
A DNA Damage Response Gene Panel for Different Histologic Types of Epithelial Ovarian Carcinomas and Their Outcomes

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Specimens
2.2. The Panel of DNA Damage Repair Genes
2.3. Genomic DNA Extraction
2.4. Library Preparation, Next-Generation Sequencing, and Sequence Mapping
2.5. Variant Classification
2.6. Statistical Analysis
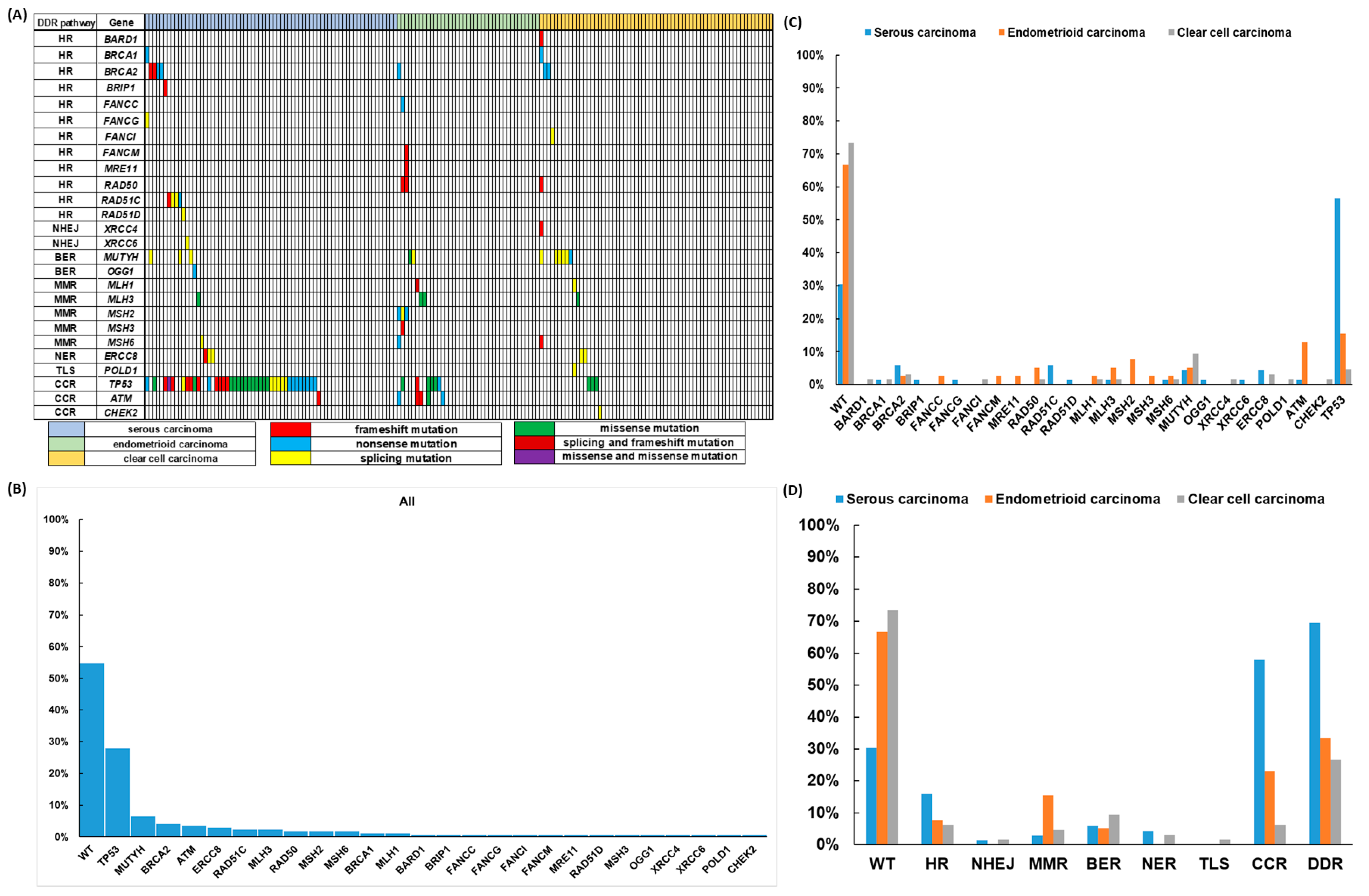
3. Results
3.1. Clinical Characteristics of the Patients
3.2. Deleterious DDR Gene Mutations
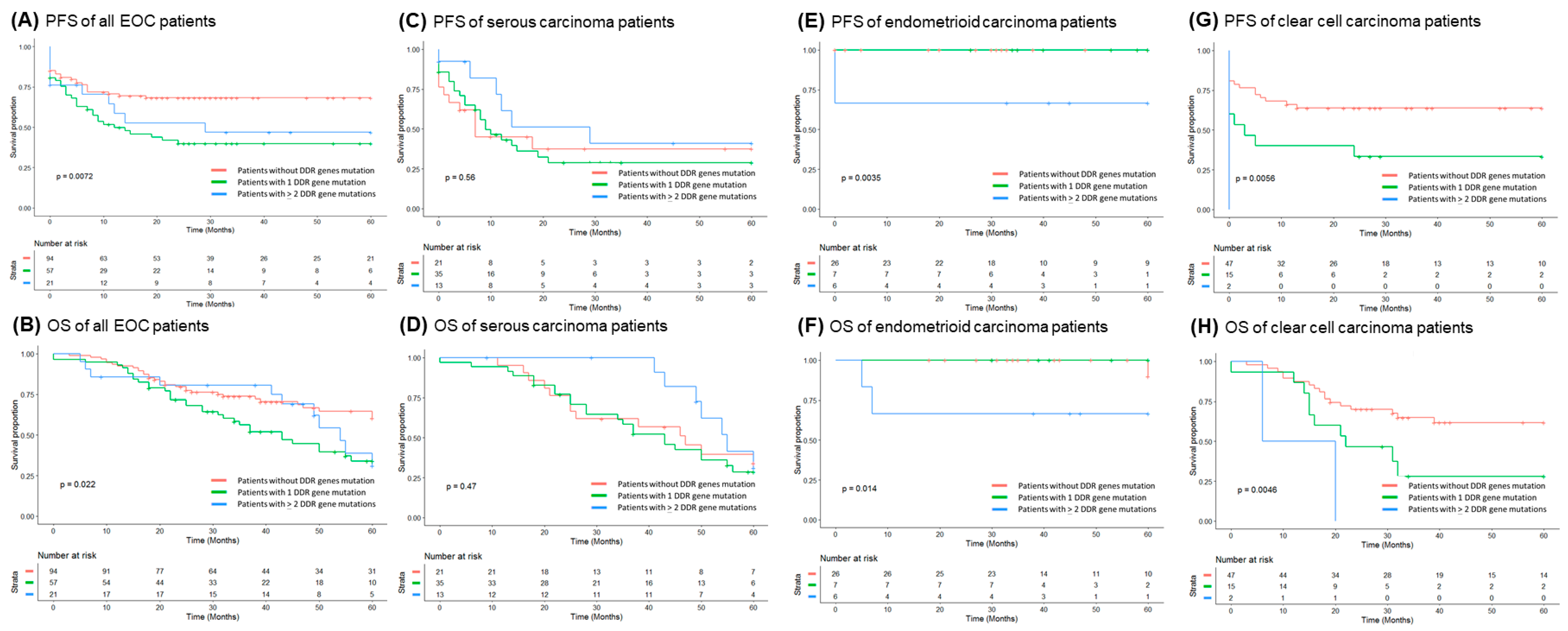
3.3. Correlation of DDR Gene Mutations with Clinical Outcomes of the EOC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Chiang, Y.C.; Chen, C.A.; Chiang, C.J.; Hsu, T.H.; Lin, M.C.; You, S.L.; Cheng, W.F.; Lai, M.S. Trends in incidence and survival outcome of epithelial ovarian cancer: 30-year national population-based registry in Taiwan. J. Gynecol. Oncol. 2013, 24, 342–351. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Coleman, R.L.; Monk, B.J.; Sood, A.K.; Herzog, T.J. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nat. Rev. Clin. Oncol. 2013, 10, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Gurung, A.; Hung, T.; Morin, J.; Gilks, C.B. Molecular abnormalities in ovarian carcinoma: Clinical, morphological and therapeutic correlates. Histopathology 2013, 62, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Colombo, P.E.; Fabbro, M.; Theillet, C.; Bibeau, F.; Rouanet, P.; Ray-Coquard, I. Sensitivity and resistance to treatment in the primary management of epithelial ovarian cancer. Crit. Rev. Oncol./Hematol. 2014, 89, 207–216. [Google Scholar] [CrossRef]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef]
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef]
- Prat, J. Staging classification for cancer of the ovary, fallopian tube, and peritoneum. Int. J. Gynaecol. Obstet. 2014, 124, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.H.; Kuo, W.H.; Huang, A.C.; Lu, Y.S.; Lin, C.H.; Kuo, S.H.; Wang, M.Y.; Liu, C.Y.; Cheng, F.T.; Yeh, M.H.; et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Mimori, T.; Nariai, N.; Kojima, K.; Takahashi, M.; Ono, A.; Sato, Y.; Yamaguchi-Kabata, Y.; Nagasaki, M. iSVP: An integrated structural variant calling pipeline from high-throughput sequencing data. BMC Syst. Biol. 2013, 7 (Suppl. S6), S8. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Mathe, E.; Olivier, M.; Kato, S.; Ishioka, C.; Hainaut, P.; Tavtigian, S.V. Computational approaches for predicting the biological effect of p53 missense mutations: A comparison of three sequence analysis based methods. Nucleic Acids Res. 2006, 34, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.B.; Pal, T.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Goggins, M.; Hutton, M.L.; et al. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2021, 19, 77–102. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Timms, K.M.; Carey, M.S.; Gutin, A.; Meyer, L.A.; Flake, D.D., 2nd; Abkevich, V.; Potter, J.; Pruss, D.; Glenn, P.; et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J. Clin. Oncol. 2010, 28, 3570–3576. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef]
- Leskela, S.; Romero, I.; Cristobal, E.; Pérez-Mies, B.; Rosa-Rosa, J.M.; Gutierrez-Pecharroman, A.; Caniego-Casas, T.; Santón, A.; Ojeda, B.; López-Reig, R.; et al. Mismatch Repair Deficiency in Ovarian Carcinoma: Frequency, Causes, and Consequences. Am. J. Surg. Pathol. 2020, 44, 649–656. [Google Scholar] [CrossRef]
