
Chronic Mexiletine Administration Increases Sodium Current in Non-Diseased Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes


 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of hiPSC-CMs
2.2. Drug Incubation
2.3. Patch Clamp Analysis
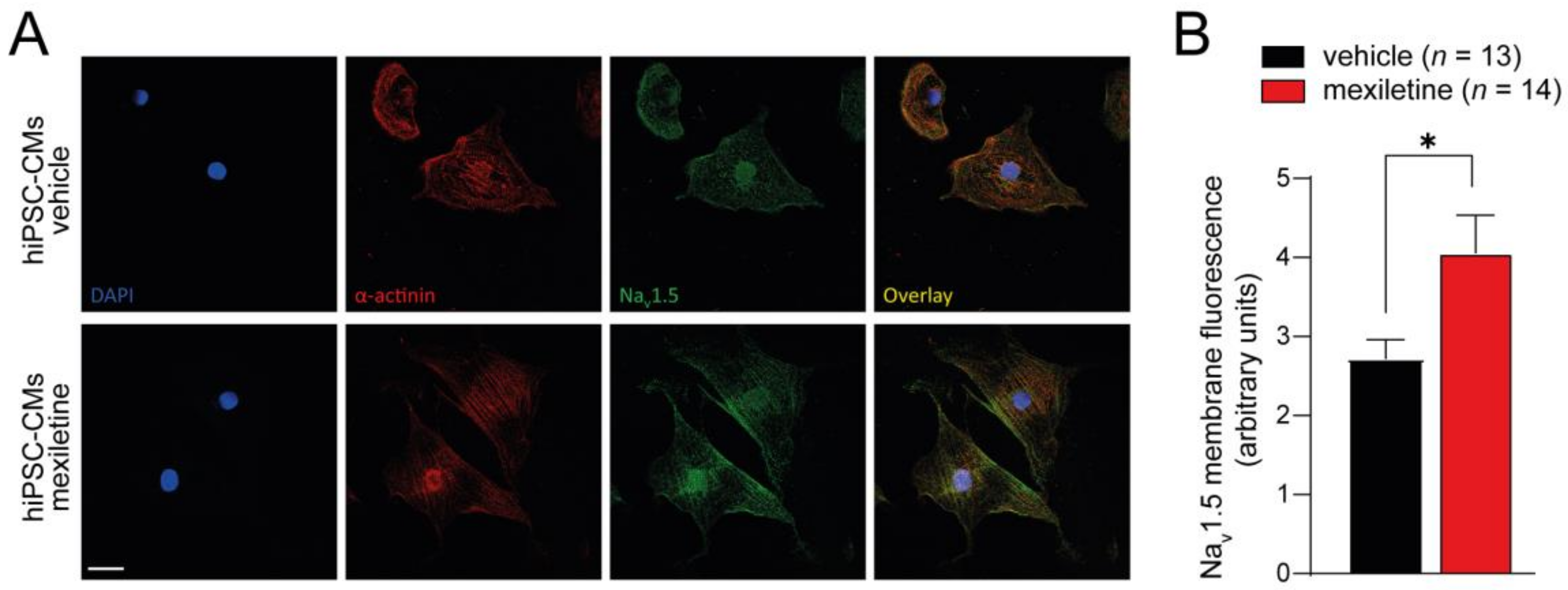
2.4. Immunocytochemistry
2.5. Statistical Analysis
3. Results
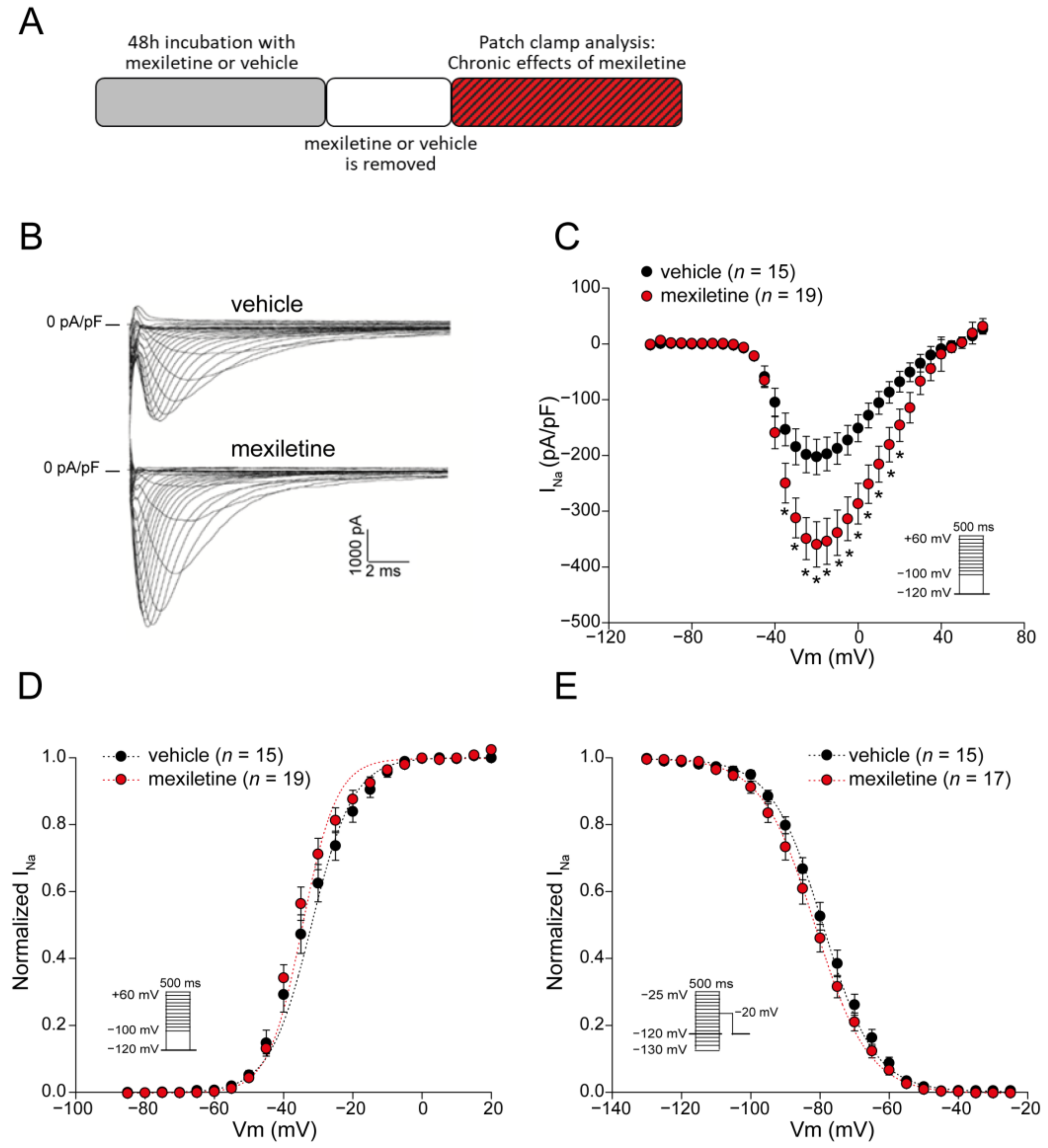
3.1. Chronic Mexiletine Treatment Increases Peak INa in hiPSC-CMs
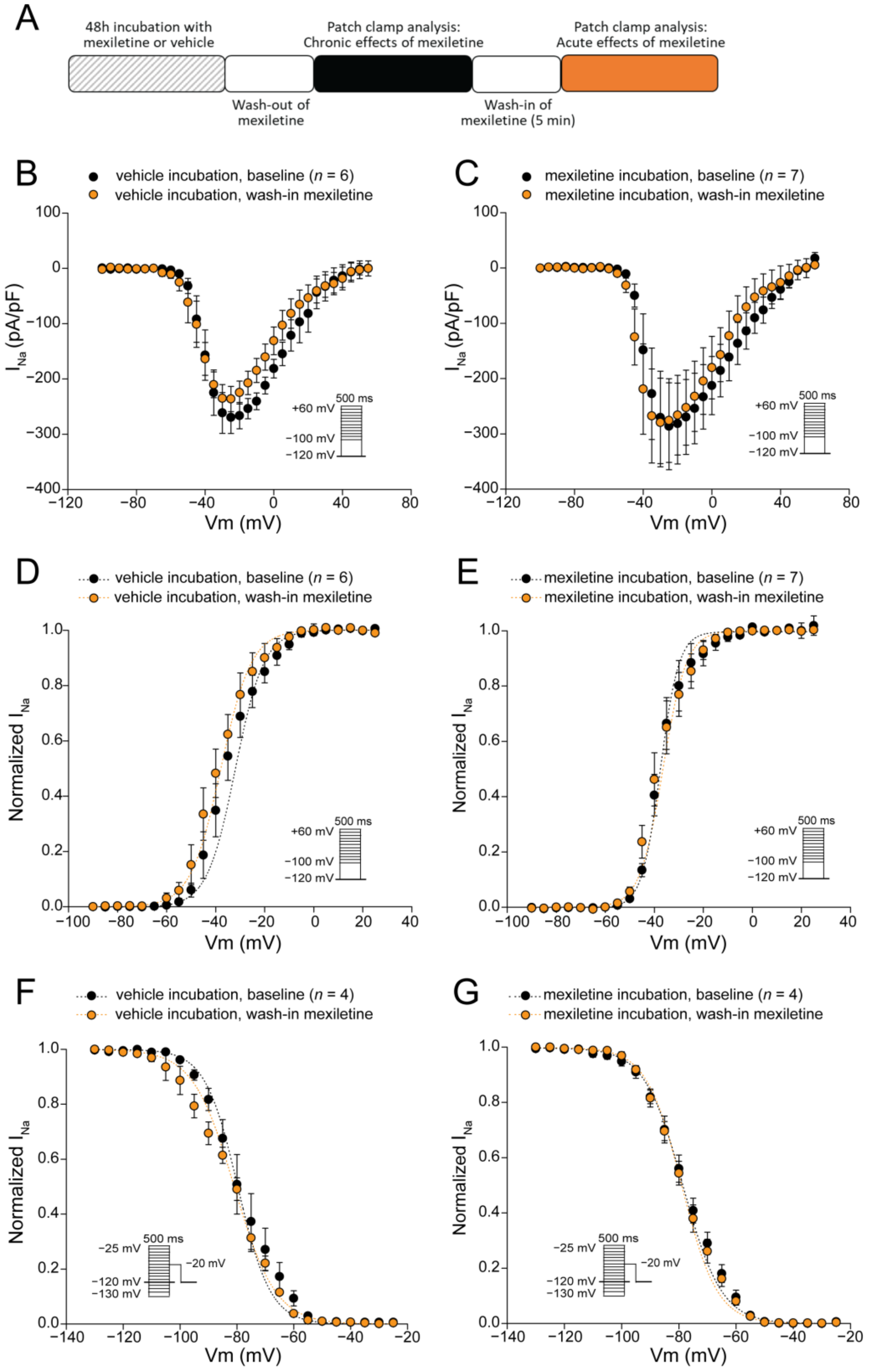
3.2. Acute Administration of Mexiletine Does Not Affect Peak INa in hiPSC-CMs
3.3. Chronic Mexiletine Increases Action Potential Upstroke Velocity in hiPSC-CMs
3.4. No Acute Effect of Mexiletine on AP Upstroke Velocity in hiPSC-CMs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Veerman, C.C.; Wilde, A.A.M.; Lodder, E.M. The Cardiac Sodium Channel Gene SCN5A and Its Gene Product NaV1.5: Role in Physiology and Pathophysiology. Gene 2015, 573, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Chen-Izu, Y.; Shaw, R.M.; Pitt, G.S.; Yarov-Yarovoy, V.; Sack, J.T.; Abriel, H.; Aldrich, R.W.; Belardinelli, L.; Cannell, M.B.; Catterall, W.A.; et al. Na+ Channel Function, Regulation, Structure, Trafficking and Sequestration. J. Physiol. 2015, 593, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, P.C.; Balser, J.R. Inherited Sodium Channelopathies s Continuum of Channel Dysfunction. Trends Cardiovasc. Med. 2004, 14, 28–35. [Google Scholar] [CrossRef]
- Rivaud, M.R.; Delmar, M.; Remme, C.A. Heritable Arrhythmia Syndromes Associated with Abnormal Cardiac Sodium Channel Function: Ionic and Non-Ionic Mechanisms. Cardiovasc. Res. 2020, 116, 1557–1570. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.D.; Clancy, C.E. Pathophysiology of the Cardiac Late Na Current and Its Potential as a Drug Target. J. Mol. Cell. Cardiol. 2012, 52, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Olleik, F.; Kamareddine, M.H.; Spears, J.; Tse, G.; Liu, T.; Yan, G.X. Mexiletine: Antiarrhythmic Mechanisms, Emerging Clinical Applications and Mortality. Pacing Clin. Electrophysiol. 