Abstract
Malignant melanoma represents the most fatal skin cancer due to its aggressive biological behavior and high metastatic potential. Treatment strategies for advanced disease have dramatically changed over the last years due to the introduction of BRAF/MEK inhibitors and immunotherapy. However, many patients either display primary (i.e., innate) or eventually develop secondary (i.e., acquired) resistance to systemic treatments. Treatment resistance depends on multiple mechanisms driven by a set of rewiring processes, which involve cancer metabolism, epigenetic, gene expression, and interactions within the tumor microenvironment. Prognostic and predictive biomarkers are needed to guide patients’ selection and treatment decisions. Indeed, there are no recognized clinical or biological characteristics that identify which patients will benefit more from available treatments, but several biomarkers have been studied with promising preliminary results. In this review, we will summarize novel tumor metabolic pathways and tumor-host metabolic crosstalk mechanisms leading to melanoma progression and drug resistance, with an overview on their translational potential as novel therapeutic targets.
1. Introduction
Cutaneous melanoma represents 5.6% of all new cancer cases in the United States (US) [1]. According to the Surveillance, Epidemiology, and End Results (SEER) database, the 5-year relative survival rate for patients with cutaneous melanoma (all stages) exceeds 93%, ranging from 99% for localized and early-stage disease to less than 30% for metastatic disease [1]. Over the last years, the prognosis of patients with advanced/metastatic melanoma has significantly improved with the introduction of novel therapeutic strategies. These include drugs targeting the mitogen activated protein-kinase (MAPK) pathway, namely, BRAF and MEK inhibitors, in patients with BRAF mutant melanoma (i.e., approximately 50% of patients with cutaneous melanoma) [2,3]. To date, three different targeted therapies against BRAF and MEK have been approved for the treatment of advanced unresectable/metastatic melanoma, namely, vemurafenib plus cobimetinib, dabrafenib plus trametinib, and encorafenib plus binimetinib [2,3,4]. The combination of dabrafenib and trametinib has recently demonstrated to provide sustained relapse-free survival improvement as an adjuvant treatment for resected stage III melanoma, thus, being approved in this therapeutic setting [5].
Another therapeutic strategy consists of the use of monoclonal antibodies targeting immune checkpoint molecules, such as the anti-cytotoxic T-lymphocyte antigen 4 (CTLA-4) antibody, ipilimumab, and the anti-programmed cell death 1 (PD-1) antibodies, nivolumab and pembrolizumab [6,7,8,9]. Given the survival advantages with a more favorable toxicity profile, anti-PD1 treatment has become the standard first line immunotherapy strategy, while anti-CTLA-4 is commonly used in further lines of treatment in case of disease progression on previous anti-PD1 therapy. The combination of ipilimumab plus nivolumab has demonstrated to provide further improvements in survival outcomes, also in challenging subpopulations of patients (e.g., patients with brain metastases), however, with a higher rate of immune-related adverse events [8]. Anti-PD1 treatment has recently become standard of care in the adjuvant setting for patients with resected stage III (pembrolizumab or nivolumab) or stage IV melanoma with no evidence of disease (nivolumab) [10,11].
Despite the undoubted therapeutic advances, the majority of patients with metastatic melanoma will still die from their disease, either because of primary (i.e., innate) or secondary (i.e., acquired/adaptive) resistance mechanisms (Figure 1) [12,13,14]. Several different predictive and prognostic biomarkers have been investigated to improve patients’ selection and risk stratification, however, with disappointing and inconsistent results [15,16,17,18]. Intense research is, therefore, required to understand the biologic and molecular features of aggressive melanoma and the mechanisms underlying treatment resistance [13] to increase therapeutic possibilities.
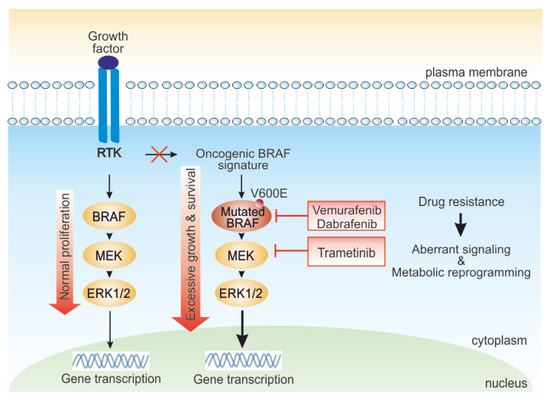
Figure 1.
Role of the mitogen activated protein kinase (MAPK) pathway in melanoma cells and targets of BRAF and MEK inhibitors. In normal cells, external growth stimuli trigger receptor tyrosine kinase (RTK), activating the MAPK pathway kinase cascade. In BRAF-driven melanoma, mutant BRAF (BRAF V600E) can start signaling independently of growth factor signal to hyperactivate cellular growth. BRAF mutated melanoma responds to BRAF/MEK inhibitors-targeted therapy. However, various intrinsic or adaptive resistance mechanisms attenuate response to targeted BRAF inactivation, deregulating signaling and rewiring cell metabolism.
Metabolic rewiring (i.e., glycolysis and/or oxidative phosphorylation OXPHOS pathways) in melanoma, and the metabolic crosstalk between melanoma cells and the components of the tumor microenvironment (TME), represent an intriguing field of research due to the role of metabolic process in supporting tumor survival and progression but also drug resistance [19,20,21,22,23] (Figure 1).
In this review, we explore the symbiotic relationship between melanoma and immune/stromal cells, focusing on metabolic plasticity of melanoma and immune cells within the TME and metabolic soluble factors that promote melanoma aggressiveness and immune evasion. We also discuss the ongoing clinical trials and future potential combination strategies exploiting metabolic players in cutaneous melanoma to further improve therapeutic outcomes.
2. Tumor Metabolic Plasticity Driving Melanoma Progression and Resistance
Metabolic reprogramming has been widely accepted as one of the hallmarks of cancer [19,24]. Tumor cells must adapt their metabolic needs to support sustained proliferation, growth, and metastatic potential [25]. To achieve this, cancer cells usually switch from a mitochondrial oxidative metabolism to a glycolysis-based metabolism, a process known as the “Warburg effect”. In 1920, Otto Warburg first defined this metabolic feature of tumors, which showed high rates of glucose uptake and lactate secretion, even in the presence of oxygen (i.e., aerobic glycolysis) [23,26,27]. This paradigm has slightly changed since it is now recognized that several tumors use OXPHOS, rather than glycolysis, as the preferential method for energy production [28,29]. Several intrinsic factors in tumor cells, mainly driven by oncogenes activation and tumor suppressor genes inactivation, together with the hypoxic condition and nutrients competition within the TME, drive the acquisition of such metabolic pathways (Figure 2) [24,30]. The BRAF oncogene has emerged as a critical regulator of these processes in melanoma cells, underlying the importance of metabolic rewiring in the pathogenesis and treatment of metastatic melanoma. In the early phase of its development, melanoma is characterized by a glycolytic metabolism [31,32,33]. BRAF mutations lead to MAPK pathway hyperactivation and subsequent stimulation of transcription factors such as MYC and hypoxia inducible factor-1α (HIF-1α), which are key regulators of glycolysis, inducing transcription of several genes involved in glucose metabolism (i.e., glucose transporter 1 (GLUT1), hexokinase (HK2), and lactate dehydrogenase (LDH)) [31,34]. In parallel, BRAF mutations actively inhibit OXPHOS, repressing expression of microphthalmia-associated transcription factor (MITF) and its target peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α), a key regulator of mitochondrial functions [35,36,37]. Treatment with BRAF and MEK inhibitors rapidly and significantly reduces the addiction of melanoma cells to glycolytic processes [31,38]. However, increasing evidence has demonstrated that melanoma is a highly heterogeneous tumor, and subsets of melanoma possess an oxidative metabolism, correlating with poorer survival, progression, and metastasis [21,22,37,39]. Higher OXPHOS is driven by elevated expression of PGC-1α. These tumors show an improved tolerance to the detrimental effects of reactive oxygen species (ROS), indicating their increased ability to survive under conditions of oxidative stress [39]. It is, therefore, clear that both glycolysis and OXPHOS play a significant role in metabolic reprogramming of melanoma cells, and that there is a dynamic switch and plasticity between these two metabolic phenotypes [21,28,40]. In addition, some melanomas show a “hybrid” glycolysis/OXPHOS metabolic phenotype, meaning that tumor cells have the flexibility to use different energy sources and nutrients to adapt their growth according to different TME conditions [33,39,41].
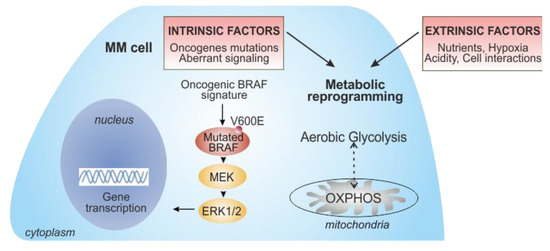
Figure 2.
Key intrinsic and extrinsic factors contributing to metabolic reprogramming in metastatic melanoma (MM) cells. The oncogenic BRAF mutated molecular pathway, leading to the overactivation of MAPK, drives metabolic reprogramming in melanoma cells, promoting glucose metabolism (intrinsic factors). However, some melanomas rely on oxidative phosphorylation (OXPHOS), suggesting a metabolic plasticity that supports melanoma progression and resistance to drugs. During the onset of BRAF resistance, prolonged inhibition of BRAF/MEK decreases glycolysis, leading to a dependence on mitochondrial metabolism. Metabolic rewiring of MM cells is also regulated by several extrinsic factors, such as the availability of nutrients, hypoxic conditions, and acidity of the TME, as well as the interplay with stromal/immune cells within the TME. The progressive metabolic reprogramming in melanoma is accompanied by a drastic increase in tumor aggressiveness.
Metabolic plasticity is essential during the onset of resistance to BRAF inhibitors. In resistant patients there is a switch from glycolysis to a mitochondrial metabolism [34,42,43]. One hypothesis to explain this phenomenon is that this metabolic oxidative profile pre-exists in some cell clones, and that treatment leads to the selection of these cell clones. Another possibility is that tumor cells acquire resistance as a consequence of treatment, following drug exposure. The main molecular player of this metabolic phenotype is the reactivation of the MITF/PCG-1α axis, driving increased mitochondrial content, mitochondrial activity, and mitochondrial oxidative capacity [37,39]. This adaptive metabolic program limits the efficacy of BRAF inhibitors and is responsible for adaptive resistance [37,39,43,44,45]. Overall, it is clear that metabolic plasticity confers to melanoma cells a significant advantage to adapt their growth to different environmental conditions and to increase their survival, even under drug-imposed selective pressures.
3. Metabolic Exchanges within the TME: Soluble Factors
Metabolic conditions in the TME are influenced by many factors, including oxygen levels, gradients of nutrients, soluble molecules, tissue vascularization, interactions between tumor and stromal/immune cells, and systemic metabolism (Figure 2). Recent detailed reviews summarized this complex network of metabolic crosstalk within the TME [46,47,48,49,50]. Here, we discuss the main players of this complex network, focusing on melanoma–TME metabolic crosstalk.