- Tan, D.S.; Miller, R.E.; Kaye, S.B. New perspectives on molecular targeted therapy in ovarian clear cell carcinoma. Br. J. Cancer 2013, 108, 1553–1559. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.L.; Shih Ie, M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, C.; Ren, Y.; Yi, H.; Luo, T.; Xing, F.; Bai, X.; Cui, L.; Zhu, L.; Ouyang, J.; et al. Genomic characterization of Chinese ovarian clear cell carcinoma identifies driver genes by whole exome sequencing. Neoplasia 2020, 22, 399–430. [Google Scholar] [CrossRef]
- Kim, S.I.; Lee, J.W.; Lee, M.; Kim, H.S.; Chung, H.H.; Kim, J.W.; Park, N.H.; Song, Y.S.; Seo, J.S. Genomic landscape of ovarian clear cell carcinoma via whole exome sequencing. Gynecol. Oncol. 2018, 148, 375–382. [Google Scholar] [CrossRef]
- Murakami, R.; Matsumura, N.; Brown, J.B.; Higasa, K.; Tsutsumi, T.; Kamada, M.; Abou-Taleb, H.; Hosoe, Y.; Kitamura, S.; Yamaguchi, K.; et al. Exome Sequencing Landscape Analysis in Ovarian Clear Cell Carcinoma Shed Light on Key Chromosomal Regions and Mutation Gene Networks. Am. J. Pathol. 2017, 187, 2246–2258. [Google Scholar] [CrossRef]
- Gounaris, I.; Brenton, J.D. Molecular pathogenesis of ovarian clear cell carcinoma. Future Oncol. (Lond. Engl.) 2015, 11, 1389–1405. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.; Köbel, M.; Garsed, D.W.; Fereday, S.; Pandey, A.; Etemadmoghadam, D.; Hendley, J.; Kawabata, A.; Noguchi, D.; Yanaihara, N.; et al. Survival Following Chemotherapy in Ovarian Clear Cell Carcinoma Is Not Associated with Pathological Misclassification of Tumor Histotype. Clin. Cancer Res. 2019, 25, 3962–3973. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.F.; Tsai, E.M.; Chen, C.C.; Wu, C.C.; Er, T.K. Targeted sequencing of a specific gene panel detects a high frequency of ARID1A and PIK3CA mutations in ovarian clear cell carcinoma. Clin. Chim. Acta Int. J. Clin. Chem. 2019, 494, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, Y.; Tokunaga, H.; Saito, S.; Shimokawa, K.; Katsuoka, F.; Bin, L.; Kojima, K.; Nagasaki, M.; Yamamoto, M.; Yaegashi, N.; et al. Identification of somatic genetic alterations in ovarian clear cell carcinoma with next generation sequencing. Genes Chromosomes Cancer 2018, 57, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y.; Tanaka, N.; Ohira, M.; Itami, M.; Hippo, Y.; Nagase, H. Identification of novel mutations in Japanese ovarian clear cell carcinoma patients using optimized targeted NGS for clinical diagnosis. Gynecol. Oncol. 2017, 144, 377–383. [Google Scholar] [CrossRef]
- Friedlander, M.L.; Russell, K.; Millis, S.; Gatalica, Z.; Bender, R.; Voss, A. Molecular Profiling of Clear Cell Ovarian Cancers: Identifying Potential Treatment Targets for Clinical Trials. Int. J. Gynecol. Cancer 2016, 26, 648–654. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Norquist, B.; Lacchetti, C.; Armstrong, D.; Grisham, R.N.; Goodfellow, P.J.; Kohn, E.C.; Levine, D.A.; Liu, J.F.; Lu, K.H.; et al. Germline and Somatic Tumor Testing in Epithelial Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 1222–1245. [Google Scholar] [CrossRef]
- Vergote, I.; Banerjee, S.; Gerdes, A.M.; van Asperen, C.; Marth, C.; Vaz, F.; Ray-Coquard, I.; Stoppa-Lyonnet, D.; Gonzalez-Martin, A.; Sehouli, J.; et al. Current perspectives on recommendations for BRCA genetic testing in ovarian cancer patients. Eur. J. Cancer 2016, 69, 127–134. [Google Scholar] [CrossRef]
- Lancaster, J.M.; Powell, C.B.; Chen, L.M.; Richardson, D.L. Society of Gynecologic Oncology statement on risk assessment for inherited gynecologic cancer predispositions. Gynecol. Oncol. 2015, 136, 3–7. [Google Scholar] [CrossRef]
