate Kinase Deficiency Natural History Study. Blood 2018, 131, 2183–2192. [Google Scholar] [CrossRef]
- Grace, R.F.; Barcellini, W. Management of pyruvate kinase deficiency in children and adults. Blood 2020, 136, 1241–1249. [Google Scholar] [CrossRef]
- Voit, R.A.; Grace, R.F. Pyruvate kinase deficiency in a newborn with extramedullary hematopoiesis in the skin. Blood 2020, 136, 770. [Google Scholar] [CrossRef]
- Christensen, R.D.; Yaish, H.M.; Gallagher, P.G. A pediatrician’s practical guide to diagnosing and treating hereditary spherocytosis in neonates. Pediatrics 2015, 135, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Polizzi, A.; Dicembre, L.P.; Failla, C.; Matola, T.D.; Moretti, M.; Ranieri, S.C.; Papa, F.; Cenci, A.M.; Buttarello, M. Overview on Hereditary Spherocytosis Diagnosis. Int. J. Lab. Hematol. 2025, 47, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liao, L.; Lin, F. The diagnostic protocol for hereditary spherocytosis-2021 update. J. Clin. Lab. Anal. 2021, 35, e24034. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, V.; Jain, S.K. Hereditary Spherocytosis. NeoReviews 2016, 17, e697–e704. [Google Scholar] [CrossRef]
- Weiss, N.M.; Kuzniewicz, M.W.; Shimano, K.A.; Walsh, E.M.; Newman, T.B. Use of Complete Blood Cell Count Components to Screen for Hereditary Spherocytosis in Neonates. Pediatrics 2021, 148, e2020021642. [Google Scholar] [CrossRef]
- Gallagher, P.G. Difficulty in Diagnosis of Hereditary Spherocytosis in the Neonate. Pediatrics 2021, 148, e2021051100. [Google Scholar] [CrossRef]
- Da Costa, L.; Galimand, J.; Fenneteau, O.; Mohandas, N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013, 27, 167–178. [Google Scholar] [CrossRef]
- Kalfa, T.A. Diagnosis and clinical management of red cell membrane disorders. Hematol. Am. Soc. Hematol. Educ. Program 2021, 2021, 331–340. [Google Scholar] [CrossRef]
- Andolfo, I.; Russo, R.; Gambale, A.; Iolascon, A. New insights on hereditary erythrocyte membrane defects. Haematologica 2016, 101, 1284–1294. [Google Scholar] [CrossRef]
- Blankenhorn, K.; Strumph, K. Hemoglobinopathies in the Neonate. NeoReviews 2024, 25, e720–e728. [Google Scholar] [CrossRef]
- Lal, A.; Vichinsky, E. The Clinical Phenotypes of Alpha Thalassemia. Hematol. Oncol. Clin. N. Am. 2023, 37, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Frömmel, C. Newborn Screening for Sickle Cell Disease and Other Hemoglobinopathies: A Short Review on Classical Laboratory Methods—Isoelectric Focusing, HPLC, and Capillary Electrophoresis. Int. J. Neonatal Screen. 2018, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, V.; Manzoni, F.; Raffaeli, G.; Cavallaro, G.; Fattizzo, B.; Amelio, G.S.; Gulden, S.; Amodeo, I.; Giannotta, J.A.; Mosca, F.; et al. Severe Presentation of Congenital Hemolytic Anemias in the Neonatal Age: Diagnostic and Therapeutic Issues. Diagnostics 2021, 11, 1549. [Google Scholar] [CrossRef] [PubMed]
- Risinger, M.; Emberesh, M.; Kalfa, T.A. Rare Hereditary Hemolytic Anemias: Diagnostic Approach and Considerations in Management. Hematol. Oncol. Clin. N. Am. 2019, 33, 373–392. [Google Scholar] [CrossRef]
- Olusanya, B.O.; Kaplan, M.; Hansen, T.W.R. Neonatal hyperbilirubinaemia: A global perspective. Lancet Child Adolesc. Health 2018, 2, 610–620. [Google Scholar] [CrossRef]
- Effenberger-Neidnicht, K.; Hartmann, M. Mechanisms of Hemolysis During Sepsis. Inflammation 2018, 41, 1569–1581. [Google Scholar] [CrossRef]
- Tsantes, A.G.; Parastatidou, S.; Tsantes, E.A.; Bonova, E.; Tsante, K.A.; Mantzios, P.G.; Vaiopoulos, A.G.; Tsalas, S.; Konstantinidi, A.; Houhoula, D.; et al. Sepsis-Induced Coagulopathy: An Update on Pathophysiology, Biomarkers, and Current Guidelines. Life 2023, 13, 350. [Google Scholar] [CrossRef]
- Hafez Ahmed, M.Z.; Saad, R.H.A.H.; Gadalla, A.; Thabet, R.H.I.; Elsisi, A.A.E.; Mohamed, A.A.N.A.; Abdallah, M.G.; Shikhon, T.; Mohmmed Maged, H.E.H.; Elfiky, M.A.H.; et al. The association of oxidative stress of neonatal hyperbilirubinemia and vitamin E supplementation. Clin. Exp. Hepatol. 2024, 10, 30–38. [Google Scholar] [CrossRef]