3.1. Lactate Shuttling in Cancer Cell Metabolism
The concept describing lactate as solely a hypoxic waste product has changed in the last decades. It is clear that lactate is both a potent fuel and a critical signaling molecule, and it is constantly being produced and circulated throughout the body, even in the presence of adequate O2 levels [51]. For these reasons, it is widely accepted that lactate is a key intermediate metabolite in cellular metabolism. The “lactate shuttle hypothesis”, originally introduced by George Brooks in 1986 [52], describes how in normal physiology, as well as in pathophysiology, lactate shuttling between and among cells satisfies at least three purposes for lactate: (i) a major energy source, (ii) the major gluconeogenic precursor in Cori cycle, and (iii) a signaling molecule with autocrine, paracrine, and endocrine-like properties [51]. Exchanges between lactate “producer” and “consumer” exist within cell “intracellular lactate shuttle” and among cells, tissues, and organs “cell–cell lactate shuttle” [53]. Lactate can be exported or up-taken across biological membranes through monocarboxylate transporters (MCTs). Importantly, MCTs are bidirectional, allowing for tissues to switch between lactate release and uptake depending on changes in concentration and pH. Usually, MCT1 is typically expressed in cells importing lactate, while MCT4 is expressed in cells exporting lactate [54].
Tumor mass, as previously discussed, is characterized by a metabolic plasticity and heterogeneity among tumor cells; moreover, different tumor types rely on multiple metabolic pathways. Lactate is an essential metabolite present in the TME and can be shuttled to and from cancer cells and is highly correlated with cancer aggressiveness and poor survival [55,56,57]. The traditional “Warburg Effect” describes glycolytic tumors relying highly on glucose uptake (via GLUTs transporters) with subsequent lactate exportation (via MCT4) in normoxia. In this context, other cancer cells within the tumor mass and/or tumor stromal cells can then take up lactate (via MCT1) to fuel their oxidative metabolism and activate signaling [57,58]. Increasing knowledge on cancer metabolism supports the idea that lactate production, “lactagenesis”, is the purpose of the Warburg Effect. As discussed by San-Millan and Brooks in 2017, lactagenesis is a highly orchestrated effort from oncogenes and tumor suppressor mutations for continuous glucose utilization to produce lactate, involving five major steps: (i) increased glucose uptake through upregulation of GLUT transporter expression; (ii) upregulation of glycolytic enzyme expression; (iii) decreased mitochondrial respiration; (iv) increased lactate production, accumulation, and release; and (v) upregulation of MCT expression for further lactate shuttling and promotion of carcinogenesis [57]. Secreted lactate is necessary for supporting angiogenesis, immune escape, cell migration, metastasis, and metabolic self-sufficiency of cancer cells [57,59]. On the contrary, in the “Reverse Warburg Effect” theory, lactate is produced by glycolytic stromal cells and utilized by cancer cells, via the tricarboxylic acid (TCA) cycle and OXPHOS, as a major source of energy. Overall, lactate plays a critical role in several aspects of tumor biology, including in melanoma, as reviewed in the next sections.
3.2. The Role of Lactate: TME Acidification
The acidification of microenvironment is a hallmark of melanoma altering metabolic adaptation, proliferation, survival, migration, and invasion [44]. The upregulation of glycolysis in melanoma leads to protons and LDH-dependent lactate generation [60]. Protons, as lactate, are transported out of cancer cells through MCT4 [54,60]. Hypoxia, resulting also both both tumor and endothelial cells’ high oxygen consumption, sustains acidosis through the upregulation of the glycolytic pathway, mostly linked to the stabilization of HIF-1α [60,61]. Over-expression of GLUT1 and MCT4 are significantly correlated with progression from primary tumor to lymph node metastasis in a cohort of patient-derived melanoma samples, suggesting that the Warburg phenotype, lactate and protons secretion, drastically alters the melanoma microenvironment, facilitating angiogenesis, promoting melanoma metastasis, and suppressing the immune system [62,63].
In order to promote melanoma invasiveness and metastasization, lactate can contribute to tumor escape from immune responses by altering cytotoxic T lymphocytes (CTLs) metabolism and function [64,65], as summarized in Figure 3. High levels of lactate are associated with a significant decrease of CD8+ T and natural killer (NK) cell number and activity, both in vitro and in vivo [66]. Furthermore, lactate can also induce macrophage phenotype plasticity, promoting pro-tumoral M2-like features [62,67]. In addition, it prevents the maturation of dendritic cells (DCs), resulting in increased immunosuppressive IL-10 cytokine levels in the TME [62].
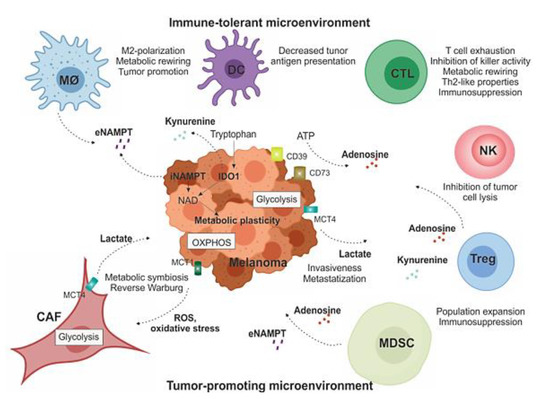
Figure 3.
Soluble factors and immunometabolic interplay within the TME. Melanoma is usually surrounded by a wide array of stromal cells (CAFs) and infiltrating immune cells of both innate and acquired immunity, such as MDSCs, MO-TAMs, DCs, NK cells, and T lymphocytes. They form complex interactions and exchange of soluble factors with melanoma cells that modulate metabolic plasticity of cellular components and support tumor growth by creating a tolerogenic environment that enables cancers to evade immune surveillance and destruction, as detailed in the text. MO-TAM: tumor-associated macrophages; DC: dentric cell; CTL: cytotoxic T lymphocytes; NK: natural killer cell; Treg: T-regulatory lymphocyte; MDSC: myeloid-derived suppressive cells; CAF: cancer-associated fibroblasts.
Melanoma microenvironment acidification hampers immunotherapy response [44]. LDH serum level is a well-known prognostic factor in melanoma, and it also affects response, progression-free survival (PFS), and overall survival (OS) of melanoma patients treated with immune-checkpoint inhibitors [68]. Therefore, the efficacy of immunotherapy could be improved by counteracting microenvironment acidification and lactate extracellular accumulation, as suggested by recent studies [68,69,70].
3.3. The Reverse Warburg Effect: Melanoma Cells and Fibroblasts Crosstalk
Metabolic reprogramming involves not only cancer cells but also cellular components of the TME, namely, cancer-associated fibroblasts (CAFs), the most abundant pro-tumoral population within tumor stroma [41,46,71]. The progressive acidification of TME and hypoxic conditions modify the metabolic interactions between cancer cells and stroma. CAFs are reprogrammed toward a glycolytic phenotype upon interaction with cancer cells, an effect called “Reverse Warburg”, Figure 3 [72,73,74]. ROS produced by cancer cells further stimulate CAFs glucose upload and lactate secretion via MCT4 [75,76]. In turn, lactate and metabolites secreted by CAFs can be taken up by tumor cells via MCT1 to feed into the TCA cycle for OXPHOS-mediated energy production [77]. This Reverse Warburg effect was initially reported in a variety of cancers, including prostate and breast [46,74], and has been confirmed also in melanoma [21,39,78]. Increasing evidence has highlighted the contribution of CAFs to disease progression, metastasis, and drug resistance in melanoma, via both direct cell–cell interaction and chemical interplay (CAF’s secretome) to distant cells [79,80,81]. CAFs-tumor crosstalk is mediated by secretion of extracellular vesicles (EVs), such as exosomes, important factors in pre-metastatic niche formation [82]. The composition of exosomes is complex and not fully elucidated; however, it includes proteins, lipids, metabolites, nucleic acids, and microRNA, which can be trafficked in circulation and internalized by recipient cells to exert their effect [46,83]. Understanding the metabolic interplay between CAFs and other stromal cells with melanoma can determine drivers of cancer progression and potentially lead to the discovery of prognostic and predictive cancer biomarkers and novel anti-cancer therapies. Moreover, these findings highlight that melanoma cells can shift from glycolysis to OXPHOS and vice versa, depending on the TME conditions and interaction with microenvironmental populations, and prompt further research to develop more effective glycolysis and/or OXPHOS inhibitors that can be associated with conventional therapies.
4. Immunometabolic Interplay within the TME
Immune cells, together with stromal cells, are critical components of the TME. There is growing interest in studying the dynamic metabolic interactions occurring between melanoma cells and the immune cells, as summarized in Figure 3, to discover potential novel targets to combine with immunotherapy. Additionally, immune cells undergo different metabolic reprogramming mechanisms and adapt their metabolic pathways to external conditions and upon interaction with cancer cells [49,84,85]. For example, M1 anti-inflammatory macrophages exhibit a glycolytic phenotype, while tumor associated macrophages (TAMs) or M2-like macrophages cells utilize OXPHOS as their main source of adenosine triphosphate (ATP), and neutrophils prevalently use glycolysis [84,85]; on the contrary, DCs extensively rely on OXPHOS to produce ATP, to switch toward a glucose metabolism upon activation [62]. T cells possess metabolic plasticity depending on their activation state and the subclass of T cells to which they belong: activated effector T cells adopt a glycolytic phenotype [77], T regulatory cells (Tregs) are highly oxidative cells, while regulatory T helper-17 (Th17) cells depend on glycolysis [84,85,86].
The crosstalk between these immune cell populations and cancer cells can impact and deregulate metabolic pathways affecting immune responses and creating an immunosuppressive TME. In addition, the availability of nutrients within the TME and the presence of soluble metabolites and enzymes secreted by both tumor and immune cells in the extracellular environment can alter the phenotype and the functionality of immune cells.
4.1. Nutrient Availability and Metabolic Competition between Tumor and Immune Cells
The nutrient competition among cells within the TME can influence tumor cell growth, survival, and aggressive features. At the same time, the abundance or the deprivation of glucose, lactate, glutamine, amino acids, fatty acids, and other metabolites, as well as growth factors, significantly affect immune cell functions, leading to cancer progression [87]. The most evident and studied effect of nutrient competition within the TME is on T cell functions [88]. Cancer cells can evade the immune system by triggering T cell dysfunction, a condition called exhaustion, or by activating immune checkpoints that inhibit T cell function. Tumors can dampen T cell function by competing for glucose [89,90]. Different papers have revealed that aerobic glycolysis in tumors results in a glucose-poor microenvironment causing T cell exhaustion, demonstrating that metabolic competition, as a distinct mechanism, can lead to T cell hyporesponsiveness. In 2015, Chang et al. demonstrated that glucose consumption by glycolytic tumors can metabolically restrict T cells, which completely depend on aerobic glycolysis to exert effector activities [91,92], directly dampening their effector function and allowing tumor progression [93]. Moreover, tumor-derived lactate can also suppress T cell function by blocking lactate export [64], which disrupts their ability to maintain aerobic glycolysis. As a mechanism of adaptation in a glucose-poor microenvironment, T cells preferentially differentiate into Treg lymphocytes, probably because their oxidative phenotype is metabolically suited to survive in this environment [94].