2023, 46, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- van der Ree, M.H.; van Dussen, L.; Rosenberg, N.; Stolwijk, N.; van den Berg, S.; van der Wel, V.; Jacobs, B.A.W.; Wilde, A.A.M.; Hollak, C.E.M.; Postema, P.G. Effectiveness and Safety of Mexiletine in Patients at Risk for (Recurrent) Ventricular Arrhythmias: A Systematic Review. Europace 2022, 24, 1809–1823. [Google Scholar] [CrossRef] [PubMed]
- Farkowski, M.M.; Karlinski, M.; Pytkowski, M.; de Asmundis, C.; Lewandowski, M.; Mugnai, G.; Conte, G.; Marijon, E.; Anic, A.; Boveda, S.; et al. Mexiletine for Recurrent Ventricular Tachycardia in Adult Patients with Structural Heart Disease and Implantable Cardioverter Defibrillator: An EHRA Systematic Review. Europace 2022, 24, 1504–1511. [Google Scholar] [CrossRef]
- Alhourani, N.; Wolfes, J.; Könemann, H.; Ellermann, C.; Frommeyer, G.; Güner, F.; Lange, P.S.; Reinke, F.; Köbe, J.; Eckardt, L. Relevance of Mexiletine in the Era of Evolving Antiarrhythmic Therapy of Ventricular Arrhythmias. Clin. Res. Cardiol. 2024, 113, 791–800. [Google Scholar] [CrossRef]
- Frommeyer, G.; Garthmann, J.; Ellermann, C.; Dechering, D.G.; Kochhäuser, S.; Reinke, F.; Köbe, J.; Wasmer, K.; Eckardt, L. Broad Antiarrhythmic Effect of Mexiletine in Different Arrhythmia Models. Europace 2018, 20, 1375–1381. [Google Scholar] [CrossRef]
- Mazzanti, A.; Maragna, R.; Faragli, A.; Monteforte, N.; Bloise, R.; Memmi, M.; Novelli, V.; Baiardi, P.; Bagnardi, V.; Etheridge, S.P.; et al. Gene-Specific Therapy with Mexiletine Reduces Arrhythmic Events in Patients with Long QT Syndrome Type 3. J. Am. Coll. Cardiol. 2016, 67, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Priori, S.G.; Locati, E.H.; Napolitano, C.; Cantu, F.; Towbin, J.A.; Keating, M.T.; Hammoude, H.; Brown, A.M.; Chen, L.S.K.; et al. Long QT Syndrome Patients with Mutations of the SCN5A and HERG Genes Have Differential Responses to Na+ Channel Blockade and to Increases in Heart Rate: Implications for Gene-Specific Therapy. Circulation 1995, 92, 3381–3386. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.D.; Zhu, W.; Mangold, K.; Chung, W.; Silva, J.R. A Molecularly Detailed NaV1.5 Model Reveals a New Class I Antiarrhythmic Target. Basic Transl. Sci. 2019, 4, 736–751. [Google Scholar] [CrossRef] [PubMed]
