Abstract
Background: The reported outcomes of pediatric dilated cardiomyopathy (DCM) have varied across studies. There are few outcome data concerning DCM in Chinese children. Therefore, we conducted a retrospective study to describe clinical features and determine risk factors for poor outcomes in children with DCM. Methods: We enrolled 121 children with DCM in our hospital from 2003 to 2021. General information and laboratory and echocardiographic data were collected and analyzed. Cox regression analysis was performed to determine risk factors for poor outcomes. Results: This study included 121 patients (69 males and 52 females). The median age at diagnosis was 10.8 years, and the follow-up time was 10.0 months. Eighty-two patients (67.8%) exhibited cardiac function classes III–IV at the time of diagnosis. Tachypnea was the most common symptom (78.5%). In echocardiography, the mean left ventricular end-diastolic dimension z score was 7.36 ± 2.73, and the left ventricular ejection fraction z score was −6.58 ± 2.17. The 1-, 2-, and 5-year survival rates were 51.2%, 43.8%, and 32.2%, respectively. Cox analysis revealed that cardiac function classes III–IV (hazard ratio [HR] = 1.801, 95% confidence interval [95% CI] = 1.030–3.149, p = 0.039) and calcium levels (HR = 0.219, 95% CI = 0.084–0.576, p = 0.002) were predictors of poor outcomes in children with DCM. Conclusions: Children with DCM are at high risk of death. Cardiac function class III–IV and calcium levels were related to the prognosis of pediatric DCM patients.
1. Introduction
Dilated cardiomyopathy (DCM) is a life-threatening disorder characterized by left ventricle dilation and impaired contractility [1]. It is the most common form of pediatric cardiomyopathy [2]. Epidemiologically, in the United States, DCM incidence was 0.57/100,000 per year for children aged 0–18 years [3]. Approximately 64% of patients with DCM died or underwent a cardiac transplant within 1 year of their first admission [4]. Despite substantial advances in treatment, the prognosis for DCM patients, especially children, remains poor [5]. The reported outcomes of DCM in children vary widely depending on the medical condition of patients in the region. One single-center retrospective study from India reported a 5-year survival rate of 59% [6], while another study from the United States estimated a 5-year survival rate of 83% with heart transplantation [7]. According to a population-based study from Australia, the survival rates of children with DCM who died or underwent cardiac transplantation over 1 year and 5 years were 72% and 63%, respectively [8]. However, few data have addressed DCM outcomes in Chinese children. Previous studies have provided great insights into the risk factors for poor outcomes in children with DCM. Age at diagnosis, male sex, familial DCM, heart failure, heart rate, left ventricular fractional shortening (LVFS) z score, left ventricular ejection fraction (LVEF) z score, uric acid, serum cholesterol, atrial filling pressure, and mixed venous saturation are considered predictors of poor outcomes in pediatric patients with DCM [3,4,8,9]. However, the results vary between different studies. A better understanding of the clinical features and risk factors for poor outcomes in children with DCM may help improve individual risk stratification and develop better treatment strategies. Therefore, we conducted this retrospective single-center study in a region tertiary hospital in Southwestern China to describe clinical features and identify risk factors for poor outcomes in children with DCM.
2. Materials and Methods
2.1. Patients and Definitions
This study was performed at the First Affiliated Hospital of Guangxi Medical University. Patients with DCM admitted from March 2003 to September 2021 were consecutively enrolled. DCM was defined as a dilation and systolic dysfunction of the LV. LV dilation was defined as an LV end-diastolic dimension (LVEDD) exceeding two standard deviations (SDs) of the body surface area-adjusted mean value (i.e., z score > 2). LV systolic dysfunction was defined as an LVEF or LVFS lower than 2 SDs for a reference population of the same age (i.e., z score < 2) [1,10]. The inclusion criteria for patients were as follows: (1) met the diagnostic criteria for DCM; (2) aged < 18 years; (3) had complete and traceable clinical data. Patients with DCM caused by arrhythmia, hypertensive heart disease, congenital heart disease, cardiac valve disease, ischemic heart disease, infection, inherited metabolic disease, and drugs were excluded. The cardiac function class was confirmed at the time of presentation by specialist pediatric cardiologists using the New York Heart Association (NYHA) class (age > 1 year) or modified Ross score (age ≤ 1 year) [11,12]. Cardiac function I was defined as NYHA class I or modified Ross scores of 0–2 points, cardiac function II as NYHA class II or modified Ross scores of 3–6 points, cardiac function III as NYHA class III or modified Ross scores of 7–9 points, and cardiac function IV as NYHA class IV or modified Ross scores of 10–12 points. The study protocols were in accordance with the Helsinki Declaration. Written informed consent from parents was obtained. Ethical approval was obtained from the Ethics Committee of the First Affiliated Hospital of Guangxi Medical University (NO. 2022(KY-E-007)).