It has been described that the competition for glucose can affect tumor infiltrating lymphocytes (TILs) activity in melanoma. Glucose deprivation increases secretion of transforming growth factor-β (TGF-β), decreasing concentration of interferon-γ (IFN-γ), which is essential for cytotoxic activity of T cells and to inhibit growth of murine B16 melanomas [95,96]. Interestingly, IFN-γ translation can be inhibited by the glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH) when it is not engaged in glycolysis, highlighting the importance of glucose metabolism for maintenance of T cell function [93]. Recently, a connection between the expression of PD-L1 and PD-1 and glucose metabolism has been evidenced in melanoma and in other solid tumors. Immune-checkpoint molecule expression not only suppresses T cell function but also enhances aerobic glycolysis in cancer cells, further limiting the availability of glucose for T cells, increasing their dysfunctions [97]. Increasing data suggest that checkpoint blockade antibodies, affecting glucose metabolism, might be more effective against tumors with higher glycolytic flux [88,92]. For these reasons, the glycolytic rate of a tumor could be used as a prognostic/predictive tool to determine the efficacy of these treatments. Therefore, glucose metabolism through the glycolytic pathway is central in shaping T cell responses and is emerging as an ideal target to improve the efficacy of cancer immunotherapy [92]. In addition, it has been shown that enhancing fatty acid (FA) catabolism in conditions of low O2 and glucose improves TILs’ ability to kill cancer cells and that the use of peroxisome proliferator-activated receptor (PPAR)-alpha agonist enhances the therapeutic effect of PD-1 blockade in melanoma [98].
Similar to glucose metabolism, amino acid metabolism can play a regulatory role in T cell activation. For example, tumor indoleamine 2,3-dioxygenase (IDO), an enzyme that converts tryptophan to kynurenine, has been shown to deplete the essential amino acid, tryptophan, in the microenvironment, resulting in T cell inhibition [99], as detailed later in this review.
4.2. Adenosine Metabolism: A Critical Immunosuppressive Metabolite
A major feature of melanoma cells is their ability to escape the immune surveillance. Adenosine (ADO) is one of the main metabolites present in the TME that generates immunosuppressive conditions [100,101,102]. Metabolic stress and cell damage caused by hypoxia and inflammation lead to an enhanced hydrolysis of ATP into ADO through the enzymatic activity of two cell-surface ectonucleotidases, CD39 and CD73 [103,104,105,106]. Either produced by tumor cells and/or by immune suppressive cells, ADO accumulates in tumor tissues where it suppresses T effector cell functions, reducing their proliferation, cytotoxic activity, and pro-inflammatory cytokine secretion, including IFN-γ, tumor necrosis factor (TNF)-β, and IL-2, by binding to purinergic receptors A2aR and, partially, A2bR [101,107,108,109,110,111,112]. These receptors are Gs-coupled receptors that, by increasing intracellular cyclic AMP (cAMP) levels, mediate the immune suppressive effects of ADO, facilitating tumor progression. In addition, A2aR stimulation reduces the expression of CD25 and CD40 ligand (CD40L) and increases the expression of PD-1 and CTLA-4 on T cells [113]. ADO also promotes immunosuppression by increasing IL-10 secretion and expression of immunosuppressive proteins, such as IDO, TGF-β, and arginase-2, promoting peripheral tolerance by inducing T-cell anergy and also metabolic dysregulation [112,114,115,116].
By inducing a dysfunction in the immune responses, the CD39/CD73/ADO axis is apparently relevant for melanogenesis [117,118,119,120]. ADO interferes with signals mediated by IL-2 receptor and exerts a direct anti-proliferative effect on naive CD4+T and CD8+T cells in melanoma TME [108]. Therefore, ADO has been suggested as a key player for melanoma cells escaping from adaptive immune control (Figure 3) [121]. Immune and melanoma cells express both CD73 and ADO receptors. A2aR expression in human melanoma cell lines was originally reported by Merighi et al. This study demonstrated that ADO enhances melanoma cell proliferation through A2aR activation [122]. The first evidence of the role of A2aR in vivo in the control of melanoma growth has been reported by Ohta et al., showing that 60% of A2aR deficient mice completely rejected established immunogenic tumors by anti-tumor CD8+T cells [107]. These results were confirmed some years later by Waickman et al. using a different disease model (i.e., lymphoma), suggesting that A2aR is an attractive target for tumor immunotherapy that synergizes with other immunomodulatory approaches [123,124].
Recent findings have demonstrated that the activation of the MAPK pathway leads to CD73 over-expression on the surface of melanoma cells, thus, promoting an invasive phenotype. Conversely, CD73 reduction is followed by blocking the BRAF/MEK signaling [125,126,127]. Moreover, recent studies have also demonstrated that Treg and myeloid-derived suppressor cells (MDSCs) lead to the up-regulation of CD73/CD39 expression in both primary melanoma and lymph nodes, therefore, enhancing an immunosuppressive function [128,129]. In addition, high basal levels of soluble CD73 have been associated with low response rates in melanoma patients receiving immunotherapy, suggesting that CD73 potentially represents a prognostic biomarker of survival during treatment [130].
Overall, these findings provide the rationale for future therapeutic strategies mostly aimed at inhibiting CD73 signaling and A2aR blocking, in combination with targeted therapies and/or immunotherapy [102,112,124].
4.3. Indoleamine-2,3-dioxygenase (IDO)-Kynurenine Metabolism
Another critical player in driving immune tolerance is IDO enzyme [131,132,133,134]. Together with extrinsic suppression of CD8+ T effector cells by Tregs and engagement of the inhibitory receptor PD-1 by the ligand PD-L1, the deregulation of IDO represents a key mechanism promoting immunosuppression in melanoma (Figure 3) [135,136]. IDO is a cytosolic, heme-dependent enzyme responsible for the rate-limiting step of de novo nicotinamide adenine dinucleotide (NAD) synthesis from tryptophan in extrahepatic tissues. By catalyzing the initial and rate-limiting step of tryptophan degradation, IDO reduces the local tryptophan concentration and produces immune modulatory tryptophan metabolites [137,138]. In particular, cells expressing IDO produce the tryptophan catabolite kynurenine, which regulates immune functions by interacting with the aryl hydrocarbon receptor (AhR) expressed by T cells, Tregs, and DCs [139]. Kynurenine was recently shown to favor immunosuppression in the TME, leading to induction of T-cell anergy, apoptosis, increased conversion of naïve CD4+ T cells into Tregs, and polarization of DCs and macrophages toward an immunosuppressive phenotype [131,134,140,141,142,143]. Expression and activity of IDO on tumor and immune cells can be modulated by several signaling systems, including the engagement of toll-like receptors (TLRs), tumor necrosis factor superfamily members (TNFRs), interferon beta receptor (IFNBR), interferon gamma receptor (IFNGR), and transforming growth factor beta receptors (TGFBRs), which are able to induce or maintain IDO expression. NF-KB activation is a central downstream signal of these pathways regulating IDO expression [144].
Increased expression of IDO was associated with poor survival outcomes in patients with ovarian, lung, colorectal, and breast cancer; brain tumors; and melanoma [140,145,146,147,148]. Melanoma cells can express IDO and directly mediate T cell and NK cell cytotoxicity [149]. Spranger et al. demonstrated that IDO expression, together with PD-L1, is mediated by signals, such as IFN-γ, derived from TILs within the TME [135]. In melanoma patients and in mouse models, IDO is expressed by antigen processing cells (APCs) in tumor draining lymph nodes [150,151]. In a cohort of patients with early stage (i.e., stage I–II) melanoma, IDO expression in the sentinel lymph node was an independent negative prognostic factor for PFS and OS [152], as well as its expression in primary melanoma [148]. Moreover, a positive correlation between the high expression of IDO and clinical response to anti-CTLA-4 therapy in melanoma has been reported [153].
According to its role in driving immunosuppression, IDO has become a valid target in cancer therapy over the last years [154,155]. Competitive inhibitors of IDO are currently being tested in clinical trials in patients with solid cancer, including melanoma, with the aim of enhancing the efficacy of conventional chemotherapy, vaccines, or checkpoint inhibitors [156]. Several IDO1 inhibitors have been developed and are currently under clinical development [157,158]. Moreno et al. demonstrated that targeting IDO with the competitive inhibitor 1-methyl-tryptophan (1-MT) retards the proliferation of melanoma cells in vitro [159,160]. Although monotherapy with 1-MT has little effect on the growth of subcutaneous melanoma B16-F10 tumors, 1-MT sensitizes the tumors to chemotherapy and whole-body radiation [161]. While promising, additional exploration is required to further define how IDO mediates immunosuppression in melanoma and whether or not 1-MT can be combined with currently approved therapies. To date, the most advanced IDO inhibitor is epacadostat, a highly specific IDO1 inhibitor, which has already been tested in several clinical trials [162,163]. However, data of epacadostat in combination with immunotherapy in patients with melanoma have failed to demonstrate a significant survival benefit (see further) [164]. A better understanding of IDO biology could lead to contrast the compensatory mechanisms unleashed in the tumor cell by blocking IDO. Clinical trials with a translational background can help improve the dynamic changes of tumor metabolic mechanisms during treatment. However, IDO1 pathway remains a relevant target to block in order to improve the efficacy of cancer immunotherapy.
4.4. Metabolic Enzymes/Cytokines: The Role of Nicotinamide Phosphoribosyltransferase (NAMPT)
NAD is an essential cofactor for redox reactions and the substrate for NAD-consuming enzymes, including sirtuins, activating genetic and epigenetic pathways [165,166]. Dysregulation of NAD levels is a contributing factor in the pathogenesis of several diseases, including cancer [167,168,169]. Given that NAD-dependent processes catabolize the molecule, permanent NAD synthesis is required in actively proliferating cells. For this reason, cancer metabolism rewiring is often accompanied by the upregulation of NAMPT, the key-limiting enzyme that catalyzes the first reversible step in NAD biosynthesis from nicotinamide (Nam) [165,168,170,171,172]. NAMPT is highly regulated at transcriptional levels; for example, the oncogene c-MYC regulates the expression of NAMPT, enhancing glycolysis and lactate production, leading to the Warburg effect [34,173,174]. Intriguingly, NAMPT has a second life outside the cell creating immune suppressive and pro-tumor conditions [175,176,177,178]. Extracellular (e)NAMPT levels are increased in many tumors, suggesting that the molecule is a novel player in tumor–host interactions. It binds TLR4, recently identified as a receptor [179,180], on tumor or immune cells and activates several signaling pathways [166,181]. For these reasons, NAMPT was the first NAD-biosynthetic enzyme (NBE) for which a clear potential as a therapeutic target in both solid and hematologic tumors was demonstrated. NAMPT-specific inhibitors reduce NAD levels by inhibiting energy metabolism pathways, such as glycolysis, TCA, and OXPHOS, contributing to the suppression of cancer cell proliferation [178,181,182]. Unfortunately, the NAMPT inhibitors (NAMPTi) FK866 and GMX-1778 both failed in clinical trials [182], likely because they were used in unselected patients, suggesting that it is very important to select tumors addicted to NAMPT activity.