- Norquist, B.M.; Brady, M.F.; Harrell, M.I.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Burger, R.A.; Tewari, K.S.; et al. Mutations in Homologous Recombination Genes and Outcomes in Ovarian Carcinoma Patients in GOG 218: An NRG Oncology/Gynecologic Oncology Group Study. Clin. Cancer Res. 2018, 24, 777–783. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Bonnet, C.; Krieger, S.; Vezain, M.; Rousselin, A.; Tournier, I.; Martins, A.; Berthet, P.; Chevrier, A.; Dugast, C.; Layet, V.; et al. Screening BRCA1 and BRCA2 unclassified variants for splicing mutations using reverse transcription PCR on patient RNA and an ex vivo assay based on a splicing reporter minigene. J. Med. Genet. 2008, 45, 438–446. [Google Scholar] [CrossRef]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef]
- Li, H.; LaDuca, H.; Pesaran, T.; Chao, E.C.; Dolinsky, J.S.; Parsons, M.; Spurdle, A.B.; Polley, E.C.; Shimelis, H.; Hart, S.N.; et al. Classification of variants of uncertain significance in BRCA1 and BRCA2 using personal and family history of cancer from individuals in a large hereditary cancer multigene panel testing cohort. Genet. Med. 2020, 22, 701–708. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Toland, A.E.; Forman, A.; Couch, F.J.; Culver, J.O.; Eccles, D.M.; Foulkes, W.D.; Hogervorst, F.B.L.; Houdayer, C.; Levy-Lahad, E.; Monteiro, A.N.; et al. Clinical testing of BRCA1 and BRCA2: A worldwide snapshot of technological practices. NPJ Genom. Med. 2018, 3, 7. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | DDR Pathway | Gene | DDR Pathway |
|---|---|---|---|
| ATM | CCR | ku70/XRCC6 | NHEJ |
| BARD1 | HR | ku80/XRCC5 | NHEJ |
| BRCA1 | HR | MDM4 | CCR |
| BRCA2/FANCD1 | HR | MLH1 | MMR |
| BRIP1/FANCJ | HR | MLH3 | MMR |
| CHEK2 | CCR | MRE11 | HR |
| DDB1 | NER | MSH2 | MMR |
| DDB2 | NER | MSH3 | MMR |
| ERCC1 | NER | MSH6 | MMR |
| ERCC2/XPD | NER | MUTYH | BER |
| ERCC3/XPB | NER | NBN | HR |
| ERCC4 | NER | NBS1 | HR |
| ERCC5/BIVM | NER | OGG1 | BER |
| ERCC6/CSB | NER | PMS1 | MMR |
| ERCC8/CSA | NER | PMS2 | MMR |
| FANCA | HR | POLD1 | TLS |
| FANCB | HR | POLE | TLS |
| FANCC | HR | POLB | TLS |
| FANCD1/BRCA2 | HR | POLH | TLS |
| FANCD2 | HR | POLK | TLS |
| FANCE | HR | RAD50 | HR |
| FANCF | HR | RAD51 | HR |
| FANCG/XRCC | HR | RAD51C/FANCO | HR |
| FANCI | HR | RAD51D | HR |
| FANCJ/BRIP1 | HR | TP53 | CCR |
| FANCL/PHF9 | HR | XPA | NER |
| FANCM | HR | XPC | NER |
| FANCN/PALB2 | HR | XRCC2 | NHEJ |
| FANCO/RAD51C | HR | XRCC3 | NHEJ |
| FANCP/SLX4 | HR | XRCC4 | NHEJ |
| Patient Numbers | 172 |
|---|---|
| Median Age (years old) | 52 (29–85) |
| Median CA 125 (U/mL) | 400 (12–7265) |
| Histology | |
| Serous carcinoma | 69 (40.1%) |
| Endometrioid carcinoma | 39 (22.7%) |
| Clear cell carcinoma | 64 (37.2%) |
| FIGO stage | |
| Early | 69 (40.1%) |
| Advanced | 103 (59.9%) |
| Grade | |
| Low | 29 (16.9%) |
| High | 143 (83.1%) |
| Debulking surgery | |
| Optimal | 112 (65.1%) |
| Suboptimal | 60 (34.9%) |
| Recurrence | |
| Yes | 102 (59.3%) |
| No | 70 (40.7%) |
| Death | |
| Yes | 76 (44.2%) |
| No | 96 (55.8%) |
| Gene | Mutation | Transcript | gDNA/cDNA | Amino Acid | Reported/Novel |
|---|---|---|---|---|---|
| ATM | frameshift deletion | NM_000051 | c.1402_1403del | p.K468fs | rs587781347 |
| ATM | frameshift deletion | NM_000051 | c.8426delA | p.Q2809fs | rs587782558 |
| ATM | frameshift insertion | NM_000051 | c.4736dupA | p.Q1579fs | rs864622164 |
| ATM | missense mutation | NM_000051 | c.C6200A | p.A2067D | rs397514577 |
| ATM | nonsense mutation | NM_000051 | c.C5188T | p.R1730X | rs764389018 |
| ATM | nonsense mutation | NM_000051 | c.C850T | p.Q284X | rs757782702 |
| BARD1 | frameshift insertion | NM_000465 | c.70_71insGT | p.P24fs | NA |
| BRCA1 | nonsense mutation | NM_007294 | c.3531dupT | p.S1178_K1179delinsX | NA |
| BRCA1 | nonsense mutation | NM_007294 | c.G2635T | p.E879X | rs80357251 |