- Taouis, M.; Sheldon, R.S.; Duff, H.J. Upregulation of the Rat Cardiac Sodium Channel by in Vivo Treatment with a Class I Antiarrhythmic Drug. J. Clin. Investig. 1991, 88, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.S.; Duff, H.J.; Thakore, E.; Hill, R.J. Class I Antiarrhythmic Drugs: Allosteric Inhibitors of [3H] Batrachotoxinin Binding to Rat Cardiac Sodium Channels. J. Pharmacol. Exp. Ther. 1994, 268, 187–194. [Google Scholar] [PubMed]
- Valdivia, C.R.; Tester, D.J.; Rok, B.A.; Porter, C.-B.J.; Munger, T.M.; Jahangir, A.; Makielski, J.C.; Ackerman, M.J. A Trafficking Defective, Brugada Syndrome-Causing SCN5A Mutation Rescued by Drugs. Cardiovasc. Res. 2004, 62, 53–62. [Google Scholar] [CrossRef]
- Moreau, A.; Keller, D.I.; Huang, H.; Fressart, V.; Schmied, C.; Timour, Q.; Chahine, M. Mexiletine Differentially Restores the Trafficking Defects Caused by Two Brugada Syndrome Mutations. Front. Pharmacol. 2012, 3, 62. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.M.; Tester, D.J.; Li, R.; Sun, T.; Peterson, B.Z.; Ackerman, M.J.; Makielski, J.C.; Tan, B.H. Mexiletine Rescues a Mixed Biophysical Phenotype of the Cardiac Sodium Channel Arising from the SCN5A Mutation, N406K, Found in LQT3 Patients. Channels 2018, 12, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.-H.; Valdivia, C.R.; Song, C.; Makielski, J.C. Partial Expression Defect for the SCN5A Missense Mutation G1406R Depends on Splice Variant Background Q1077 and Rescue by Mexiletine. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1822–H1828. [Google Scholar] [CrossRef]
- Nei, S.D.; Danelich, I.M.; Lose, J.M.; Leung, L.Y.T.; Asirvatham, S.J.; McLeod, C.J. Therapeutic Drug Monitoring of Mexiletine at a Large Academic Medical Center. SAGE Open Med. 2016, 4. [Google Scholar] [CrossRef]