In the last 10 years, increasing evidence has supported a driving role of NAMPT in melanoma progression and drug resistance [45]. NAMPT was first identified as over-expressed in melanoma lesions compared to benign lesions at the transcriptional and protein levels [183,184,185].
Audrito et al. extensively demonstrated the driver role of the NAMPT/NAD axis in the acquisition of resistance to BRAF inhibitors: (i) NAMPT appeared to be the master regulator of NAD biosynthesis in resistant melanoma cells, a key element involved in metabolic reprogramming [45,186]; (ii) melanoma patients, including those resistant to BRAF inhibitors, showed increased tissue and serum expression of NAMPT as compared to healthy controls or to patients with localized disease. Furthermore, patients with high eNAMPT levels have an overall reduced survival [186,187]; (iii) NAMPT over-expression recapitulates BRAF inhibitors resistance phenotype plasticity [188], data confirmed also by Ohanna et al. [189]; (iv) NAMPT targeting leads to NAD and ATP depletion, decreasing cell survival and reduced tumor growth in vitro and in melanoma xenografts in immunocompromised mice [186]. The over-expression of NAMPT is associated with the oncogenic activation of MAPK pathways due to BRAF mutations. Several transcription factors activated by this oncogenic signaling (including MYC, STAT3 and 5, NK-kB, and others) can bind NAMPT promoter and induce its transcription [173,186,189]. Overall, these data confirm that NAMPT plays a central role in the phenotypic plasticity of melanoma, becoming a novel therapeutic target in the clinical setting.
The role of eNAMPT in melanoma TME remains an open issue in this field. eNAMPT can act on tumor cells activating signaling pathways that support their proliferation, including MAPK, AKT, NF-kB, as shown in [190]. Paracrine effects of eNAMPT on stromal and immune cells were described in other tumor models. For example, eNAMPT is able to polarize macrophages in M2-like type in chronic lymphocytic leukemia [191,192] and forces the mobilization of immature MDSC and enhances their production of suppressive nitric oxide in fibrosarcoma and breast carcinoma mouse models [193]. A recent paper highlights NAMPT as a critical molecule in priming pro-tumor functions of tumor-associated neutrophils (TANs) in melanoma [194], including tumorigenic conversion of TANs and their pro-angiogenic switch [194]. Lastly, a recent paper first provided evidence for a direct functional correlation in liver cancer between the expression of PD-L1 and NAD-NAMPT axis, suggesting an association between NAMPT expression and an immune escape signature [195]. NAMPT could become a predictive marker of anti-PD-L1 therapy, and this would be an important finding if confirmed also in melanoma, thinking to combination therapy with NAMPT inhibitor and/or blocking antibody [175,196,197] with targeted and immunotherapy.
5. Therapeutic Perspectives: Targeting Melanoma Metabolism
To date, several metabolic targets have been investigated in advanced/metastatic melanoma, most of which are in combination with standard therapeutic strategies, with the aim to avoid or delay the onset of resistance mechanisms to immunotherapy and targeted therapies. However, despite some promising preclinical results, the efforts to target metabolic pathways for the treatment of melanoma are still in a preliminary phase and have not modified the available treatment strategies.
5.1. Immunotherapy and Metabolic Targets
There is a strong link between cancer cell metabolism and T cell functions, as previously discussed. Several metabolic alterations and adaptive mechanisms lead to T cell differentiation, proliferation, and tumor-promoting phenotypes. For these reasons, increased attention is focused on the combination of immune checkpoint inhibitors with agents targeting metabolic reprogramming [88].
CTLA-4 signaling inhibits glycolysis, preventing activation and differentiation of naive CD8+ T cells [198]. PD-1/PD-L1 signaling has important metabolic effects on T cells, leading to impaired mechanisms of energy generation and macromolecules synthesis, and reduced cytokine secretion through the inhibition of glycolysis and upregulation of FA oxidation [93]. PD-L1 signaling has direct metabolic effects on cancer cells: in response to anti-PD-L1 agents, glucose uptake and lactate extrusion are decreased, suggesting that altered PD-L1 expression directly impairs T-cell metabolism, along with favoring cancer cell metabolic reprogramming. As a consequence, treatment with anti-PD1/PD-L1 antibodies succeeds in restoring the metabolic balance in favor of T cells [88,92], as previously mentioned. Moreover, immune checkpoint molecule expression largely depends on the TME metabolic conditions and, specifically, on extracellular adenosine levels, tissue hypoxia, and T cells metabolic stress [199]. This evidence suggests that immunotherapy has the potential ability to differentially target cancer and T cells through shared metabolic requirements.
Focusing on the CD73-adenosine axis, two different therapeutic strategies have been investigated in melanoma, namely, small molecule inhibitors and monoclonal antibodies. Combining CD73 inhibitors with both anti-CTLA-4 and anti-PD1 antibodies significantly increased the effect of immunotherapy through an increased frequency of tumor-infiltrating CD8+ T cells, suppressed Treg accumulation within tumor tissues in murine melanoma models, and enhanced T cell responses [200,201]. This effect was more pronounced in tumors with high expression levels of CD73, suggesting that CD73 could be used as a potential biomarker to select patients who are more likely to benefit from combination therapy. However, these findings should still be verified and confirmed in the clinical setting.
In addition, as previously discussed, targeting IDO could also be a novel therapeutic strategy in combination with immunotherapy. The largest phase III trial to date evaluating the IDO inhibitor, epacadostat, in combination with the anti-PD1 antibody, pembrolizumab, in advanced melanoma patients, was the ECHO-301/KEYNOTE-252 [163]. This double-blind trial randomized over 700 patients with unresectable or metastatic melanoma in a 1:1 ratio to receive pembrolizumab in combination with epacadostat or placebo. Despite the promising preclinical findings and early-stage data from the phase I/II trial ECHO-202/KEYNOTE-037, the final results of the phase III trial did not show any survival benefit in patients treated with the combination therapy as compared to pembrolizumab alone [163,164]. These disappointing results suggest that some points still need to be elucidated, mainly, which is the best clinical setting for IDO1 inhibition to be used.
5.2. Targeted Therapy and Metabolic Targets
As discussed before, BRAF oncogene plays a key role in the metabolic reprogramming of melanoma cells. Moreover, metabolic adaptation both occurs in response to MAPK pathway inhibition and also contributes to resistance mechanisms during treatment with BRAF and MEK inhibitors [202].
The combination of BRAF and MEK inhibitors with drugs targeting OXPHOS is a promising strategy to enhance the effect of MAPK pathway inhibition and overcome drug resistance [43,203,204,205]. Several drugs increase the efficacy of MAPK inhibitors to induce cell death in melanoma by acting on mitochondrial bioenergetics of tumor cells. Nonsteroidal anti-inflammatory drugs (NSAID), diclofenac and lumiracoxib, increased the anti-glycolytic impact of BRAF inhibitors and prevented RAF-inhibitor induced metabolic reprogramming towards OXPHOS, thus, leading to an enhanced sensitivity of melanoma cells to BRAF inhibitors and delayed onset of treatment resistance [206]. Gamitrinib, an inhibitor of heat shock protein (HSP)90, represents another possible strategy to overcome drug resistance to MAPK inhibitors [203] and has also shown activity in NRAS mutant melanoma in combination with 6-thio-2′-deoxyguanosine [207]. Phenformin and metformin, biguanides used in the treatment of type 2 diabetes, have shown antitumor activity in vitro and in vivo. The use of phenformin has shown not only to enhance the therapeutic effect of BRAF inhibitors [208] but also to inhibit MDSC activity and increase the efficacy of anti-PD1 in melanoma [209]. Benserazide, an inhibitor of the M2 splice isoform of pyruvate kinase (PK2M), which is a key enzyme for generating pyruvate and ATP in the glycolytic pathway, leads to the inhibition of aerobic glycolysis and concurrent upregulation of OXPHOS. Since PK2M activity and aerobic glycolysis are upregulated in BRAF inhibitors resistant melanoma cells, treatment with benserazide results in a heightened sensitivity to suppressed PK2M expression both in vitro and in vivo, with potential therapeutic applications [210]. Recently, the glutamine pathway also emerged as a possible metabolic reprogramming strategy in melanoma resistant to targeted therapies [211]. In fact, BRAF-inhibitors-resistant melanoma cells increase uptake of glutamine and show overexpression of glutaminase (GLS). The mechanisms that drive this switch from glucose utilization to glutamine remain unclear, but treatment with GLS inhibitors re-sensitizes resistant cells to BRAF inhibitors [211] and also to the chemotherapeutic drug temozolomide (TMZ) [212]. Consistently, high-OXPHOS melanoma could be supported by glutamine and fatty acid oxidation via PGC-1α axis, as demonstrated also in other cancer types [213]. Preclinical evidence has shown that the inhibition of mTORC1/2 can decrease PGC-1α expression and inhibit OXPHOS. Moreover, resistance to MAPK inhibitors can be overcome by mTORC1/2 through the nuclear exclusion of MITF, and this combination has a synergistic effect only in the OXPHOS-high phenotype of melanomas [205].
Metabolic reprogramming represents an intriguing target also in uveal melanoma. Unlike its cutaneous counterpart, uveal melanoma has not gained any therapeutic benefit over the last years, and the median survival for patients with metastatic disease is less than 12 months [214]. More than 90% of uveal melanomas show monosomy of chromosome 3, a genetic feature typically associated with metastasis and poor prognosis [215]. Molecular studies have shown that elevated levels of non-mutant succinate dehydrogenase A (SDHA), a fundamental link between the TCA and OXPHOS, are the core of a distinct metabolic program that finally leads to increased biologic aggressiveness and resistance to OXPHOS inhibition [216]. Uveal melanomas frequently show guanine nucleotide-binding protein G(q) GNAQ and GNA11 mutations, which enhance MEK-ERK1/2 signaling. Adaptive upregulation of OXPHOS has been described as a mechanism of resistance during treatment with MEK inhibitors in combination with CDK4/6 inhibitors. The addition of the OXPHOS inhibitor, IACS-010759, to CDK4/6 plus MEK inhibitors decreased cell growth and enhanced apoptosis, suggesting that direct OXPHOS inhibition could be an approach to optimize targeted therapy treatment in uveal melanoma [217]. In this setting, targeting the metabolic substrates and OXPHOS is an appealing therapeutic strategy since targeted therapies and immunotherapy have led to disappointing results in uveal melanoma.
6. Conclusions
Melanoma is a metabolic heterogeneous disease with the ability to adapt its metabolism in order to utilize a variety of fuels for energy production, facilitating tumor progression and metastasis. The significance of metabolic rewiring in melanoma is supported by growing evidence of the impact, in terms of increased efficacy, of therapeutic strategies targeting metabolic molecules in combination with standard therapies. Increasing data support the notion that metabolic phenotypes of melanoma cells depend on the contribution from both tumor intrinsic factors and extrinsic factors deriving from the TME conditions. The complex interplay between tumor metabolism and the immune system emerges as the most important aspect, as highlighted also by the numerous ongoing clinical trials specifically focused on the combination of agents targeting metabolic pathways altered in melanoma and immunotherapy (Table 1). Overall, available evidence suggests that targeting metabolic rewiring mechanisms and the metabolic crosstalk within TME represents one of the most promising and novel therapeutic strategies to overcome drug resistance or to increase therapeutic efficacy of standard treatments in specific subsets of melanoma. Prospective trials with a strong background derived from translational studies exploring both circulating and tissue metabolic biomarkers will lead to the discovery of better and more effective therapeutic combinations for the treatment of metastatic melanoma patients.