| BRCA2 | frameshift deletion | NM_000059 | c.1585delT | p.F529fs | NA |
| BRCA2 | frameshift insertion | NM_000059 | c.7407dupT | p.T2469fs | rs397507916 |
| BRCA2 | nonsense mutation | NM_000059 | c.4965delC | p.Y1655X | rs80359475 |
| BRCA2 | nonsense mutation | NM_000059 | c.A5623T | p.K1875X | NA |
| BRCA2 | nonsense mutation | NM_000059 | c.C2590T | p.Q864X | rs1060502414 |
| BRCA2 | nonsense mutation | NM_000059 | c.C6952T | p.R2318X | rs80358920 |
| BRCA2 | nonsense mutation | NM_000059 | c.G3922T | p.E1308X | rs80358638 |
| BRIP1 | frameshift insertion | NM_032043 | c.394dupA | p.T132fs | rs587781416 |
| CHEK2 | splicing | NM_007194 | g. 29130716 C>G | NA | |
| ERCC8 | frameshift deletion | NM_000082 | c.191_195del | p.S64fs | NA |
| ERCC8 | splicing | NM_000082 | c.1123-2->T | NA | |
| ERCC8 | splicing | NM_000082 | c.1123-2->T | NA | |
| ERCC8 | splicing | NM_000082 | c.1123-2->T | NA | |
| ERCC8 | splicing | NM_000082 | c.1123-2->T | rs777444521 | |
| FANCC | nonsense mutation | NM_000136 | c.G1225T | p.E409X | NA |
| FANCG | splicing | NM_004629 | c.511-2->C | NA | |
| FANCI | splicing | NM_001113378 | c.3187-2A>G | NA | |
| FANCM | frameshift deletion | NM_020937 | c.3998delA | p.Q1333fs | rs746983128 |
| MLH1 | frameshift deletion | NM_000249 | c.1771delG | p.D591fs | NA |
| MLH1 | splicing | NM_000249 | c.2104-2A>G | rs267607889 | |
| MLH1 | splicing | NM_000249 | c.790+2T>C | rs267607790 | |
| MLH3 | missense mutation | NM_001040108 | c.G2221T | p.V741F | rs28756990 |
| MLH3 | missense mutation | NM_001040108 | c.G2221T | p.V741F | rs28756990 |
| MLH3 | missense mutation | NM_001040108 | c.G2221T | p.V741F | rs28756990 |
| MLH3 | missense mutation | NM_001040108 | c.G2221T | p.V741F | rs28756990 |
| MRE11 | frameshift insertion | NM_005590 | c.1222dupA | p.T408fs | rs774440500 |
| MSH2 | nonsense mutation | NM_000251 | c.C226T | p.Q76X | rs63750042 |
| MSH2 | nonsense mutation | NM_000251 | c.G1738T | p.E580X | rs63751411 |
| MSH2 | splicing | NM_000251 | c.943-1G>C | rs12476364 | |
| MSH3 | frameshift deletion | NM_002439 | c.1141delA | p.K381fs | rs587776701 |
| MSH6 | frameshift insertion | NM_001281492 | c.2916dupT | p.T972fs | NA |
| MSH6 | nonsense mutation | NM_001281492 | c.G726A | p.W242X | NA |
| MSH6 | splicing | NM_001281492 | g. 48033792 _ 48033795 del TAAC | NA | |
| MUTYH | missense mutation | NM_001128425 | c.G1187A | p.G396D | rs36053993 |
| MUTYH | nonsense mutation | NM_001128425 | c.G467A | p.W156X | rs762307622 |
| MUTYH | splicing | NM_001128425 | c.576+1G>C | NA | |
| MUTYH | splicing | NM_001128425 | c.934-2A>G | rs77542170 | |
| MUTYH | splicing | NM_001128425 | c.934-2A>G | rs77542170 | |
| MUTYH | splicing | NM_001128425 | c.934-2A>G | rs77542170 | |
| MUTYH | splicing | NM_001128425 | c.934-2A>G | rs77542170 | |
| MUTYH | splicing | NM_001128425 | c.934-2A>G | rs77542170 | |
| MUTYH | splicing | NM_001128425 | c.934-2A>G | rs77542170 | |
| MUTYH | splicing | NM_001128425 | c.934-2A>G | rs77542170 | |
| MUTYH | splicing | NM_001128425 | c.934-2A>G | rs77542170 | |
| OGG1 | nonsense mutation | NM_016819 | c.A974G | p.X325W | NA |
| POLD1 | splicing | NM_002691 | c.2954-1G>- | NA | |
| RAD50 | frameshift deletion | NM_005732 | c.2157delA | p.L719fs | NA |
| RAD50 | frameshift deletion | NM_005732 | c.536delT | p.I179fs | NA |
| RAD50 | frameshift insertion | NM_005732 | exon13:c.2157dupA | p.L719fs | rs397507178 |
| RAD51C | frameshift insertion | NM_058216 | c.390dupA | p.G130fs | rs730881940 |
| RAD51C | nonsense mutation | NM_058216 | c.T833G | p.L278X | NA |
| RAD51C | splicing | NM_058216 | c.905-2A>C | NA | |
| RAD51C | splicing | NM_058216 | c.905-2A>C | NA | |
| RAD51D | splicing | NM_002878 | c.480+1G>A | NA | |