- Nasilli, G.; Yiangou, L.; Palandri, C.; Cerbai, E.; Davis, R.P.; Verkerk, A.O.; Casini, S.; Remme, C.A. Beneficial Effects of Chronic Mexiletine Treatment in a Human Model of SCN5A Overlap Syndrome. Europace 2023, 25, euad154. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Wilders, R. Injection of IK1 through Dynamic Clamp Can Make All the Difference in Patch-Clamp Studies on HiPSC-Derived Cardiomyocytes. Front. Physiol. 2023, 14, 1326160. [Google Scholar] [CrossRef]
- Dhamoon, A.S.; Jalife, J. The Inward Rectifier Current (IK1) Controls Cardiac Excitability and Is Involved in Arrhythmogenesis. Heart Rhythm 2005, 2, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Van Putten, R.M.E.M.; Mengarelli, I.; Guan, K.; Zegers, J.G.; Van Ginneken, A.C.G.; Verkerk, A.O.; Wilders, R. Ion Channelopathies in Human Induced Pluripotent Stem Cell Derived Cardiomyocytes: A Dynamic Clamp Study with Virtual IK1. Front. Physiol. 2015, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Janse, M.J.; Wit, A.L. Electrophysiological Mechanisms of Ventricular Arrhythmias Resulting from Myocardial Ischemia and Infarction. Physiol. Rev. 1989, 69, 1049–1169. [Google Scholar] [CrossRef]
- Rivaud, M.R.; Agullo-Pascual, E.; Lin, X.; Leo-Macias, A.; Zhang, M.; Rothenberg, E.; Bezzina, C.R.; Delmar, M.; Remme, C.A. Sodium Channel Remodeling in Subcellular Microdomains of Murine Failing Cardiomyocytes. J. Am. Heart Assoc. 2017, 6, e007622. [Google Scholar] [CrossRef]
- Makielski, J.C. Late Sodium Current: A Mechanism for Angina, Heart Failure, and Arrhythmia. Trends Cardiovasc. Med. 2016, 26, 115–122. [Google Scholar] [CrossRef]
- Rivaud, M. Functional Modulation of Atrio-Ventricular Conduction by Enhanced Late Sodium Current and Calcium-Dependent Mechanisms in Scn5a1798insD/+ Mice. Europace 2020, 22, 1579–1589. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Amin, A.S. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef]
- Portero, V.; Casini, S.; Hoekstra, M.; Verkerk, A.O.; Mengarelli, I.; Belardinelli, L.; Rajamani, S.; Wilde, A.A.M.; Bezzina, C.R.; Veldkamp, M.W.; et al. Anti-Arrhythmic Potential of the Late Sodium Current Inhibitor GS-458967 in Murine Scn5a-1798insD+/- and Human SCN5A-1795insD+/- IPSC-Derived Cardiomyocytes. Cardiovasc. Res. 2017, 113, 829–838. [Google Scholar] [CrossRef]