2.2. Data Collection
General data, including age, sex, race, family history of DCM, body mass index, cardiac function class, heart rate, systolic blood pressure (SBP) and diastolic blood pressure (DBP), main syndromes and signs, symptom duration, duration of first hospitalization, admission to the intensive care unit, and multiple hospitalizations (≥2 visits), were obtained from the medical records. We collected laboratory data, including white blood cells, red cell distribution width, hemoglobin, troponin I, creatine kinase-MB, electrolytes, serum creatinine, blood urea nitrogen, and uric acid levels, within 24 h after the first admission. Each patient underwent echocardiography at admission. We recorded echocardiographic parameters such as LVEDD, left ventricular end-diastolic posterior wall thickness, left ventricular end-systolic dimension (LVESD), LVEF, and LVFS. These echocardiographic measurements were normalized by the z score to adjust for body surface area and age. The treatments administered to patients, including digoxin, β-blockers, angiotensin-converting enzyme inhibitors (ACEIs), and diuretics, were also noted. Follow-up was performed via medical records or telephone contact. The endpoint was all-cause death during follow-up.
2.3. Statistical Analysis
The Shapiro–Wilk test was used for the normality test. Categorical variables were expressed as frequencies (percentages) and intergroup comparisons were performed with chi-square tests or Fisher’s exact test. Quantitative variables are presented as the mean ± SD (normal distribution) or median with an interquartile range (skewed distribution). Student’s t-test or the Mann–Whitney U test was used to compare quantitative data between the groups, as appropriate. A Kaplan–Meier curve was plotted for survival analysis and analyzed by the log-rank test. We performed Cox regression analysis to determine risk factors for poor outcomes in children with DCM and calculate hazard ratios (HRs) and 95% confidence intervals (95% CIs). Factors with a p-value < 0.1 in the univariate Cox regression were included in the multivariate Cox regression model. The cutoff value for calcium concentrations was determined using the receiver operating characteristic (ROC) curve. p < 0.05 was considered to indicate statistical significance. The data were analyzed with SPSS software (version 24.0 for Windows, SPSS, Inc., Chicago, IL, USA).
3. Results
3.1. Baseline Characteristics
This study involved 121 patients, including 69 males and 52 females. The median age at diagnosis was 10.8 (5.3–14.0) years. Thirty-nine patients (32.2%) exhibited cardiac function classes I–II, and eighty-two (67.8%) had cardiac function classes III–IV at the time of diagnosis. Ten (8.3%) patients had a family history of DCM. Tachypnea was the most common symptom detected in 78.5% of patients, followed by fatigue, hepatomegaly, edema, and chest distress. The median symptom duration was 1 (0.5–3.0) month, and the median length of the first hospitalization was 9 (6.0–15.0) days. Fifty-three (43.8%) patients were admitted to the ICU when diagnosed. Fifty-one (42.1%) patients needed multiple hospitalizations due to poor disease control. At the time of diagnosis, 94.2% of the patients (114/121) were prescribed a loop diuretic and digoxin, and 91.7% (111/121) were prescribed spironolactone, with 85.1% (103/121) receiving ACEIs, 50.4% (61/121) using intravenous inotropes, and 30.5% (37/121) receiving β-blockers. The prescription rates of spironolactone increased significantly after 2010 compared to before 2010, while the prescription rates of ACEIs and β-blockers remained stable (Figure 1). Compared with survivors, patients who died from all causes had a higher heart rate and lower SBP and DBP (both p < 0.05). The median follow-up period was 10 (3.0–36.5) months. A summary of the baseline characteristics is shown in Table 1.
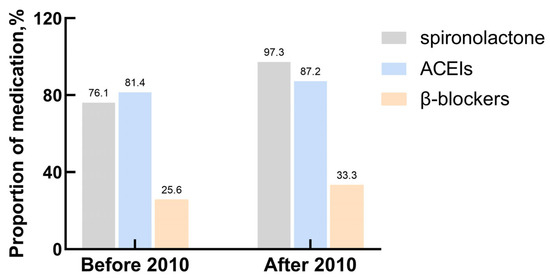
Figure 1.