| TP53 | frameshift deletion | NM_000546 | c.102delC | p.P34fs | NA |
| TP53 | frameshift deletion | NM_000546 | c.121delG | p.D41fs | NA |
| TP53 | frameshift deletion | NM_000546 | c.216delC | p.P72fs | NA |
| TP53 | frameshift deletion | NM_000546 | c.257_272del | p.A86fs | NA |
| TP53 | frameshift deletion | NM_000546 | c.501delG | p.Q167fs | NA |
| TP53 | frameshift deletion | NM_000546 | c.539_549del | p.E180fs | NA |
| TP53 | frameshift insertion | NM_000546 | c.102dupC | p.L35fs | NA |
| TP53 | frameshift insertion | NM_000546 | c.455dupC | p.P152fs | NA |
| TP53 | frameshift insertion | NM_000546 | c.498dupA | p.Q167fs | NA |
| TP53 | frameshift insertion | NM_000546 | c.889dupC | p.H297fs | NA |
| TP53 | missense mutation | NM_000546 | c.A488G | p.Y163C | rs148924904 |
| TP53 | missense mutation | NM_000546 | c.A578G | p.H193R | rs786201838 |
| TP53 | missense mutation | NM_000546 | c.A659C | p.Y220S | rs121912666 |
| TP53 | missense mutation | NM_000546 | c.A659G | p.Y220C | rs121912666 |
| TP53 | missense mutation | NM_000546 | c.A659G | p.Y220C | rs121912666 |
| TP53 | missense mutation | NM_000546 | c.A736G | p.M246V | rs483352695 |
| TP53 | missense mutation | NM_000546 | c.A838G | p.R280G | rs753660142 |
| TP53 | missense mutation | NM_000546 | c.C380T | p.S127F | rs730881999 |
| TP53 | missense mutation | NM_000546 | c.C451T | p.P151S | rs28934874 |
| TP53 | missense mutation | NM_000546 | c.C844T | p.R282W | rs28934574 |
| TP53 | missense mutation | NM_000546 | c.C844T | p.R282W | rs28934574 |
| TP53 | missense mutation | NM_000546 | c.G412C | p.A138P | rs28934875 |
| TP53 | missense mutation | NM_000546 | c.G524A | p.R175H | rs28934578 |
| TP53 | missense mutation | NM_000546 | c.G524A | p.R175H | rs28934578 |
| TP53 | missense mutation | NM_000546 | c.G638T | p.R213L | rs587778720 |
| TP53 | missense mutation | NM_000546 | c.G730A | p.G244S | rs1057519989 |
| TP53 | missense mutation | NM_000546 | c.G743A | p.R248Q | rs11540652 |
| TP53 | missense mutation | NM_000546 | c.G743A | p.R248Q | rs11540652 |
| TP53 | missense mutation | NM_000546 | c.G818A | p.R273H | rs28934576 |
| TP53 | missense mutation | NM_000546 | c.G818A | p.R273H | rs28934576 |
| TP53 | missense mutation | NM_000546 | c.G836A | p.G279E | rs1064793881 |
| TP53 | missense mutation | NM_000546 | c.G856A | p.E286K | rs786201059 |
| TP53 | nonsense mutation | NM_000546 | c.588_589insTGA | p.V197delinsX | NA |
| TP53 | nonsense mutation | NM_000546 | c.912dupT | p.K305_R306delinsX | NA |
| TP53 | nonsense mutation | NM_000546 | c.C430T | p.Q144X | NA |
| TP53 | nonsense mutation | NM_000546 | c.C499T | p.Q167X | NA |
| TP53 | nonsense mutation | NM_000546 | c.C574T | p.Q192X | NA |
| TP53 | nonsense mutation | NM_000546 | c.C586T | p.R196X | rs397516435 |
| TP53 | nonsense mutation | NM_000546 | c.C637T | p.R213X | rs397516436 |
| TP53 | nonsense mutation | NM_000546 | c.G272A | p.W91X | NA |
| TP53 | nonsense mutation | NM_000546 | c.G438A | p.W146X | NA |
| TP53 | nonsense mutation | NM_000546 | c.G859T | p.E287X | NA |
| TP53 | nonsense mutation | NM_000546 | c.G880T | p.E294X | rs1057520607 |
| TP53 | splicing | NM_000546 | c.376-1G>T | NA | |
| TP53 | splicing | NM_000546 | c.672+1G>A | rs863224499 | |
| TP53 | splicing | NM_000546 | c.993+2T>G | NA | |
| TP53 | splicing | NM_000546 | c.993+2T>G | NA | |
| TP53 | splicing | NM_000546 | c.993+1G>T | rs11575997 | |
| TP53 | splicing | NM_000546 | g.7577493_7577497 del CCTGA | NA | |
| XRCC4 | frameshift deletion | NM_003401 | c.810delA | p.R270fs | NA |
| XRCC6 | splicing | NM_001469 | c.589+1G>T | NA |