- Cutler, M.J.; Eckhardt, L.L.; Kaufman, E.S.; Arbelo, E.; Behr, E.R.; Brugada, P.; Cerrone, M.; Crotti, L.; DeAsmundis, C.; Gollob, M.H.; et al. Clinical Management of Brugada Syndrome: Commentary from the Experts. Circ. Arrhythm. Electrophysiol. 2024, 17, e012072. [Google Scholar] [CrossRef]
- Grasso, D.; Galderisi, S.; Santucci, A.; Bernini, A. Pharmacological Chaperones and Protein Conformational Diseases: Approaches of Computational Structural Biology. Int. J. Mol. Sci. 2023, 24, 5819. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.S.; Ulloa-Aguirre, A.; Tao, Y.X. Pharmacoperone Drugs: Targeting Misfolded Proteins Causing Lysosomal Storage-, Ion Channels-, and G Protein-Coupled Receptors-Associated Conformational Disorders. Expert Rev. Clin. Pharmacol. 2018, 11, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.X.; Conn, P.M. Pharmacoperones as Novel Therapeutics for Diverse Protein Conformational Diseases. Physiol. Rev. 2018, 98, 697–725. [Google Scholar] [CrossRef]
- Vauthier, V.; Housset, C.; Falguières, T. Targeted Pharmacotherapies for Defective ABC Transporters. Biochem. Pharmacol. 2017, 136, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Heard, A.; Thompson, J.; Carver, J.; Bakey, M.; Wang, X. Targeting Molecular Chaperones for the Treatment of Cystic Fibrosis: Is It a Viable Approach? Curr. Drug Targets 2015, 16, 958–964. [Google Scholar] [CrossRef]
- Mehta, A.; Ramachandra, C.J.A.; Singh, P.; Chitre, A.; Lua, C.H.; Mura, M.; Crotti, L.; Wong, P.; Schwartz, P.J.; Gnecchi, M.; et al. Identification of a Targeted and Testable Antiarrhythmic Therapy for Long-QT Syndrome Type 2 Using a Patient-Specific Cellular Model. Eur. Heart J. 2018, 39, 1446–1455. [Google Scholar] [CrossRef]
- Smith, J.L.; Reloj, A.R.; Nataraj, P.S.; Bartos, D.C.; Schroder, E.A.; Moss, A.J.; Ohno, S.; Horie, M.; Anderson, C.L.; January, C.T.; et al. Pharmacological Correction of Long QT-Linked Mutations in KCNH2 (HERG) Increases the Trafficking of Kv11.1 Channels Stored in the Transitional Endoplasmic Reticulum. Am. J. Physiol. Cell Physiol. 2013, 305, C919–C930. [Google Scholar] [CrossRef]