Table 1.
Overview of the main active and recently concluded clinical trials of drugs targeting tumor metabolism in melanoma (source: clinicaltrials.gov; accessed on 17 April 2021).
Author Contributions
Conceptualization, V.A.; writing—review and editing, A.I. and V.A., review and editing, F.G., M.M. and D.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Associazione Italiana Ricerca sul Cancro (AIRC), Investigator Grant—IG 2017 #20258 to D.T., and partially funded by Current Research RC 21/18501 to A.I.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
A.I. reports personal fees and honoraria for lectures from Bristol Myers Squibb and Merck Sharp & Dohme and advisory board for Novartis. F.G. reports honoraria for lectures and advisory board for Novartis, Pfizer, Merck, Pierre Fabre, and Boehringer Ingelheim; honoraria for lectures from Amgen, AstraZeneca, and Roche; and research funding, honoraria for lectures and advisory board from Bristol Myers Squibb. M.M., D.T., and V.A. declare no conflict of interest.
References
- Cancer Stat Facts: Melanoma of the Skin. Available online: https://seer.cancer.gov/statfacts/html/melan.html (accessed on 16 April 2021).
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Sileni, V.C.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage IIIBRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma withoutBRAFMutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Weber, J.; Mandalà, M.; Del Vecchio, M.; Gogas, H.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Blank, C.U.; Mandalà, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef]
- Nowicki, T.; Hu-Lieskovan, S.; Ribas, A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J. 2018, 24, 47–53. [Google Scholar] [CrossRef]
- Martens, A.; Wistuba-Hamprecht, K.; Foppen, M.G.; Yuan, J.; Postow, M.A.; Wong, P.; Romano, E.; Khammari, A.; Dreno, B.; Capone, M.; et al. Baseline Peripheral Blood Biomarkers Associated with Clinical Outcome of Advanced Melanoma Patients Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 2908–2918. [Google Scholar] [CrossRef] [PubMed]
- Syeda, M.M.; Wiggins, J.M.; Corless, B.C.; Long, G.V.; Flaherty, K.T.; Schadendorf, D.; Nathan, P.D.; Robert, C.; Ribas, A.; Davies, M.A.; et al. Circulating tumour DNA in patients with advanced melanoma treated with dabrafenib or dabrafenib plus trametinib: A clinical validation study. Lancet Oncol. 2021, 22, 370–380. [Google Scholar] [CrossRef]
- Jessurun, C.A.C.; Vos, J.A.M.; Limpens, J.; Luiten, R.M. Biomarkers for Response of Melanoma Patients to Immune Checkpoint Inhibitors: A Systematic Review. Front. Oncol. 2017, 7, 233. [Google Scholar] [CrossRef]
- Ribas, A.; Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Carlino, M.S.; Grob, J.J. Tumor mutational burden (TMB), T cell-inflamed gene expression profile (GEP) and PD-L1 are independently associated with response to pembrolizumab (Pembro) in patients with advanced melanoma in the KEYNOTE (KN)-006 study. Cancer Res. 2019, 79, 4217. [Google Scholar]
- Ward, P.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Bristot, I.J.; Dias, C.K.; Chapola, H.; Parsons, R.B.; Klamt, F. Metabolic rewiring in melanoma drug-resistant cells. Crit. Rev. Oncol. 2020, 153, 102995. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.M.; Gopal, Y.N.V.; McQuade, J.L.; Peng, W.; DeBerardinis, R.J.; Davies, M.A. Metabolic strategies of melanoma cells: Mechanisms, interactions with the tumor microenvironment, and therapeutic implications. Pigment. Cell Melanoma Res. 2017, 31, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, A.; Fiume, G.; Pelagalli, A.; Sanità, G.; Ruocco, M.R.; Montagnani, S.; Arcucci, A. Metabolic Plasticity of Melanoma Cells and Their Crosstalk With Tumor Microenvironment. Front. Oncol. 2020, 10, 722. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, C.; Guldberg, P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol. Med. 2015, 21, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta (BBA) 2011, 1807, 552–561. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef]
- Parmenter, T.J.; Kleinschmidt, M.; Kinross, K.M.; Bond, S.T.; Li, J.; Kaadige, M.R.; Rao, A.; Sheppard, K.E.; Hugo, W.; Pupo, G.M.; et al. Response of BRAF-Mutant Melanoma to BRAF Inhibition Is Mediated by a Network of Transcriptional Regulators of Glycolysis. Cancer Discov. 2014, 4, 423–433. [Google Scholar] [CrossRef]
- Hall, A.; Meyle, K.D.; Lange, M.K.; Klima, M.; Sanderhoff, M.; Dahl, C.; Abildgaard, C.; Thorup, K.; Moghimi, S.M.; Jensen, P.B.; et al. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the V600EBRAF oncogene. Oncotarget 2013, 4, 584–599. [Google Scholar] [CrossRef]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative Metabolic Flux Profiling of Melanoma Cell Lines. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef]
- Tarrado-Castellarnau, M.; De Atauri, P.; Cascante, M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget 2016, 7, 62726–62753. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.; Fisher, D.E.; Widlund, H.R. Molecular Pathways: BRAF Induces Bioenergetic Adaptation by Attenuating Oxidative Phosphorylation. Clin. Cancer Res. 2014, 20, 2257–2263. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF Regulates Oxidative Metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α Expression Defines a Subset of Human Melanoma Tumors with Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Trinh, A.; Khamari, R.; Kluza, J. Melanoma metabolism contributes to the cellular responses to MAPK/ERK pathway inhibitors. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2018, 1862, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.R.; Moore, J.A.; Bowles, K.M.; Rushworth, S.A.; Moncrieff, M.D. Mitochondrial oxidative phosphorylation in cutaneous melanoma. Br. J. Cancer 2021, 124, 115–123. [Google Scholar] [CrossRef] [PubMed]
- De Moura, M.B.; Vincent, G.; Fayewicz, S.L.; Bateman, N.W.; Hood, B.L.; Sun, M.; Suhan, J.; Duensing, S.; Yin, Y.; Sander, C.; et al. Mitochondrial Respiration—An Important Therapeutic Target in Melanoma. PLoS ONE 2012, 7, e40690. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef]
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.A. Metabolic rewiring in melanoma. Oncogene 2017, 36, 147–157. [Google Scholar] [CrossRef]
- Corazao-Rozas, P.; Guerreschi, P.; André, F.; Gabert, P.-E.; Lancel, S.; Dekiouk, S.; Fontaine, D.; Tardivel, M.; Savina, A.; Quesnel, B.; et al. Mitochondrial oxidative phosphorylation controls cancer cell’s life and death decisions upon exposure to MAPK inhibitors. Oncotarget 2016, 7, 39473–39485. [Google Scholar] [CrossRef]
- Ruocco, M.R.; Avagliano, A.; Granato, G.; Vigliar, E.; Masone, S.; Montagnani, S.; Arcucci, A. Metabolic flexibility in melanoma: A potential therapeutic target. Semin. Cancer Biol. 2019, 59, 187–207. [Google Scholar] [CrossRef]
- Audrito, V.; Managò, A.; Gaudino, F.; Deaglio, S. Targeting metabolic reprogramming in metastatic melanoma: The key role of nicotinamide phosphoribosyltransferase (NAMPT). Semin. Cell Dev. Biol. 2020, 98, 192–201. [Google Scholar] [CrossRef]
- Chiarugi, P.; Cirri, P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016, 380, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Kaymak, I.; Williams, K.S.; Cantor, J.R.; Jones, R.G. Immunometabolic Interplay in the Tumor Microenvironment. Cancer Cell 2021, 39, 28–37. [Google Scholar] [CrossRef]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef]
- Brooks, G.A. Lactate production under fully aerobic conditions: The lactate shuttle during rest and exercise. Fed. Proc. 1986, 45, 2924–2929. [Google Scholar]
- Gladden, L.B. Current Trends in Lactate Metabolism: Introduction. Med. Sci. Sports Exerc. 2008, 40, 475–476. [Google Scholar] [CrossRef]
- Dimmer, K.S.; Friedrich, B.; Lang, F.; Deitmer, J.W.; Broer, S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem. J. 2000, 350 Pt 1, 219–227. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Goodwin, M.L.; Gladden, L.B.; Nijsten, M.W.N.; Jones, K.B. Lactate and Cancer: Revisiting the Warburg Effect in an Era of Lactate Shuttling. Front. Nutr. 2015, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2016, 38, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Végran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frérart, F.; et al. Targeting the Lactate Transporter MCT1 in Endothelial Cells Inhibits Lactate-Induced HIF-1 Activation and Tumor Angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef] [PubMed]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A Metabolic Key Player in Cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.B.; Prado-Garcia, H.; Sánchez-García, F.J. Lactate Contribution to the Tumor Microenvironment: Mechanisms, Effects on Immune Cells and Therapeutic Relevance. Front. Immunol. 2016, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Longatto-Filho, A.; Azevedo-Silva, J.; Casal, M.; Schmitt, F.C.; Baltazar, F. Role of monocarboxylate transporters in human cancers: State of the art. J. Bioenerg. Biomembr. 2012, 44, 127–139. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Völkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Feichtinger, R.G.; Lang, R. Targeting L-Lactate Metabolism to Overcome Resistance to Immune Therapy of Melanoma and Other Tumor Entities. J. Oncol. 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- Doherty, J.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Kouidhi, S.; Ayed, F.B.; Elgaaied, A.B. Targeting Tumor Metabolism: A New Challenge to Improve Immunotherapy. Front. Immunol. 2018, 9, 353. [Google Scholar] [CrossRef]