- Drouilly, M.; Jourdan, L.; Gerard, D.; Russello, J.; Bobee, V.; Audouy, A.; Phulpin, A.; Perrin, J. Infantile pyknocytosis, a neonatal hemolytic anemia with Heinz bodies: A cohort study. Pediatr. Blood Cancer 2024, 71, e31078. [Google Scholar] [CrossRef]
- Alkén, J.; Håkansson, S.; Ekéus, C.; Gustafson, P.; Norman, M. Rates of Extreme Neonatal Hyperbilirubinemia and Kernicterus in Children and Adherence to National Guidelines for Screening, Diagnosis, and Treatment in Sweden. JAMA Netw. Open 2019, 2, e190858. [Google Scholar] [CrossRef]
- Bahr, T.M.; Christensen, R.D.; Kaplan, M. Hemolytic causes of neonatal jaundice: Diagnosis and treatment. Pediatr. Med. 2021, 4, 1–14. [Google Scholar] [CrossRef]
- Christensen, R.D.; Bahr, T.M.; Ohls, R.K.; Moise, K.J., Jr. Neonatal/perinatal diagnosis of hemolysis using ETCOc. Semin. Fetal Neonatal Med. 2024, 30, 101547. [Google Scholar] [CrossRef] [PubMed]
- Christensen, R.D.; Yaish, H.M.; Lemons, R.S. Neonatal Hemolytic Jaundice: Morphologic Features of Erythrocytes That Will Help You Diagnose the Underlying Condition. Neonatology 2014, 105, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Maurice, P.; McCallion, J.; Fitzgibbon, M.; Barthelmes, J.N.; Karmous, W.; Hardy, E.J.; Mitchell, S.A.; Mitchell, C.R.; Lee, J.; Noel, W.; et al. Patient experience and burden of haemolytic disease of the foetus and newborn: A systematic review. BMC Pregnancy Childbirth 2025, 25, 114. [Google Scholar] [CrossRef]
- Beal, J.A. American Academy of Pediatrics’ Updated Clinical Guidelines for Managing Neonatal Hyperbilirubinemia. MCN Am. J. Matern. Child Nurs. 2023, 48, 49. [Google Scholar] [CrossRef]
- Nouri, S.; Zarkesh, M. Recent Advances in Adjuvant Pharmacotherapy for Neonatal Indirect Hyperbilirubinemia: A Narrative Review. J. Compr. Pediatr. 2023; in press. [Google Scholar] [CrossRef]
- Li, Y.; Yadav, U.; Zhu, X.; Liu, J.; Liu, H.; Yi, X. Treatment of jaundice in newborn recent progress. Int. J. Sci. Invent. Today 2021, 10, 398–406. [Google Scholar]
- Pan, F.; Wang, Y.; Xie, J.; Liu, Y.; He, X. Effect of Pre-exchange Transfusion Albumin Infusion on Neonatal Hyperbilirubinemia: A Meta-Analysis of Randomized Clinical Trials. Iran. J. Pediatr. 2022; in press. [Google Scholar] [CrossRef]
- Khafaga, K.A.; Alsaid, L.M.; Salama, R.H.; Abougabal, M.T. Fenofibrate As an Adjuvant to Phototherapy in Term Neonates with Hyperbilirubinemia; A Randomized Controlled Clinical Trial. Egypt. J. Hosp. Med. 2022, 89, 4439–4443. [Google Scholar] [CrossRef]

{kind=link}
| Immune-mediated hemolysis ABO incompatibility Rh incompatibility Other RBC antigen incompatibility |
| Non-immune hemolytic causes Deficiencies of RBC enzymes (G6PD deficiency, pyruvate kinase deficiency) Defects of RBC membrane (hereditary spherocytosis, elliptocytosis, pyropoikilocytosis, xerocytosis, stomatocytosis) Hemoglobinopathies Congenital Dyserythropoietic Anemia Sepsis Extravasated blood collection Vitamin E deficiency Infantile pyknocytosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parastatidou, S.; Tsantes, A.G.; Emmanouil, C.-C.; Konstantinidi, A.; Kapetanaki, A.; Sokou, R. Neonatal Hemolytic Jaundice: Causes, Diagnostic Approach, and Management. Children 2025, 12, 666. https://doi.org/10.3390/children12060666
Parastatidou S, Tsantes AG, Emmanouil C-C, Konstantinidi A, Kapetanaki A, Sokou R. Neonatal Hemolytic Jaundice: Causes, Diagnostic Approach, and Management. Children. 2025; 12(6):666. https://doi.org/10.3390/children12060666
Chicago/Turabian StyleParastatidou, Stavroula, Andreas G. Tsantes, Chrysoula-Christina Emmanouil, Aikaterini Konstantinidi, Anastasia Kapetanaki, and Rozeta Sokou. 2025. "Neonatal Hemolytic Jaundice: Causes, Diagnostic Approach, and Management" Children 12, no. 6: 666. https://doi.org/10.3390/children12060666
APA StyleParastatidou, S., Tsantes, A. G., Emmanouil, C.-C., Konstantinidi, A., Kapetanaki, A., & Sokou, R. (2025). Neonatal Hemolytic Jaundice: Causes, Diagnostic Approach, and Management. Children, 12(6), 666. https://doi.org/10.3390/children12060666