The explanation of prescription rates of major drugs.

Table 1.
Baseline characteristics of patients with dilated cardiomyopathy.
3.2. Laboratory and Echocardiographic Examination
The results of the laboratory and echocardiographic examinations at the time of diagnosis are shown in Table 2. Echocardiography revealed an enlarged LV dimension, with a mean LVEDD z score of 7.36 ± 2.73 and a mean LVESD z score of 10.42 ± 3.35. In addition, patients in the death group had a greater LVESD z score than the survivors (p = 0.038). The mean LVEF z score was −6.58 ± 2.17, and the median LVFS z score was 9.45 (−11.55 to −6.36), indicating severely reduced systolic function. A lower serum calcium concentration was observed in the death group than in the survivor group (p = 0.002). However, neither group significantly differed in white blood cell count, red cell distribution width, hemoglobin, troponin I, creatine kinase-MB, sodium, potassium, uric acid, blood urea nitrogen, or serum creatinine levels (p > 0.05).
Table 2.
Laboratory and echocardiographic examinations of patients with DCM.
3.3. Survival Analysis
The survival rates at one, two, and five years were 51.2%, 43.8%, and 32.2%, respectively. The highest mortality was observed during the first year after diagnosis, at which time 48.8% of the children reached the endpoint (Figure 2a). Univariate Cox analysis revealed that SBP (HR = 0.968, 95% CI = 0.949–0.987, p = 0.001), DBP (HR = 0.977, 95% CI = 0.959–0.966, p = 0.018), calcium level (HR = 0.222, 95% CI = 0.084–0.574, p = 0.002), and cardiac function classes III–IV (HR = 2.516, 95% CI = 1.488–4.255, p = 0.001) were predictors of death in patients with DCM. According to the multivariate Cox analysis, only cardiac function classes III–IV and calcium levels remained significant (Table 3). Patients with cardiac function classes III–IV at diagnosis were 1.801 times more likely to die than those with cardiac function classes I–II (HR = 1.801, 95% CI = 1.030–3.149; p = 0.039). Increased calcium concentration of 1 mmol/L reduced the risk of death by 78.1% (HR = 0.219, 95% CI = 0.084–0.576, p = 0.002). The area under the curve for calcium concentration was 0.678 to distinguish between patients who died from all causes and survivors (95% CI = 0.581–0.775, p = 0.002; Figure 3). The sensitivity was 0.48, and the specificity was 0.84. The cutoff value was 2.26 mmol/L. Patients were categorized into a calcium concentration cutoff group (≤2.26 mmol/L) and a calcium concentration >2.26 mmol/L group. According to the Kaplan–Meier survival analysis, patients with a calcium concentration ≤2.26 mmol/L had a lower cumulative survival rate (p = 0.001; Figure 2b). Patients with cardiac function classes III–IV had a significantly lower survival rate than those with cardiac function classes I–II (p < 0.001; Figure 2c). Among the 83 patients who died from all causes, the median survival time (defined as the time from diagnosis to death) for those diagnosed before 2010 was 6.0 (2.0–12.5) months, while for those diagnosed after 2010 it was 7.0 (2.0–28.3) months (Figure 4).
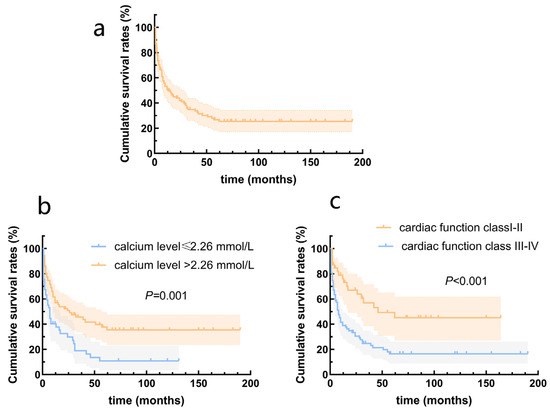
Figure 2.
Survival rates from the Kaplan–Meier estimates in patients with dilated cardiomyopathy. (a) Total patients; (b) patients grouped by serum calcium levels; (c) patients grouped by cardiac function class.
Table 3.
Predictors of death in patients with DCM according to Cox regression analysis.