- Alkasalias, T.; Moyano-Galceran, L.; Arsenian-Henriksson, M.; Lehti, K. Fibroblasts in the Tumor Microenvironment: Shield or Spear? Int. J. Mol. Sci. 2018, 19, 1532. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Shanghelin, Y.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal Metabolic Reprogramming through Lactate Shuttle Coordinately Influences Tumor-Stroma Interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg Meets Autophagy: Cancer-Associated Fibroblasts Accelerate Tumor Growth and Metastasis via Oxidative Stress, Mitophagy, and Aerobic Glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef]
- González, C.D.; Alvarez, S.; Ropolo, A.; Rosenzvit, C.; Bagnes, M.F.G.; Vaccaro, M.I. Autophagy, Warburg, and Warburg Reverse Effects in Human Cancer. BioMed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Ho, J.; De Moura, M.B.; Lin, Y.; Vincent, G.; Thorne, S.; Duncan, L.M.; Hui-Min, L.; Kirkwood, J.M.; Becker, D.; Van Houten, B.; et al. Importance of glycolysis and oxidative phosphorylation in advanced melanoma. Mol. Cancer 2012, 11, 76. [Google Scholar] [CrossRef]
- Gallagher, P.G.; Bao, Y.; Prorock, A.; Zigrino, P.; Nischt, R.; Politi, V.; Mauch, C.; Dragulev, B.; Fox, J.W. Gene Expression Profiling Reveals Cross-talk between Melanoma and Fibroblasts: Implications for Host-Tumor Interactions in Metastasis. Cancer Res. 2005, 65, 4134–4146. [Google Scholar] [CrossRef]
- Flach, E.H.; Rebecca, V.W.; Herlyn, M.; Smalley, K.S.M.; Anderson, A.R.A. Fibroblasts Contribute to Melanoma Tumor Growth and Drug Resistance. Mol. Pharm. 2011, 8, 2039–2049. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, K.; Andl, T.; Wickett, R.R.; Zhang, Y. Perspective of Targeting Cancer-Associated Fibroblasts in Melanoma. J. Cancer 2015, 6, 717–726. [Google Scholar] [CrossRef]
- Guo, Y.; Ji, X.; Liu, J.; Fan, D.; Zhou, Q.; Chen, C.; Wang, W.; Wang, G.; Wang, H.; Yuan, W.; et al. Effects of exosomes on pre-metastatic niche formation in tumors. Mol. Cancer 2019, 18, 39. [Google Scholar] [CrossRef]
- Shelton, M.; Anene, C.; Nsengimana, J.; Roberts, W.; Newton-Bishop, J.; Boyne, J. The role of CAF derived exosomal MicroRNAs in the tumour microenvironment of melanoma. Biochim. Biophys. Acta 2020, 1875, 188456. [Google Scholar] [CrossRef]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunology 2013, 38, 633–643. [Google Scholar] [CrossRef]
- McCarthy, S.A.; Mufson, R.A.; Pearce, E.J.; Rathmell, J.C.; Howcroft, T.K. Metabolic reprogramming of the immune response in the tumor microenvironment. Cancer Biol. Ther. 2013, 14, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.-H.; Jones, R.G. Fueling Immunity: Insights into Metabolism and Lymphocyte Function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef]
- Sukumar, M.; Roychoudhuri, R.; Restifo, N.P. Nutrient Competition: A New Axis of Tumor Immunosuppression. Cell 2015, 162, 1206–1208. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.E.; Coomber, B.L.; Bridle, B.W. Metabolic reprogramming in the tumour microenvironment: A hallmark shared by cancer cells and T lymphocytes. Immunology 2017, 152, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.-C.; Liu, P.-S. Metabolic communication in tumors: A new layer of immunoregulation for immune evasion. J. Immunother. Cancer 2016, 4, 4. [Google Scholar] [CrossRef]
- Zhang, L.; Romero, P. Metabolic Control of CD8+ T Cell Fate Decisions and Antitumor Immunity. Trends Mol. Med. 2018, 24, 30–48. [Google Scholar] [CrossRef]
- Cham, C.M.; Driessens, G.; O’Keefe, J.P.; Gajewski, T.F. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol. 2008, 38, 2438–2450. [Google Scholar] [CrossRef]
- Marchesi, F.; Vignali, D.; Manini, B.; Rigamonti, A.; Monti, P. Manipulation of Glucose Availability to Boost Cancer Immunotherapies. Cancers 2020, 12, 2940. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Molon, B.; Calì, B.; Viola, A. T Cells and Cancer: How Metabolism Shapes Immunity. Front. Immunol. 2016, 7, 20. [Google Scholar] [CrossRef]
- Matsushita, H.; Hosoi, A.; Ueha, S.; Abe, J.; Fujieda, N.; Tomura, M.; Maekawa, R.; Matsushima, K.; Ohara, O.; Kakimi, K. Cytotoxic T Lymphocytes Block Tumor Growth Both by Lytic Activity and IFNgamma-Dependent Cell-Cycle Arrest. Cancer Immunol. Res. 2015, 3, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Phillips, J.B.; Da Silva, L.M.; Zhou, M.; Fodstad, O.; Owen, L.B.; Tan, M. Interplay between Immune Checkpoint Proteins and Cellular Metabolism. Cancer Res. 2017, 77, 1245–1249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e9. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. [Google Scholar] [CrossRef]
- Hasko, G.; Cronstein, B.N. Adenosine: An endogenous regulator of innate immunity. Trends Immunol. 2004, 25, 33–39. [Google Scholar] [CrossRef]
- Stagg, J.; Smyth, M. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 2010, 29, 5346–5358. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 765. [Google Scholar] [CrossRef]
- Feng, L.; Sun, X.; Csizmadia, E.; Han, L.; Bian, S.; Murakami, T.; Wang, X.; Robson, S.C.; Wu, Y. Vascular CD39/ENTPD1 Directly Promotes Tumor Cell Growth by Scavenging Extracellular Adenosine Triphosphate. Neoplasia 2011, 13, 206-IN2. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.-F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Bono, M.R.; Fernández, D.; Flores-Santibáñez, F.; Rosemblatt, M.; Sauma, D. CD73 and CD39 ectonucleotidases in T cell differentiation: Beyond immunosuppression. FEBS Lett. 2015, 589, 3454–3460. [Google Scholar] [CrossRef]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef]
- Raskovalova, T.; Lokshin, A.; Huang, X.; Su, Y.; Mandic, M.; Zarour, H.M.; Jackson, E.K.; Gorelik, E. Inhibition of Cytokine Production and Cytotoxic Activity of Human Antimelanoma Specific CD8+ and CD4+ T Lymphocytes by Adenosine-Protein Kinase A Type I Signaling. Cancer Res. 2007, 67, 5949–5956. [Google Scholar] [CrossRef]
- Gessi, S.; Merighi, S.; Sacchetto, V.; Simioni, C.; Borea, P.A. Adenosine receptors and cancer. Biochim. Biophys. Acta 2011, 1808, 1400–1412. [Google Scholar] [CrossRef]
- Haskó, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef]
- Ernst, P.B.; Garrison, J.C.; Thompson, L.F. Much Ado about Adenosine: Adenosine Synthesis and Function in Regulatory T Cell Biology. J. Immunol. 2010, 185, 1993–1998. [Google Scholar] [CrossRef]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Zarek, P.E.; Huang, C.-T.; Lutz, E.R.; Kowalski, J.; Horton, M.R.; Linden, J.; Drake, C.G.; Powell, J.D. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 2008, 111, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Mastelic-Gavillet, B.; Rodrigo, B.N.; Décombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derré, L.; Valerio, M.; et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8+ T cells. J. Immunother. Cancer 2019, 7, 257. [Google Scholar] [CrossRef]
- Novitskiy, S.V.; Ryzhov, S.; Zaynagetdinov, R.; Goldstein, A.E.; Huang, Y.; Tikhomirov, O.Y.; Blackburn, M.R.; Biaggioni, I.; Carbone, D.P.; Feoktistov, I.; et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 2008, 112, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, A.; Tucci, M.; Mannavola, F.; Felici, C.; Silvestris, F. The metabolic milieu in melanoma: Role of immune suppression by CD73/adenosine. Tumor Biol. 2019, 41, 1010428319837138. [Google Scholar] [CrossRef] [PubMed]
- Umansky, V.; Shevchenko, I.; Bazhin, A.V.; Utikal, J. Extracellular adenosine metabolism in immune cells in melanoma. Cancer Immunol. Immunother. 2014, 63, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Burghoff, S.; Gong, X.; Viethen, C.; Jacoby, C.; Flögel, U.; Bongardt, S.; Schorr, A.; Hippe, A.; Homey, B.; Schrader, J. Growth and metastasis of B16-F10 melanoma cells is not critically dependent on host CD73 expression in mice. BMC Cancer 2014, 14, 1–11. [Google Scholar] [CrossRef]
- Kazemi, M.H.; Mohseni, S.R.; Hojjat-Farsangi, M.; Anvari, E.; Ghalamfarsa, G.; Mohammadi, H.; Jadidi-Niaragh, F. Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J. Cell. Physiol. 2018, 233, 2032–2057. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Morandi, B.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Zaccarello, G.; Carrega, P.; Ferlazzo, G.; Mingari, M.C.; Moretta, L.; et al. A non-canonical adenosinergic pathway led by CD38 in human melanoma cells induces suppression of T cell proliferation. Oncotarget 2015, 6, 25602–25618. [Google Scholar] [CrossRef]
- Merighi, S.; Varani, K.; Gessi, S.; Cattabriga, E.; Iannotta, V.; Ulouglu, C.; Leung, E.; Borea, P.A. Pharmacological and biochemical characterization of adenosine receptors in the human malignant melanoma A375 cell line. Br. J. Pharmacol. 2001, 134, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Waickman, A.T.; Alme, A.; Senaldi, L.; Zarek, P.E.; Horton, M.; Powell, J.D. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol. Immunother. 2011, 61, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Montinaro, A.; Iannone, R.; Pinto, A.; Morello, S. Adenosine receptors as potential targets in melanoma. Pharmacol. Res. 2013, 76, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Döbbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo Switching of Human Melanoma Cells between Proliferative and Invasive States. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef]
- Reinhardt, J.; Landsberg, J.; Schmid-Burgk, J.L.; Ramis, B.B.; Bald, T.; Glodde, N.; Lopez-Ramos, D.; Young, A.; Ngiow, S.F.; Nettersheim, D.; et al. MAPK Signaling and Inflammation Link Melanoma Phenotype Switching to Induction of CD73 during Immunotherapy. Cancer Res. 2017, 77, 4697–4709. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Ngiow, S.F.; Barkauskas, D.S.; Sult, E.; Hay, C.; Blake, S.J.; Huang, Q.; Liu, J.; Takeda, K.; Teng, M.; et al. Co-inhibition of CD73 and A2AR Adenosine Signaling Improves Anti-tumor Immune Responses. Cancer Cell 2016, 30, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Umansky, V.; Abschuetz, O.; Osen, W.; Ramacher, M.; Zhao, F.; Kato, M.; Schadendorf, D. Melanoma-Specific Memory T Cells Are Functionally Active in Ret Transgenic Mice without Macroscopic Tumors. Cancer Res. 2008, 68, 9451–9458. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Stucci, L.S.; Mannavola, F.; Passarelli, A.; D’Oronzo, S.; Lospalluti, L.; Giudice, G.; Silvestris, F. Defective levels of both circulating dendritic cells and T-regulatory cells correlate with risk of recurrence in cutaneous melanoma. Clin. Transl. Oncol. 2019, 21, 845–854. [Google Scholar] [CrossRef]