| Genes | Histology | Type | FIGO Stage | Tumor Grade | Recurrence | Death | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OSA | OEA | OCCA | I | II | Early | Advanced | Low | High | No | Yes | No | Yes | ||
| Total | 172 | 69 | 39 | 64 | 104 | 68 | 69 | 103 | 29 | 143 | 70 | 102 | 96 | 76 |
| HR | ||||||||||||||
| Wild type | 154 | 58 | 36 | 60 | 97 | 57 | 64 | 90 | 26 | 128 | 64 | 90 | 89 | 65 |
| 89.53% | 84.06% | 92.31% | 93.75% | 93.27% | 83.82% | 92.75% | 87.38% | 89.66% | 89.51% | 91.43% | 88.24% | 92.71% | 85.53% | |
| Mutation | 18 | 11 | 3 | 4 | 7 | 11 | 5 | 13 | 3 | 15 | 6 | 12 | 7 | 11 |
| 10.47% | 15.94% | 7.69% | 6.25% | 6.73% | 16.18% | 7.25% | 12.62% | 10.34% | 10.49% | 8.57% | 11.76% | 7.29% | 14.47% | |
| p value * | 0.154 | 0.048 | 0.259 | 0.981 | 0.502 | 0.126 | ||||||||
| NHEJ | ||||||||||||||
| Wild type | 170 | 68 | 39 | 63 | 103 | 67 | 69 | 101 | 29 | 141 | 70 | 100 | 96 | 74 |
| 98.84% | 98.55% | 100.00% | 98.44% | 99.04% | 98.53% | 100.00% | 98.06% | 100.00% | 98.60% | 100.00% | 98.04% | 100.00% | 97.37% | |
| Mutation | 2 | 1 | 0 | 1 | 1 | 1 | 0 | 2 | 0 | 2 | 0 | 2 | 0 | 2 |
| 1.16% | 1.45% | 0.00% | 1.56% | 0.96% | 1.47% | 0.00% | 1.94% | 0.00% | 1.40% | 0.00% | 1.96% | 0.00% | 2.63% | |
| p value* | 0.742 | 0.761 | 0.244 | 0.522 | 0.239 | 0.11 | ||||||||
| MMR | ||||||||||||||
| Wild type | 161 | 67 | 33 | 61 | 95 | 66 | 65 | 96 | 24 | 137 | 66 | 95 | 91 | 70 |
| 93.60% | 97.10% | 84.62% | 95.31% | 91.35% | 97.06% | 94.20% | 93.20% | 82.76% | 95.80% | 94.29% | 93.14% | 94.79% | 92.11% | |
| Mutation | 11 | 2 | 6 | 3 | 9 | 2 | 4 | 7 | 5 | 6 | 4 | 7 | 5 | 6 |
| 6.40% | 2.90% | 15.38% | 4.69% | 8.65% | 2.94% | 5.80% | 6.80% | 17.24% | 4.20% | 5.71% | 6.86% | 5.21% | 7.89% | |
| p value * | 0.03 | 0.134 | 0.793 | 0.009 | 0.762 | 0.475 | ||||||||
| BER | ||||||||||||||
| Wild type | 160 | 65 | 37 | 58 | 96 | 64 | 65 | 95 | 27 | 133 | 66 | 94 | 91 | 69 |
| 93.02% | 94.20% | 94.87% | 90.63% | 92.31% | 94.12% | 94.20% | 92.23% | 93.10% | 93.01% | 94.29% | 92.16% | 94.79% | 90.79% | |
| Mutation | 12 | 4 | 2 | 6 | 8 | 4 | 4 | 8 | 2 | 10 | 4 | 8 | 5 | 7 |
| 6.98% | 5.80% | 5.13% | 9.38% | 7.69% | 5.88% | 5.80% | 7.77% | 6.90% | 6.99% | 5.71% | 7.84% | 5.21% | 9.21% | |
| p value * | 0.631 | 0.649 | 0.619 | 0.985 | 0.59 | 0.306 | ||||||||
| NER | ||||||||||||||
| Wild type | 167 | 66 | 39 | 62 | 102 | 65 | 67 | 100 | 29 | 138 | 67 | 100 | 93 | 74 |
| 97.09% | 95.65% | 100.00% | 96.88% | 98.08% | 95.59% | 97.10% | 97.09% | 100.00% | 96.50% | 95.71% | 98.04% | 96.88% | 97.37% | |
| Mutation | 5 | 3 | 0 | 2 | 2 | 3 | 2 | 3 | 0 | 5 | 3 | 2 | 3 | 2 |
| 2.91% | 4.35% | 0.00% | 3.13% | 1.92% | 4.41% | 2.90% | 2.91% | 0.00% | 3.50% | 4.29% | 1.96% | 3.13% | 2.63% | |
| p value * | 0.43 | 0.342 | 0.996 | 0.307 | 0.373 | 0.848 | ||||||||
| TLS | ||||||||||||||
| Wild type | 171 | 69 | 39 | 63 | 103 | 68 | 69 | 102 | 29 | 142 | 70 | 101 | 96 | 75 |
| 99.42% | 100.00% | 100.00% | 98.44% | 99.04% | 100.00% | 100.00% | 99.03% | 100.00% | 99.30% | 100.00% | 99.02% | 100.00% | 98.68% | |
| Mutation | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 |
| 0.58% | 0.00% | 0.00% | 1.56% | 0.96% | 0.00% | 0.00% | 0.97% | 0.00% | 0.70% | 0.00% | 0.98% | 0.00% | 1.32% | |
| p value * | 0.428 | 0.417 | 0.412 | 0.652 | 0.406 | 0.26 | ||||||||
| DSBR | ||||||||||||||
| Wild type | 153 | 57 | 36 | 60 | 97 | 56 | 64 | 89 | 26 | 127 | 64 | 89 | 89 | 64 |
| 88.95% | 82.61% | 92.31% | 93.75% | 93.27% | 82.35% | 92.75% | 86.41% | 89.66% | 88.81% | 91.43% | 87.25% | 92.71% | 84.21% | |
| Mutation | 19 | 12 | 3 | 4 | 7 | 12 | 5 | 14 | 3 | 16 | 6 | 13 | 7 | 12 |
| 11.05% | 17.39% | 7.69% | 6.25% | 6.73% | 17.65% | 7.25% | 13.59% | 10.34% | 11.19% | 8.57% | 12.75% | 7.29% | 15.79% | |
| p value * | 0.092 | 0.026 | 0.193 | 0.895 | 0.391 | 0.077 | ||||||||