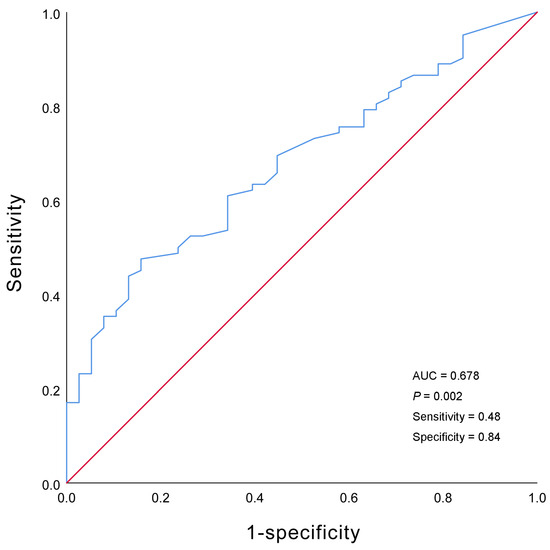
Figure 3.
Receiver operating characteristic curve of the serum calcium concentration.
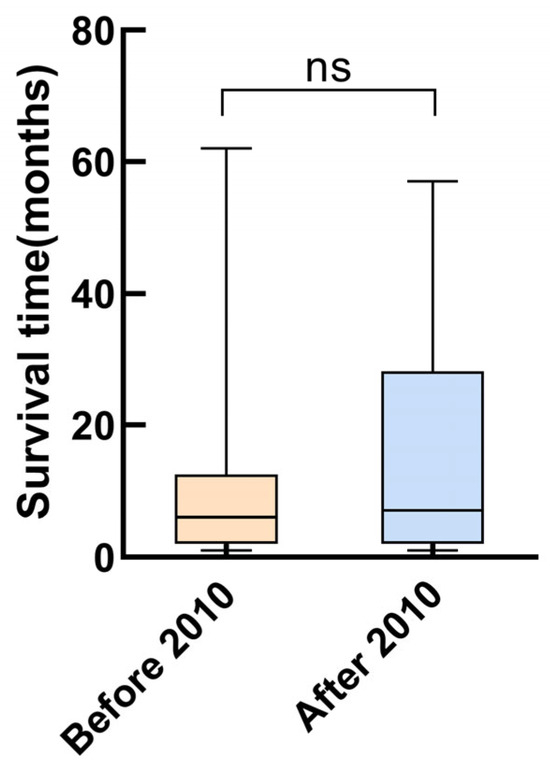
Figure 4.
Survival time of the patients who died from all causes at different recruitment periods.
4. Discussion
We retrospectively analyzed the clinical features and outcomes of DCM patients in a large academic tertiary center in Guangxi, China. The results revealed that most patients were symptomatic at the time of diagnosis and usually severely ill. For instance, 67.8% of the patients presented with cardiac function classes III–IV and the majority needed medication. The mortality rate was relatively high in children with DCM admitted to the hospital. Cardiac function classes III–IV and serum calcium levels were found to be independent predictive factors of death in pediatric DCM patients.
The reported survival rates of DCM patients have varied across studies. One-year survival rates of 63–90% and 5-year survival rates of 34–83% have been described in institutional reviews [3,4,5,7,9,13,14]. In our cohort study, the 1-year survival rate was 51.2%, and the 5-year survival rate was 32.2%, suggesting a greater risk of death than in other studies. One possible explanation for this difference might be that none of the patients in our study underwent cardiac transplantation. Cardiac transplantation is a final therapeutic strategy for DCM patients and can improve patient prognosis. One study reported that 1- and 5-year survival rates were 90% and 83%, respectively, with heart transplants [7]. However, the percentage of patients free from “cardiac death” (death or cardiac transplant) was only 70% at 1 year and 58% at 5 years. Another study reported by Hollander et al. [4] showed that the total survival rate at 1 year was 84.5% in 83 pediatric patients with DCM. However, the transplantation-free survival rate was only 46% at 1 year, which was similar to our total survival rate in the population without cardiac transplants. Another reason for the high mortality could be that most of the patients in this study were severely ill. Our hospital is a tertiary-care medical institution that receives patients with severe conditions from local hospitals. In this study, the majority (67.8%) of patients presented with advanced heart failure at the time of onset. Almost half of our patients (43.8%) had to be admitted to the intensive care unit, indicating a poor prognosis.