- Morello, S.; Capone, M.; Sorrentino, C.; Giannarelli, D.; Madonna, G.; Mallardo, D.; Grimaldi, A.M.; Pinto, A.; Ascierto, P.A. Soluble CD73 as biomarker in patients with metastatic melanoma patients treated with nivolumab. J. Transl. Med. 2017, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Takikawa, O. Biochemical and medical aspects of the indoleamine 2,3-dioxygenase-initiated l-tryptophan metabolism. Biochem. Biophys. Res. Commun. 2005, 338, 12–19. [Google Scholar] [CrossRef]
- King, N.J.; Thomas, S.R. Molecules in focus: Indoleamine 2,3-dioxygenase. Int. J. Biochem. Cell Biol. 2007, 39, 2167–2172. [Google Scholar] [CrossRef]
- Katz, J.B.; Muller, A.J.; Prendergast, G.C. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol. Rev. 2008, 222, 206–221. [Google Scholar] [CrossRef]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-Regulation of PD-L1, IDO, and Tregs in the Melanoma Tumor Microenvironment Is Driven by CD8+ T Cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Routy, J.-P.; Routy, B.; Graziani, G.M.; Mehraj, V. The Kynurenine Pathway is a Double-Edged Sword in Immune-Privileged Sites and in Cancer: Implications for Immunotherapy. Int. J. Tryptophan Res. 2016, 9, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Munn, D.H. Ido expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef]
- Mandi, Y.; Vécsei, L. The kynurenine system and immunoregulation. J. Neural. Transm. 2012, 119, 197–209. [Google Scholar] [CrossRef]
- Sica, A.; Strauss, L.; Consonni, F.M.; Travelli, C.; Genazzani, A.; Porta, C. Metabolic regulation of suppressive myeloid cells in cancer. Cytokine Growth Factor Rev. 2017, 35, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Wick, W.; Eynde, B.J.V.D. Tryptophan Catabolism in Cancer: Beyond IDO and Tryptophan Depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef]
- Puccetti, P.; Grohmann, U. IDO and regulatory T cells: A role for reverse signalling and non-canonical NF-κB activation. Nat. Rev. Immunol. 2007, 7, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Mbongue, J.C.; Nicholas, D.A.; Torrez, T.W.; Kim, N.-S.; Firek, A.F.; Langridge, W.H.R. The role of indoleamine 2, 3-dioxygenase in immune suppression and autoimmunity. Vaccines 2015, 3, 703–729. [Google Scholar] [CrossRef] [PubMed]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef]
- Yu, C.-P.; Fu, S.-F.; Chen, X.; Ye, J.; Ye, Y.; Kong, L.-D.; Zhu, Z. The Clinicopathological and Prognostic Significance of IDO1 Expression in Human Solid Tumors: Evidence from a Systematic Review and Meta-Analysis. Cell. Physiol. Biochem. 2018, 49, 134–143. [Google Scholar] [CrossRef]
- Rubel, F.; Kern, J.S.; Technau-Hafsi, K.; Uhrich, S.; Thoma, K.; Häcker, G.; Von Bubnoff, N.; Meiss, F.; Von Bubnoff, D. Indoleamine 2,3-Dioxygenase Expression in Primary Cutaneous Melanoma Correlates with Breslow Thickness and Is of Significant Prognostic Value for Progression-Free Survival. J. Investig. Dermatol. 2018, 138, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Pietra, G.; Manzini, C.; Rivara, S.; Vitale, M.; Cantoni, C.; Petretto, A.; Balsamo, M.; Conte, R.; Benelli, R.; Minghelli, S.; et al. Melanoma Cells Inhibit Natural Killer Cell Function by Modulating the Expression of Activating Receptors and Cytolytic Activity. Cancer Res. 2012, 72, 1407–1415. [Google Scholar] [CrossRef]
- Sharma, M.D.; Hou, D.-Y.; Liu, Y.; Koni, P.A.; Metz, R.; Chandler, P.; Mellor, A.L.; He, Y.; Munn, D.H. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood 2009, 113, 6102–6111. [Google Scholar] [CrossRef] [PubMed]
- Gerlini, G.; Di Gennaro, P.; Mariotti, G.; Urso, C.; Chiarugi, A.; Pimpinelli, N.; Borgognoni, L. Indoleamine 2,3-Dioxygenase+ Cells Correspond to the BDCA2+ Plasmacytoid Dendritic Cells in Human Melanoma Sentinel Nodes. J. Investig. Dermatol. 2010, 130, 898–901. [Google Scholar] [CrossRef]
- Speeckaert, R.; Vermaelen, K.; van Geel, N.; Autier, P.; Lambert, J.; Haspeslagh, M.; van Gele, M.; Thielemans, K.; Neyns, B.; Roche, N.; et al. Indoleamine 2,3-dioxygenase, a new prognostic marker in sentinel lymph nodes of melanoma patients. Eur. J. Cancer 2012, 48, 2004–2011. [Google Scholar] [CrossRef]
- Hamid, O.; Schmidt, H.; Nissan, A.; Ridolfi, L.; Aamdal, S.; Hansson, J.; Guida, M.; Hyams, D.M.; Gómez, H.; Bastholt, L.; et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J. Transl. Med. 2011, 9, 204. [Google Scholar] [CrossRef]
- Löb, S.; Königsrainer, A.; Rammensee, H.-G.; Opelz, G.; Terness, P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: Can we see the wood for the trees? Nat. Rev. Cancer 2009, 9, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Brochez, L.; Chevolet, I.; Kruse, V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Patterson, L.F.S.; Mohapatra, S.R.; Dewi, D.L.; Sadik, A.; Platten, M.; Trump, S. The therapeutic potential of targeting tryptophan catabolism in cancer. Br. J. Cancer 2020, 122, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.C.R.; Clara, R.O.; Coimbra, J.B.; Júlio, A.R.; Albuquerque, R.C.; Oliveira, E.M.; Campa, A.; Maria-Engler, S.S. The expanding roles of 1-methyl-tryptophan (1-MT): In addition to inhibiting kynurenine production, 1-MT activates the synthesis of melatonin in skin cells. FEBS J. 2013, 280, 4782–4792. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, D.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable Therapeutic Efficacy Utilizing Combinatorial Blockade against IDO, CTLA-4, and PD-L1 in Mice with Brain Tumors. Clin. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef]
- Hou, D.-Y.; Muller, A.J.; Sharma, M.D.; DuHadaway, J.; Banerjee, T.; Johnson, M.; Mellor, A.L.; Prendergast, G.C.; Munn, D.H. Inhibition of Indoleamine 2,3-Dioxygenase in Dendritic Cells by Stereoisomers of 1-Methyl-Tryptophan Correlates with Antitumor Responses. Cancer Res. 2007, 67, 792–801. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Dwyer, P.J.; Clark, J.; Shi, J.G.; Bowman, K.J.; Scherle, P.A.; Newton, R.C.; Schaub, R.; Maleski, J.; Leopold, L.; et al. First-in-Human Phase I Study of the Oral Inhibitor of Indoleamine 2,3-Dioxygenase-1 Epacadostat (INCB024360) in Patients with Advanced Solid Malignancies. Clin. Cancer Res. 2017, 23, 3269–3276. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients with Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Amjad, S.; Nisar, S.; Bhat, A.A.; Shah, A.R.; Frenneaux, M.P.; Fakhro, K.; Haris, M.; Reddy, R.; Patay, Z.; Baur, J.; et al. Role of NAD+ in regulating cellular and metabolic signaling pathways. Mol. Metab. 2021, 49, 101195. [Google Scholar] [CrossRef]
- Audrito, V.; Managò, A.; Gaudino, F.; Sorci, L.; Messana, V.G.; Raffaelli, N.; Deaglio, S. NAD-Biosynthetic and Consuming Enzymes as Central Players of Metabolic Regulation of Innate and Adaptive Immune Responses in Cancer. Front. Immunol. 2019, 10, 1720. [Google Scholar] [CrossRef]
- Chiarugi, A.; Dölle, C.; Felici, R.; Ziegler, M. The NAD metabolome—A key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef] [PubMed]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Burgos, E.S. NAMPT in regulated NAD biosynthesis and its pivotal role in human metabolism. Curr. Med. Chem. 2011, 18, 1947–1961. [Google Scholar] [CrossRef]
- Dahl, T.B.; Holm, S.; Aukrust, P.; Halvorsen, B. Visfatin/NAMPT: A Multifaceted Molecule with Diverse Roles in Physiology and Pathophysiology. Annu. Rev. Nutr. 2012, 32, 229–243. [Google Scholar] [CrossRef]
- Garten, A.; Schuster, S.; Penke, M.; Gorski, T.; De Giorgis, T.; Kiess, W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 2015, 11, 535–546. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zanca, C.; Rajkumar, U.; Koga, T.; Diao, Y.; Raviram, R.; Liu, F.; Turner, K.; Yang, H.; Brunk, E.; et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nat. Cell Biol. 2019, 569, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Menssen, A.; Hydbring, P.; Kapelle, K.; Vervoorts, J.; Diebold, J.; Luscher, B.; Larsson, L.-G.; Hermeking, H. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc. Natl. Acad. Sci. USA 2012, 109, E187–E196. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V. The dual face of NAMPT: Intracellular/extracellular protein and diagnostic/therapeutic target in cancer. EBioMedicine 2020, 62, 103109. [Google Scholar] [CrossRef]
- Audrito, V.; Messana, V.G.; Deaglio, S. NAMPT and NAPRT: Two Metabolic Enzymes With Key Roles in Inflammation. Front. Oncol. 2020, 10, 358. [Google Scholar] [CrossRef]
- Grolla, A.A.; Travelli, C.; Genazzani, A.A.; Sethi, J.K. Extracellular nicotinamide phosphoribosyltransferase, a new cancer metabokine. Br. J. Pharmacol. 2016, 173, 2182–2194. [Google Scholar] [CrossRef] [PubMed]
- Heske, C.M. Beyond Energy Metabolism: Exploiting the Additional Roles of NAMPT for Cancer Therapy. Front. Oncol. 2020, 9, 1514. [Google Scholar] [CrossRef] [PubMed]
- Camp, S.M.; Ceco, E.; Evenoski, C.L.; Danilov, S.M.; Zhou, T.; Chiang, E.T.; Moreno-Vinasco, L.; Mapes, B.; Zhao, J.; Gursoy, G.; et al. Unique Toll-Like Receptor 4 Activation by NAMPT/PBEF Induces NFκB Signaling and Inflammatory Lung Injury. Sci. Rep. 2015, 5, 13135. [Google Scholar] [CrossRef]