| SSBR | ||||||||||||||
| Wild type | 145 | 60 | 31 | 54 | 86 | 59 | 59 | 86 | 22 | 123 | 59 | 86 | 83 | 62 |
| 84.30% | 86.96% | 79.49% | 84.38% | 82.69% | 86.76% | 85.51% | 83.50% | 75.86% | 86.01% | 84.29% | 84.31% | 86.46% | 81.58% | |
| Mutation | 27 | 9 | 8 | 10 | 18 | 9 | 10 | 17 | 7 | 20 | 11 | 16 | 13 | 14 |
| 15.70% | 13.04% | 20.51% | 15.63% | 17.31% | 13.24% | 14.49% | 16.50% | 24.14% | 13.99% | 15.71% | 15.69% | 13.54% | 18.42% | |
| p value * | 0.591 | 0.473 | 0.722 | 0.171 | 0.996 | 0.382 | ||||||||
| CCR | ||||||||||||||
| Wild type | 119 | 29 | 30 | 60 | 91 | 28 | 60 | 59 | 24 | 95 | 57 | 62 | 74 | 45 |
| 69.19% | 42.03% | 76.92% | 93.75% | 87.50% | 41.18% | 86.96% | 57.28% | 82.76% | 66.43% | 81.43% | 60.78% | 77.08% | 59.21% | |
| Mutation | 53 | 40 | 9 | 4 | 13 | 40 | 9 | 44 | 5 | 48 | 13 | 40 | 22 | 31 |
| 30.81% | 57.97% | 23.08% | 6.25% | 12.50% | 58.82% | 13.04% | 42.72% | 17.24% | 33.57% | 18.57% | 39.22% | 22.92% | 40.79% | |
| p value * | <0.001 | <0.001 | <0.001 | 0.083 | 0.004 | 0.012 | ||||||||
| DDR | ||||||||||||||
| Wild type | 94 | 21 | 26 | 47 | 74 | 20 | 50 | 44 | 20 | 74 | 47 | 47 | 63 | 31 |
| 54.65% | 30.43% | 66.67% | 73.44% | 71.15% | 29.41% | 72.46% | 42.72% | 68.97% | 51.75% | 67.14% | 46.08% | 65.63% | 40.79% | |
| 1 gene | 57 | 35 | 7 | 15 | 22 | 35 | 14 | 43 | 5 | 52 | 16 | 41 | 24 | 33 |
| mutation | 33.14% | 50.72% | 17.95% | 23.44% | 21.15% | 51.47% | 20.29% | 41.75% | 17.24% | 36.36% | 22.86% | 40.20% | 25.00% | 43.42% |
| 2 gene | 15 | 12 | 2 | 1 | 3 | 12 | 2 | 13 | 0 | 15 | 4 | 11 | 6 | 9 |
| mutations | 8.72% | 17.39% | 5.13% | 1.56% | 2.88% | 17.65% | 2.90% | 12.62% | 0.00% | 10.49% | 5.71% | 10.78% | 6.25% | 11.84% |
| 3 gene | 2 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| mutations | 1.16% | 1.45% | 2.56% | 0.00% | 0.96% | 1.47% | 1.45% | 0.97% | 3.45% | 0.70% | 1.43% | 0.98% | 1.04% | 1.32% |
| 4 gene | 2 | 0 | 2 | 0 | 2 | 0 | 2 | 0 | 2 | 0 | 2 | 0 | 2 | 0 |
| mutations | 1.16% | 0.00% | 5.13% | 0.00% | 1.92% | 0.00% | 2.90% | 0.00% | 6.90% | 0.00% | 2.86% | 0.00% | 2.08% | 0.00% |
| 5 gene | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 1 |
| Mutations | 0.58% | 0.00% | 2.56% | 0.00% | 0.96% | 0.00% | 0.00% | 0.97% | 3.45% | 0.00% | 0.00% | 0.98% | 0.00% | 1.32% |
| 6 gene | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 |
| mutations | 0.58% | 0.00% | 0.00% | 1.56% | 0.96% | 0.00% | 0.00% | 0.97% | 0.00% | 0.70% | 0.00% | 0.98% | 0.00% | 1.32% |
| Total | 78 | 48 | 13 | 17 | 30 | 48 | 19 | 59 | 9 | 69 | 23 | 55 | 33 | 45 |
| mutations | 45.35% | 69.57% | 33.33% | 26.56% | 28.85% | 70.59% | 27.54% | 57.28% | 31.03% | 48.25% | 32.86% | 53.92% | 34.38% | 59.21% |
| p value * | <0.001 | <0.001 | <0.001 | 0.089 | 0.006 | 0.001 | ||||||||
| Factors | Recurrence | Death | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | ||||||
| n | Hazard Ratio (95% CI) | p | Hazard Ratio (95% CI) | p | Hazard Ratio (95% CI) | p | Hazard Ratio (95% CI) | p | |
| Histology | |||||||||
| OSA | 69 | 1 (reference) | 1 (reference) | 1 (reference) | 1 (reference) | ||||
| OEA | 39 | 0.17 (0.08–0.37) | <0.001 | 0.42 (0.16–1.12) | 0.082 | 0.12 (0.04–0.38) | <0.001 | 0.45 (0.13–1.55) | 0.205 |
| OCCA | 64 | 0.96 (0.64–1.44) | 0.835 | 1.37 (0.86–2.18) | 0.188 | ||||
| Type | |||||||||
| I | 104 | 1 (reference) | 1 (reference) | 1 (reference) | 1 (reference) | ||||
| II | 68 | 2.69 (1.81–4.00) | <0.001 | 0.77 (0.46–1.28) | 0.311 | 1.88 (1.19–2.96) | 0.007 | 0.35 (0.20–0.60) | <0.001 |
| FIGO stage | |||||||||
| Early | 69 | 1 (reference) | 1 (reference) | 1 (reference) | 1 (reference) | ||||
| Advanced | 103 | 5.29 (3.16–8.85) | <0.001 | 3.08 (1.63–5.80) | 0.001 | 6.84 (3.28–14.25) | <0.001 | 4.82 (2.09–11.09) | <0.001 |