The NYHA class is usually used to evaluate the severity of HF. However, this grading system is based on the limitations of physical activity in adults and may not apply to children, especially infants. Therefore, we used a modified Ross score rather than the NYHA class in patients aged ≤1 year. We concluded that cardiac function III–IV at the time of diagnosis increased the risk of all-cause death by 80.1%, which was consistent with previous studies. Castelli et al. [15] reported that a high NYHA class was associated with cardiovascular mortality and heart transplantation. Similarly, Christ et al. [16] reported that a high NYHA class increased the risk of death or heart transplant in patients with idiopathic DCM. Research has also shown that primary outcomes at 1 year are predicted by the NYHA class in both familial and sporadic DCM patients [17]. Heart failure at the time of diagnosis was correlated with a 3.18-fold increased risk of transplantation or death in patients with both familial DCM and idiopathic DCM [18]. These results revealed that patients with a higher cardiac function class are more severely ill and likely to have poor outcomes. A key goal of DCM therapy may be to reduce heart failure.
To the best of our knowledge, this is the first study to show the prognostic value of serum calcium concentration in DCM patients. Calcium is a crucial mediator of cardiac contraction and diastole, playing a key role in regulating excitation–contraction coupling [19]. In recent years, several studies have reported that hypocalcemia is related to poor prognosis in patients with HF. Liu et al. [20] demonstrated that hypocalcemia predicted cardiac readmission and death within 1 year. Additionally, Jensen et al. [21] proposed that an imbalance in calcium homeostasis results in a greater mortality rate in chronic heart failure patients. Furthermore, Miura et al. [22] reported that hypocalcemia was an independent risk factor for death in patients with heart failure and chronic kidney disease. However, the etiologies of heart failure are quite diverse. The primary causes of heart failure were not considered in previous studies. To date, serum calcium concentration has not been reported to predict the prognosis of DCM patients. In this study, we found a negative correlation between high serum calcium levels and poor prognosis in patients with DCM. Specifically, for each unit increase in calcium concentration, the risk of death falls by 78.1%. Patients with calcium levels ≤2.26 mmol/L had worse survival outcomes than those with calcium levels >2.26 mmol/L. Extracellular calcium concentration may affect myocardial contractility by influencing the membrane potential of cardiac cells [23]. Serum calcium influxes into the cytosol when the membrane potential changes, leading to an increased release of calcium from the sarcoplasmic reticulum. In such circumstances, calcium binds to cardiac troponin C and causes the myocardium to contract [24,25]. Therefore, low serum calcium levels may reduce cardiac contractility. Yang et al. [26] reported that cardiac systolic function rapidly improved in a 56-year-old DCM patient with recurrent hypocalcemia after serum calcium level restoration, indicating that hypocalcemia is an important cause of DCM. However, the mechanism responsible for calcium’s impact on DCM has not been elucidated.
Some predictors of poor prognosis for patients with DCM in the literature are age at diagnosis, male sex, the LVFS z score, the LVEF z score, heart rate, and BP at diagnosis [3,8,27,28,29,30]. However, age at diagnosis, sex, the LVFS z score, and the LVEF z score were not related to the risk of death in our study. Consistent with findings in the literature [30,31], univariate Cox regression analysis revealed that SBP and DBP were protective factors against death in patients with DCM. This outcome is likely because patients with low BP are more likely to have lower cardiac output and neurohormonal activation, which may exacerbate their symptoms [32,33]. However, the correlation was not significant according to the multivariate Cox regression model, likely due to the effects of confounding factors.
DCM treatment is aimed at alleviating symptoms, improving quality of life, preventing disease progression, and increasing survival. Most patients in our cohort received medical therapy, which has been shown to decrease mortality and cardiac remodeling. This therapy included ACEIs, β-blockers, and mineralocorticoid receptor antagonists [34,35]. Digoxin, a medicine commonly used for heart failure and DCM, can reduce hospitalization and mortality in patients at high risk of heart failure [36,37]. In the present study, 94.2% of the patients received loop diuretics and digoxin, 91.7% received spironolactone, 85.1% received ACEIs, and 30.5% received β-blockers. These results were comparable with other studies [8,14,29]. In our study, the prescription rates of spironolactone increased significantly after 2010, while the prescription rates of ACEIs and β-blockers remained stable. The survival time in this study showed no evident temporal trend toward improvement. No significant associations were observed between the use of medical drugs and patients’ prognosis. Although medical therapy has been reported to be beneficial for children with DCM in the past, we could not conclude from the current study that medical therapy modified the outcome of children with DCM. The children included in this study were quite ill and at high risk of death, and may have benefited from effective early treatment.
Limitations
This study has several limitations. First, selection bias was inevitable due to the retrospective nature of the study. Second, this was a single-center study with a relatively small sample size. Third, genetic tests were not routinely available in our study. Due to the limited availability of gene data, gene analysis was not performed in this study. Finally, DCM etiologies were heterogeneous in our study. However, too few patients were included in the subgroup analysis.