- Managò, A.; Audrito, V.; Mazzola, F.; Sorci, L.; Gaudino, F.; Gizzi, K.; Vitale, N.; Incarnato, D.; Minazzato, G.; Ianniello, A.; et al. Extracellular nicotinate phosphoribosyltransferase binds Toll like receptor 4 and mediates inflammation. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Dalamaga, M.; Christodoulatos, G.S.; Mantzoros, C.S. The role of extracellular and intracellular Nicotinamide phosphoribosyl-transferase in cancer: Diagnostic and therapeutic perspectives and challenges. Metabolism 2018, 82, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Galli, U.; Colombo, G.; Travelli, C.; Tron, G.C.; Genazzani, A.A.; Grolla, A.A. Recent Advances in NAMPT Inhibitors: A Novel Immunotherapic Strategy. Front. Pharmacol. 2020, 11, 656. [Google Scholar] [CrossRef]
- Wachsman, W.; Morhenn, V.; Palmer, T.; Walls, L.; Hata, T.; Zalla, J.; Scheinberg, R.; Sofen, H.; Mráz, S.; Gross, K.; et al. Noninvasive genomic detection of melanoma. Br. J. Dermatol. 2011, 164, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Maldi, E.; Travelli, C.; Caldarelli, A.; Agazzone, N.; Cintura, S.; Galli, U.; Scatolini, M.; Ostano, P.; Miglino, B.; Chiorino, G.; et al. Nicotinamide phosphoribosyltransferase (NAMPT) is over-expressed in melanoma lesions. Pigment. Cell Melanoma Res. 2013, 26, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Tang, W.; Chen, X.; Wang, S.; Wang, X.; Xu, H.; Li, L. The NAMPT/E2F2/SIRT1 axis promotes proliferation and inhibits p53-dependent apoptosis in human melanoma cells. Biochem. Biophys. Res. Commun. 2017, 493, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V.; Managò, A.; La Vecchia, S.; Zamporlini, F.; Vitale, N.; Baroni, G.; Cignetto, S.; Serra, S.; Bologna, C.; Stingi, A.; et al. Nicotinamide Phosphoribosyltransferase (NAMPT) as a Therapeutic Target in BRAF-Mutated Metastatic Melanoma. J. Natl. Cancer Inst. 2018, 110, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V.; Managò, A.; Zamporlini, F.; Rulli, E.; Gaudino, F.; Madonna, G.; D’Atri, S.; Cappellini, G.C.A.; Ascierto, P.A.; Massi, D.; et al. Extracellular nicotinamide phosphoribosyltransferase (eNAMPT) is a novel marker for patients with BRAF-mutated metastatic melanoma. Oncotarget 2018, 9, 18997–19005. [Google Scholar] [CrossRef]
- Audrito, V.; Messana, V.G.; Moiso, E.; Vitale, N.; Arruga, F.; Brandimarte, L.; Gaudino, F.; Pellegrino, E.; Vaisitti, T.; Riganti, C.; et al. NAMPT Over-Expression Recapitulates the BRAF Inhibitor Resistant Phenotype Plasticity in Melanoma. Cancers 2020, 12, 3855. [Google Scholar] [CrossRef]
- Ohanna, M.; Cerezo, M.; Nottet, N.; Bille, K.; Didier, R.; Beranger, G.; Mograbi, B.; Rocchi, S.; Yvan-Charvet, L.; Ballotti, R.; et al. Pivotal role of NAMPT in the switch of melanoma cells toward an invasive and drug-resistant phenotype. Genes Dev. 2018, 32, 448–461. [Google Scholar] [CrossRef]
- Grolla, A.A.; Torretta, S.; Gnemmi, I.; Amoruso, A.; Orsomando, G.; Gatti, M.; Caldarelli, A.; Lim, D.; Penengo, L.; Brunelleschi, S.; et al. Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is a tumoural cytokine released from melanoma. Pigment. Cell Melanoma Res. 2015, 28, 718–729. [Google Scholar] [CrossRef]
- Audrito, V.; Serra, S.; Brusa, D.; Mazzola, F.; Arruga, F.; Vaisitti, T.; Coscia, M.; Maffei, R.; Rossi, D.; Wang, T.; et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood 2015, 125, 111–123. [Google Scholar] [CrossRef]
- Travelli, C.; Colombo, G.; Mola, S.; Genazzani, A.A.; Porta, C. NAMPT: A pleiotropic modulator of monocytes and macrophages. Pharmacol. Res. 2018, 135, 25–36. [Google Scholar] [CrossRef]
- Travelli, C.; Consonni, F.M.; Sangaletti, S.; Storto, M.; Morlacchi, S.; Grolla, A.A.; Galli, U.; Tron, G.C.; Portararo, P.; Rimassa, L.; et al. Nicotinamide Phosphoribosyltransferase Acts as a Metabolic Gate for Mobilization of Myeloid-Derived Suppressor Cells. Cancer Res. 2019, 79, 1938–1951. [Google Scholar] [CrossRef]
- Pylaeva, E.; Harati, M.D.; Spyra, I.; Bordbari, S.; Strachan, S.; Thakur, B.K.; Höing, B.; Franklin, C.; Skokowa, J.; Welte, K.; et al. NAMPT signaling is critical for the proangiogenic activity of tumor-associated neutrophils. Int. J. Cancer 2018, 144, 136–149. [Google Scholar] [CrossRef]
- Lv, H.; Lv, G.; Chen, C.; Zong, Q.; Jiang, G.; Ye, D.; Cui, X.; He, Y.; Xiang, W.; Han, Q.; et al. NAD+ Metabolism Maintains Inducible PD-L1 Expression to Drive Tumor Immune Evasion. Cell Metab. 2021, 33, 110–127.e5. [Google Scholar] [CrossRef] [PubMed]
- Quijada, H.; Bermudez, T.; Kempf, C.L.; Valera, D.G.; Garcia, A.N.; Camp, S.M.; Song, J.H.; Franco, E.; Burt, J.K.; Sun, B.; et al. Endothelial eNAMPT Amplifies Preclinical Acute Lung Injury: Efficacy of an eNAMPT-Neutralising mAb. Eur. Respir. J. 2021, 57, 2002536. [Google Scholar] [CrossRef] [PubMed]
- Colombo, G.; Clemente, N.; Zito, A.; Bracci, C.; Colombo, F.S.; Sangaletti, S.; Jachetti, E.; Ribaldone, D.G.; Caviglia, G.P.; Pastorelli, L.; et al. Neutralization of extracellular NAMPT (nicotinamide phosphoribosyltransferase) ameliorates experimental murine colitis. J. Mol. Med. 2020, 98, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; Rathmell, J.C. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015, 36, 257–264. [Google Scholar] [CrossRef]
- Eohta, A. A Metabolic Immune Checkpoint: Adenosine in Tumor Microenvironment. Front. Immunol. 2016, 7, 109. [Google Scholar] [CrossRef]
- Iannone, R.; Miele, L.; Maiolino, P.; Pinto, A.; Morello, S. Adenosine limits the therapeutic effectiveness of anti-CTLA4 mAb in a mouse melanoma model. Am. J. Cancer Res. 2014, 4, 172–181. [Google Scholar]
- Beavis, P.A.; Milenkovski, N.; Henderson, M.A.; John, L.B.; Allard, B.; Loi, S.; Kershaw, M.H.; Stagg, J.; Darcy, P.K. Adenosine Receptor 2A Blockade Increases the Efficacy of Anti–PD-1 through Enhanced Antitumor T-cell Responses. Cancer Immunol. Res. 2015, 3, 506–517. [Google Scholar] [CrossRef]
- Smith, L.K.; Rao, A.D.; McArthur, G.A. Targeting metabolic reprogramming as a potential therapeutic strategy in melanoma. Pharmacol. Res. 2016, 107, 42–47. [Google Scholar] [CrossRef]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef]
- Livingstone, E.; Swann, S.; Lilla, C.; Schadendorf, D.; Roesch, A. Combining BRAF V600E inhibition with modulators of the mitochondrial bioenergy metabolism to overcome drug resistance in metastatic melanoma. Exp. Dermatol. 2015, 24, 709–710. [Google Scholar] [CrossRef]
- McQuade, J.L.; Gopal, Y.V. Counteracting oxidative phosphorylation-mediated resistance of melanomas to MAPK pathway inhibition. Mol. Cell. Oncol. 2015, 2, e991610. [Google Scholar] [CrossRef] [PubMed]
- Brummer, C.; Faerber, S.; Bruss, C.; Blank, C.; Lacroix, R.; Haferkamp, S.; Herr, W.; Kreutz, M.; Renner, K. Metabolic targeting synergizes with MAPK inhibition and delays drug resistance in melanoma. Cancer Lett. 2019, 442, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Uribe, P.; Adrianzen-Ruesta, M.P.; Deng, Z.; Echevarría-Vargas, I.; Mender, I.; Saheb, S.; Liu, Q.; Altieri, D.C.; Murphy, M.E.; Shay, J.W.; et al. Exploiting TERT dependency as a therapeutic strategy for NRAS-mutant melanoma. Oncogene 2018, 37, 4058–4072. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Ito, K.; Perez-Lorenzo, R.; Del Guzzo, C.; Lee, J.H.; Shen, C.-H.; Bosenberg, M.W.; McMahon, M.; Cantley, L.C.; Zheng, B. Phenformin enhances the therapeutic benefit of BRAFV600E inhibition in melanoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18226–18231. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Li, M.; Trousil, S.; Zhang, Y.; di Magliano, M.P.; Swanson, K.D.; Zheng, B. Phenformin Inhibits Myeloid-Derived Suppressor Cells and Enhances the Anti-Tumor Activity of PD-1 Blockade in Melanoma. J. Investig. Dermatol. 2017, 137, 1740–1748. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, Z.; Su, J.; Li, J.; Zhao, S.; Wu, L.; Zhang, J.; He, Y.; Zhang, G.; Tao, J.; et al. Benserazide is a novel inhibitor targeting PKM2 for melanoma treatment. Int. J. Cancer 2020, 147, 139–151. [Google Scholar] [CrossRef]
- Baenke, F.; Chaneton, B.; Smith, M.; Van Den Broek, N.; Hogan, K.; Tang, H.; Viros, A.; Martin, M.; Galbraith, L.; Girotti, M.R.; et al. Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol. Oncol. 2016, 10, 73–84. [Google Scholar] [CrossRef]
- Chang, X.; Zhu, W.; Zhang, H.; Lian, S. Sensitization of melanoma cells to temozolomide by overexpression of microRNA 203 through direct targeting of glutaminase-mediated glutamine metabolism. Clin. Exp. Dermatol. 2017, 42, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Gentric, G.; Kieffer, Y.; Mieulet, V.; Goundiam, O.; Bonneau, C.; Nemati, F.; Hurbain, I.; Raposo, G.; Popova, T.; Stern, M.-H.; et al. PML-Regulated Mitochondrial Metabolism Enhances Chemosensitivity in Human Ovarian Cancers. Cell Metab. 2019, 29, 156–173.e10. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, C.; Kim, D.W.; Gombos, D.S.; Oba, J.; Qin, Y.; Williams, M.D.; Esmaeli, B.; Grimm, E.A.; Wargo, J.A.; Woodman, S.E.; et al. Uveal melanoma: From diagnosis to treatment and the science in between. Cancer 2016, 122, 2299–2312. [Google Scholar] [CrossRef] [PubMed]
- Ewens, K.G.; Kanetsky, P.A.; Richards-Yutz, J.; Purrazzella, J.; Shields, C.L.; Ganguly, T.; Ganguly, A. Chromosome 3 Status Combined with BAP1 and EIF1AX Mutation Profiles Are Associated With Metastasis in Uveal Melanoma. Investig. Opthalmol. Vis. Sci. 2014, 55, 5160–5167. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, C.; Oba, J.; Roszik, J.; Marszalek, J.R.; Chen, K.; Qi, Y.; Eterovic, K.; Robertson, A.G.; Burks, J.K.; McCannel, T.A.; et al. Elevated Endogenous SDHA Drives Pathological Metabolism in Highly Metastatic Uveal Melanoma. Investig. Opthalmol. Vis. Sci. 2019, 60, 4187–4195. [Google Scholar] [CrossRef] [PubMed]
- Teh, J.L.; Purwin, T.J.; Han, A.; Chua, V.; Patel, P.; Baqai, U.; Liao, C.; Bechtel, N.; Sato, T.; Davies, M.A.; et al. Metabolic Adaptations to MEK and CDK4/6 Cotargeting in Uveal Melanoma. Mol. Cancer Ther. 2020, 19, 1719–1726. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).