- O’Hare, B.J.; Kim, C.S.J.; Hamrick, S.K.; Ye, D.; Tester, D.J.; Ackerman, M.J. Promise and Potential Peril with Lumacaftor for the Trafficking Defective Type 2 Long-QT Syndrome-Causative Variants, p.G604S, p.N633S, and p.R685P, Using Patient-Specific Re-Engineered Cardiomyocytes. Circ. Genomic Precis. Med. 2020, 13, 466–475. [Google Scholar] [CrossRef]
- Zhao, J.; Ziane, R.; Chatelier, A.; O’Leary, M.E.; Chahine, M. Lidocaine Promotes the Trafficking and Functional Expression of Na(v)1.8 Sodium Channels in Mammalian Cells. J. Neurophysiol. 2007, 98, 467–477. [Google Scholar] [CrossRef]
- Duff, H.J.; Offord, J.; West, J.; Catterall, W.A. Class I and IV Antiarrhythmic Drugs and Cytosolic Calcium Regulate MRNA Encoding the Sodium Channel α Subunit in Rat Cardiac Muscle. Mol. Pharmacol. 1992, 42, 570–574. [Google Scholar] [PubMed]
- Gualdani, R.; Tadini-Buoninsegni, F.; Roselli, M.; Defrenza, I.; Contino, M.; Colabufo, N.A.; Lentini, G. Inhibition of HERG Potassium Channel by the Antiarrhythmic Agent Mexiletine and Its Metabolite M-Hydroxymexiletine. Pharmacol. Res. Perspect. 2015, 3, e00160. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Kiyosue, T.; Arita, M. Comparison of the Inhibitory Effects of Mexiletine and Lidocaine on the Calcium Current of Single Ventricular Cells. Life Sci. 1986, 39, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Postema, P.G.; Schwartz, P.J.; Arbelo, E.; Bannenberg, W.J.; Behr, E.R.; Belhassen, B.; Brugada, J.; Brugada, P.; Camm, A.J.; Casado-Arroyo, R.; et al. Continued Misuse of Orphan Drug Legislation: A Life-Threatening Risk for Mexiletine. Eur. Heart J. 2020, 41, 614–617. [Google Scholar] [CrossRef]
- van den Berg, S.; van der Wel, V.; de Visser, S.J.; Stunnenberg, B.C.; Timmers, L.; van der Ree, M.H.; Postema, P.G.; Hollak, C.E.M. Cost-Based Price Calculation of Mexiletine for Nondystrophic Myotonia. Value Health 2021, 24, 925–929. [Google Scholar] [CrossRef]