5. Conclusions
In summary, the findings of this study demonstrated that children with DCM are at high risk of death, with the highest risk occurring during the first year after diagnosis. Survival was lower in patients with cardiac function III–IV and low serum calcium levels at the time of diagnosis. Efficient risk stratification, early individualized therapy, and close outpatient observation may improve these patients’ outcomes.
Author Contributions
C.C. and Y.H. (Yanyun Huang) contributed to the study design and drafted the article. D.S., S.Q. and Y.H. (Yuqin Huang) contributed to the statistical analysis. B.Y. and D.L. collected the data. Y.P. contributed to the research and edited the manuscript. All the authors took part in drafting the article and approved the submitted version. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by a grant from the Scientific Research Project of Guangxi Health Planning Commission (No. Z20210993) and Guangxi Clinical Research Center for Pediatric Disease (No. AD22035219).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the research ethics committee of the First Affiliated Hospital of Guangxi Medical University (approval number: 2022(KY-E-007) and date of approval 7 January 2022.
Informed Consent Statement
Informed consent was obtained from the parents or guardians of all study participants.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy reasons.
Acknowledgments
We are grateful for the help of all the working partners of the First Ward of Pediatrics in our hospital and http://zscore.chboston.org (accessed on 8 October 2021) for z-score calculations during the preparation of this manuscript. We would also like to thank the Key Laboratory of Children’s Disease Research in Guangxi’s Colleges and Universities, Education Department of Guangxi Zhuang Autonomous Region.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; Austin, E.D.; Dipchand, A.I.; Everitt, M.D.; Hsu, D.T.; Lin, K.Y.; Price, J.F.; Wilkinson, J.D.; et al. Cardiomyopathy in children: Classification and diagnosis: A scientific statement from the american heart association. Circulation 2019, 140, e9–e68. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, H.P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Primers 2019, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Lowe, A.M.; Colan, S.D.; Sleeper, L.A.; Orav, E.J.; Clunie, S.; Messere, J.; Cox, G.F.; Lurie, P.R.; Hsu, D.; et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006, 296, 1867–1876. [Google Scholar] [CrossRef]
- Hollander, S.A.; Bernstein, D.; Yeh, J.; Dao, D.; Sun, H.Y.; Rosenthal, D. Outcomes of children following a first hospitalization for dilated cardiomyopathy. Circ. Heart Fail. 2012, 5, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Puggia, I.; Merlo, M.; Barbati, G.; Rowland, T.J.; Stolfo, D.; Gigli, M.; Ramani, F.; Di Lenarda, A.; Mestroni, L.; Sinagra, G. Natural history of dilated cardiomyopathy in children. J. Am. Heart Assoc. 2016, 5, e003450. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, G.; Sasidharan, B.; Krishnamoorthy, K.M.; Kurup, H.K.N.; Gopalakrishnan, A.; Sasikumar, D.; Sarma, S.; Valaparambil, A.K.; Sivasubramonian, S. Clinical profile and outcomes of childhood dilated cardiomyopathy-A single-center three-decade experience. Ann. Pediatr. Cardiol. 2023, 16, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Tsirka, A.E.; Trinkaus, K.; Chen, S.C.; Lipshultz, S.E.; Towbin, J.A.; Colan, S.D.; Exil, V.; Strauss, A.W.; Canter, C.E. Improved outcomes of pediatric dilated cardiomyopathy with utilization of heart transplantation. J. Am. Coll. Cardiol. 2004, 44, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Daubeney, P.E.; Nugent, A.W.; Chondros, P.; Carlin, J.B.; Colan, S.D.; Cheung, M.; Davis, A.M.; Chow, C.W.; Weintraub, R.G. Clinical features and outcomes of childhood dilated cardiomyopathy: Results from a national population-based study. Circulation 2006, 114, 2671–2678. [Google Scholar] [CrossRef]