| Tumor grade | |||||||||
| Low | 29 | 1 (reference) | 1 (reference) | 1 (reference) | 1 (reference) | ||||
| High | 143 | 5.57 (2.26–13.70) | <0.001 | 1.68 (0.55–5.15) | 0.366 | 17.97 (2.50–129.29) | 0.004 | 7.38 (0.93–58.28) | 0.058 |
| Debulking surgery | |||||||||
| Suboptimal | 60 | 1 (reference) | 1 (reference) | 1 (reference) | 1 (reference) | ||||
| Optimal | 112 | 0.28 (0.18–0.41) | <0.001 | 0.51 (0.32–0.80) | 0.004 | 0.26 (0.16–0.41) | <0.001 | 0.38 (0.22–0.64) | <0.001 |
| HR | |||||||||
| Wild type | 154 | 1 (reference) | 1 (reference) | ||||||
| Mutation | 18 | 1.22 (0.67–2.23) | 0.516 | 1.15 (0.59–2.25) | 0.674 | ||||
| NHEJ | |||||||||
| Wild type | 170 | 1 (reference) | 1 (reference) | ||||||
| Mutation | 2 | 2.04 (0.50–8.28) | 0.319 | 2.52 (0.62–10.32) | 0.197 | ||||
| MMR | |||||||||
| Wild type | 161 | 1 (reference) | 1 (reference) | ||||||
| Mutation | 11 | 1.31 (0.61–2.83) | 0.487 | 1.88 (0.81–4.33) | 0.139 | ||||
| BER | |||||||||
| Wild type | 160 | 1 (reference) | 1 (reference) | ||||||
| Mutation | 12 | 1.32 (0.64–2.71) | 0.454 | 1.70 (0.78–3.72) | 0.185 | ||||
| NER | |||||||||
| Wild type | 167 | 1 (reference) | 1 (reference) | ||||||
| Mutation | 5 | 0.58 (0.14–2.36) | 0.449 | 0.71 (0.18–2.91) | 0.639 | ||||
| TLS | |||||||||
| Wild type | 171 | 1 (reference) | 1 (reference) | 1 (reference) | |||||
| Mutation | 1 | 5.19 (0.71–37.89) | 0.104 | 33.76 (3.95–289.00) | 0.001 | 9.57 (1.08–84.83) | 0.042 | ||
| DSBR | |||||||||
| Wild type | 153 | 1 (reference) | 1 (reference) | ||||||
| Mutation | 19 | 1.23 (0.69–2.20) | 0.488 | 1.20 (0.63–2.27) | 0.584 | ||||
| SSBR | |||||||||
| Wild type | 145 | 1 (reference) | 1 (reference) | ||||||
| Mutation | 27 | 1.10 (0.64–1.87) | 0.736 | 1.46 (0.82–2.61) | 0.202 | ||||
| CCR | |||||||||
| Wild type | 119 | 1 (reference) | 1 (reference) | 1 (reference) | |||||
| Mutation | 53 | 1.68 (1.12–2.50) | 0.011 | 0.98 (0.58–1.66) | 0.939 | 1.54 (0.97–2.45) | 0.066 | ||
| DDR | |||||||||
| Wild type | 94 | 1 (reference) | 1 (reference) | 1 (reference) | 1 (reference) | ||||
| 1 gene mutation | 57 | 1.71 (1.12–2.60) | 0.013 | 1.18 (0.73–1.91) | 0.496 | 1.96 (1.20–3.20) | 0.007 | 1.57 (0.97–2.54) | 0.062 |
| ≥2 gene mutations | 21 | 1.52 (0.84–2.76) | 0.171 | 1.56 (0.78–3.11) | 0.207 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, Y.-C.; Lin, P.-H.; Lu, T.-P.; Kuo, K.-T.; Tai, Y.-J.; Hsu, H.-C.; Wu, C.-Y.; Lee, C.-Y.; Shen, H.; Chen, C.-A.; et al. A DNA Damage Response Gene Panel for Different Histologic Types of Epithelial Ovarian Carcinomas and Their Outcomes. Biomedicines 2021, 9, 1384. https://doi.org/10.3390/biomedicines9101384
Chiang Y-C, Lin P-H, Lu T-P, Kuo K-T, Tai Y-J, Hsu H-C, Wu C-Y, Lee C-Y, Shen H, Chen C-A, et al. A DNA Damage Response Gene Panel for Different Histologic Types of Epithelial Ovarian Carcinomas and Their Outcomes. Biomedicines. 2021; 9(10):1384. https://doi.org/10.3390/biomedicines9101384
Chicago/Turabian StyleChiang, Ying-Cheng, Po-Han Lin, Tzu-Pin Lu, Kuan-Ting Kuo, Yi-Jou Tai, Heng-Cheng Hsu, Chia-Ying Wu, Chia-Yi Lee, Hung Shen, Chi-An Chen, and et al. 2021. "A DNA Damage Response Gene Panel for Different Histologic Types of Epithelial Ovarian Carcinomas and Their Outcomes" Biomedicines 9, no. 10: 1384. https://doi.org/10.3390/biomedicines9101384
APA StyleChiang, Y.-C., Lin, P.-H., Lu, T.-P., Kuo, K.-T., Tai, Y.-J., Hsu, H.-C., Wu, C.-Y., Lee, C.-Y., Shen, H., Chen, C.-A., & Cheng, W.-F. (2021). A DNA Damage Response Gene Panel for Different Histologic Types of Epithelial Ovarian Carcinomas and Their Outcomes. Biomedicines, 9(10), 1384. https://doi.org/10.3390/biomedicines9101384