- Postema, P.G. About the Different Faces of Mexiletine. Heart Rhythm 2020, 17, 1951–1952. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vehicle | Mexiletine | |
|---|---|---|
| Activation V1/2 (mV) | n = 15 −33.0 ± 1.6 | n = 19 −35.1 ± 1.2 |
| k (mV) Inactivation V1/2 (mV) k (mV) | 4.3 ± 0.4 n = 15 −78.2 ± 1.4 −7.5 ± 0.2 | 5.1 ± 0.4 n = 17 −80.9 ± 1.5 −7.5 ± 0.3 |
| hiPSC-CMs Incubated with the Vehicle | hiPSC-CMs Incubated with Mexiletine | |||
|---|---|---|---|---|
| Baseline | Acute Mexiletine Wash-In | Baseline | Acute Mexiletine Wash-In | |
| Activation V1/2 (mV) | n = 6 −35.0 ± 2.4 | n = 6 −38.4 ± 2.9 | n = 7 −36.7 ± 2.1 | n = 7 −37.4 ± 2.4 |
| k (mV) Inactivation V1/2 (mV) k (mV) | 4.5 ± 0.7 n = 4 −78.9 ± 2.6 −7.5 ± 0.7 | 4.0 ± 0.5 n = 4 −81.4 ± 1.3 −8.7 ± 0.8 | 6.5 ± 0.9 n = 4 −77.3 ± 1.6 −8.1 ± 0.2 | 5.4 ± 0.6 n = 4 −77.6 ± 1.5 −7.5 ± 0.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasilli, G.; Verkerk, A.O.; O’Reilly, M.; Yiangou, L.; Davis, R.P.; Casini, S.; Remme, C.A. Chronic Mexiletine Administration Increases Sodium Current in Non-Diseased Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Biomedicines 2024, 12, 1212. https://doi.org/10.3390/biomedicines12061212
Nasilli G, Verkerk AO, O’Reilly M, Yiangou L, Davis RP, Casini S, Remme CA. Chronic Mexiletine Administration Increases Sodium Current in Non-Diseased Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Biomedicines. 2024; 12(6):1212. https://doi.org/10.3390/biomedicines12061212
Chicago/Turabian StyleNasilli, Giovanna, Arie O. Verkerk, Molly O’Reilly, Loukia Yiangou, Richard P. Davis, Simona Casini, and Carol Ann Remme. 2024. "Chronic Mexiletine Administration Increases Sodium Current in Non-Diseased Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes" Biomedicines 12, no. 6: 1212. https://doi.org/10.3390/biomedicines12061212
APA StyleNasilli, G., Verkerk, A. O., O’Reilly, M., Yiangou, L., Davis, R. P., Casini, S., & Remme, C. A. (2024). Chronic Mexiletine Administration Increases Sodium Current in Non-Diseased Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Biomedicines, 12(6), 1212. https://doi.org/10.3390/biomedicines12061212