- Addin, M.B.J.; Young, D.; McCarrison, S.; Hunter, L. Dilated cardiomyopathy in a national paediatric population. Eur. J. Pediatr. 2019, 178, 1229–1235. [Google Scholar] [CrossRef]
- Singh, R.K.; Canter, C.E.; Shi, L.; Colan, S.D.; Dodd, D.A.; Everitt, M.D.; Hsu, D.T.; Jefferies, J.L.; Kantor, P.F.; Pahl, E.; et al. Survival without cardiac transplantation among children with dilated cardiomyopathy. J. Am. Coll. Cardiol. 2017, 70, 2663–2673. [Google Scholar] [CrossRef]
- Ross, R.D.; Bollinger, R.O.; Pinsky, W.W. Grading the severity of congestive heart failure in infants. Pediatr. Cardiol. 1992, 13, 72–75. [Google Scholar] [CrossRef]
- Reithmann, C.; Reber, D.; Kozlik-Feldmann, R.; Netz, H.; Pilz, G.; Welz, A.; Werdan, K. A post-receptor defect of adenylyl cyclase in severely failing myocardium from children with congenital heart disease. Eur. J. Pharmacol. 1997, 330, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Taliercio, C.P.; Seward, J.B.; Driscoll, D.J.; Fisher, L.D.; Gersh, B.J.; Tajik, A.J. Idiopathic dilated cardiomyopathy in the young: Clinical profile and natural history. J. Am. Coll. Cardiol. 1985, 6, 1126–1131. [Google Scholar] [CrossRef]
- Mori, H.; Yoshikawa, T.; Kimura, H.; Ono, H.; Kato, H.; Ono, Y.; Nii, M.; Shindo, T.; Inuzuka, R.; Horigome, H.; et al. Outcomes of dilated cardiomyopathy in japanese children-a retrospective cohort study. Circ. J. 2021, 86, 109–115. [Google Scholar] [CrossRef]
- Castelli, G.; Fornaro, A.; Ciaccheri, M.; Dolara, A.; Troiani, V.; Tomberli, B.; Olivotto, I.; Gensini, G.F. Improving survival rates of patients with idiopathic dilated cardiomyopathy in Tuscany over 3 decades: Impact of evidence-based management. Circ. Heart Fail. 2013, 6, 913–921. [Google Scholar] [CrossRef]
- Christ, M.; Klima, T.; Grimm, W.; Mueller, H.H.; Maisch, B. Prognostic significance of serum cholesterol levels in patients with idiopathic dilated cardiomyopathy. Eur. Heart J. 2006, 27, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Asselbergs, F.W.; Sammani, A.; Elliott, P.; Gimeno, J.R.; Tavazzi, L.; Tendera, M.; Kaski, J.P.; Maggioni, A.P.; Rubis, P.P.; Jurcut, R.; et al. Differences between familial and sporadic dilated cardiomyopathy: ESC EORP Cardiomyopathy & Myocarditis registry. ESC Heart Fail. 2021, 8, 95–105. [Google Scholar] [CrossRef]
- Rusconi, P.; Wilkinson, J.D.; Sleeper, L.A.; Lu, M.; Cox, G.F.; Towbin, J.A.; Colan, S.D.; Webber, S.A.; Canter, C.E.; Ware, S.M.; et al. Differences in presentation and outcomes between children with familial dilated cardiomyopathy and children with idiopathic dilated cardiomyopathy: A report from the pediatric cardiomyopathy registry study group. Circ. Heart Fail. 2017, 10, e002637. [Google Scholar] [CrossRef] [PubMed]
- Zaffran, S.; Kraoua, L.; Jaouadi, H. Calcium handling in inherited cardiac diseases: A focus on catecholaminergic polymorphic ventricular tachycardia and hypertrophic cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 3365. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, H.; Li, Y.; Lu, X. Hypocalcaemia predicts 12-month re-hospitalization in heart failure. Eur. J. Clin. Investig. 2020, 50, e13261. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.C.; Polcwiartek, C.; Søgaard, P.; Mortensen, R.N.; Davidsen, L.; Aldahl, M.; Eriksen, M.A.; Kragholm, K.; Torp-Pedersen, C.; Hansen, S.M. The association between serum calcium levels and short-term mortality in patients with chronic heart failure. Am. J. Med. 2019, 132, 200–208.e1. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Yoshihisa, A.; Takiguchi, M.; Shimizu, T.; Nakamura, Y.; Yamauchi, H.; Iwaya, S.; Owada, T.; Miyata, M.; Abe, S.; et al. Association of hypocalcemia with mortality in hospitalized patients with heart failure and chronic kidney disease. J. Card. Fail. 2015, 21, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zheng, Q.; Zhou, J.; Zhang, Q.; Gao, X.; Liu, Y.; Li, S.; Shan, W.; Liu, L.; Guo, N.; et al. Associations between serum electrolyte and short-term outcomes in patients with acute decompensated heart failure. Ann. Med. 2023, 55, 155–167. [Google Scholar] [CrossRef]
- Keefe, J.A.; Moore, O.M.; Ho, K.S.; Wehrens, X.H.T. Role of Ca(2+) in healthy and pathologic cardiac function: From normal excitation-contraction coupling to mutations that cause inherited arrhythmia. Arch. Toxicol. 2023, 97, 73–92. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and excitation-contraction coupling in the heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Shen, F.R.; Lu, Y.Q. Hypocalcemia: A reversible cause of T wave alternans and heart failure. J. Zhejiang Univ. Sci. B 2014, 15, 598–600. [Google Scholar] [CrossRef]
- Cannatà, A.; Fabris, E.; Merlo, M.; Artico, J.; Gentile, P.; Loco, C.P.; Ballaben, A.; Ramani, F.; Barbati, G.; Sinagra, G. Sex differences in the long-term prognosis of dilated cardiomyopathy. Can. J. Cardiol. 2020, 36, 37–44. [Google Scholar] [CrossRef]
- Rossano, J.W.; Kantor, P.F.; Shaddy, R.E.; Shi, L.; Wilkinson, J.D.; Jefferies, J.L.; Czachor, J.D.; Razoky, H.; Wirtz, H.S.; Depre, C.; et al. Elevated heart rate and survival in children with dilated cardiomyopathy: A multicenter study from the pediatric cardiomyopathy registry. J. Am. Heart Assoc. 2020, 9, e015916. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, B.; Fan, Y.; Yi, Y.; Lv, J.; Wang, J.; Yang, X.; Jiang, D.; Zhao, L.; Zhang, J.; et al. Clinical profile and risk factors for cardiac death in pediatric patients with primary dilated cardiomyopathy at a tertiary medical center in China. Front. Pediatr. 2022, 10, 833434. [Google Scholar] [CrossRef]
- Li, X.; Luo, R.; Jiang, R.; Kong, H.; Tang, Y.; Shu, Y.; Hua, W. The prognostic use of serum concentrations of cardiac troponin-I, CK-MB and myoglobin in patients with idiopathic dilated cardiomyopathy. Heart Lung 2014, 43, 219–224. [Google Scholar] [CrossRef]
- Wang, H.Y.; Huang, Y.; Chen, X.Z.; Zhang, Z.L.; Gui, C. Prognostic potential of liver injury in patients with dilated cardiomyopathy: A retrospective study. Eur. J. Med. Res. 2022, 27, 237. [Google Scholar] [CrossRef] [PubMed]
- Tsimploulis, A.; Lam, P.H.; Arundel, C.; Singh, S.N.; Morgan, C.J.; Faselis, C.; Deedwania, P.; Butler, J.; Aronow, W.S.; Yancy, C.W.; et al. Systolic blood pressure and outcomes in patients with heart failure with preserved ejection fraction. JAMA Cardiol. 2018, 3, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Arundel, C.; Lam, P.H.; Gill, G.S.; Patel, S.; Panjrath, G.; Faselis, C.; White, M.; Morgan, C.J.; Allman, R.M.; Aronow, W.S.; et al. Systolic blood pressure and outcomes in patients with heart failure with reduced ejection fraction. J. Am. Coll. Cardiol. 2019, 73, 3054–3063. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; Lakdawala, N.K.; Tschöpe, C.; Klingel, K. Dilated cardiomyopathy: Causes, mechanisms, and current and future treatment approaches. Lancet 2023, 402, 998–1011. [Google Scholar] [CrossRef] [PubMed]
- Rupp, S.; Jux, C. Advances in heart failure therapy in pediatric patients with dilated cardiomyopathy. Heart Fail. Rev. 2018, 23, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Rahim, A.H.; Shen, L.; Rush, C.J.; Jhund, P.S.; Lees, K.R.; McMurray, J.J.V. Effect of digoxin in patients with heart failure and mid-range (borderline) left ventricular ejection fraction. Eur. J. Heart Fail. 2018, 20, 1139–1145. [Google Scholar] [CrossRef]
- Gheorghiade, M.; Patel, K.; Filippatos, G.; Anker, S.D.; Van Veldhuisen, D.J.; Cleland, J.G.; Metra, M.; Aban, I.B.; Greene, S.J.; Adams, K.F.; et al. Effect of oral digoxin in high-risk heart failure patients: A pre-specified subgroup analysis of the DIG trial. Eur. J. Heart Fail. 2013, 15, 551–559. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).