
Which Comes First, Nonalcoholic Fatty Liver Disease or Arterial Hypertension?

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Methods
- Article type: Classical Article, Clinical Study, Clinical Trial, Comparative Study, Con- trolled Clinical Trial, Multicenter Study, Meta-Analysis, Observational Study, and Randomized Controlled Trial, Preprint;
- Species: Humans, Other Animals;
- Article language: English;
- Age: Adult;
- Publication date: 5 years.
3. Prognoses of Patients with NAFLD and AH
3.1. NAFLD as Independent Risk Factor for AH Development
3.2. AH as Independent Risk Factor for NAFLD Development
3.3. Bidirectional Relationship
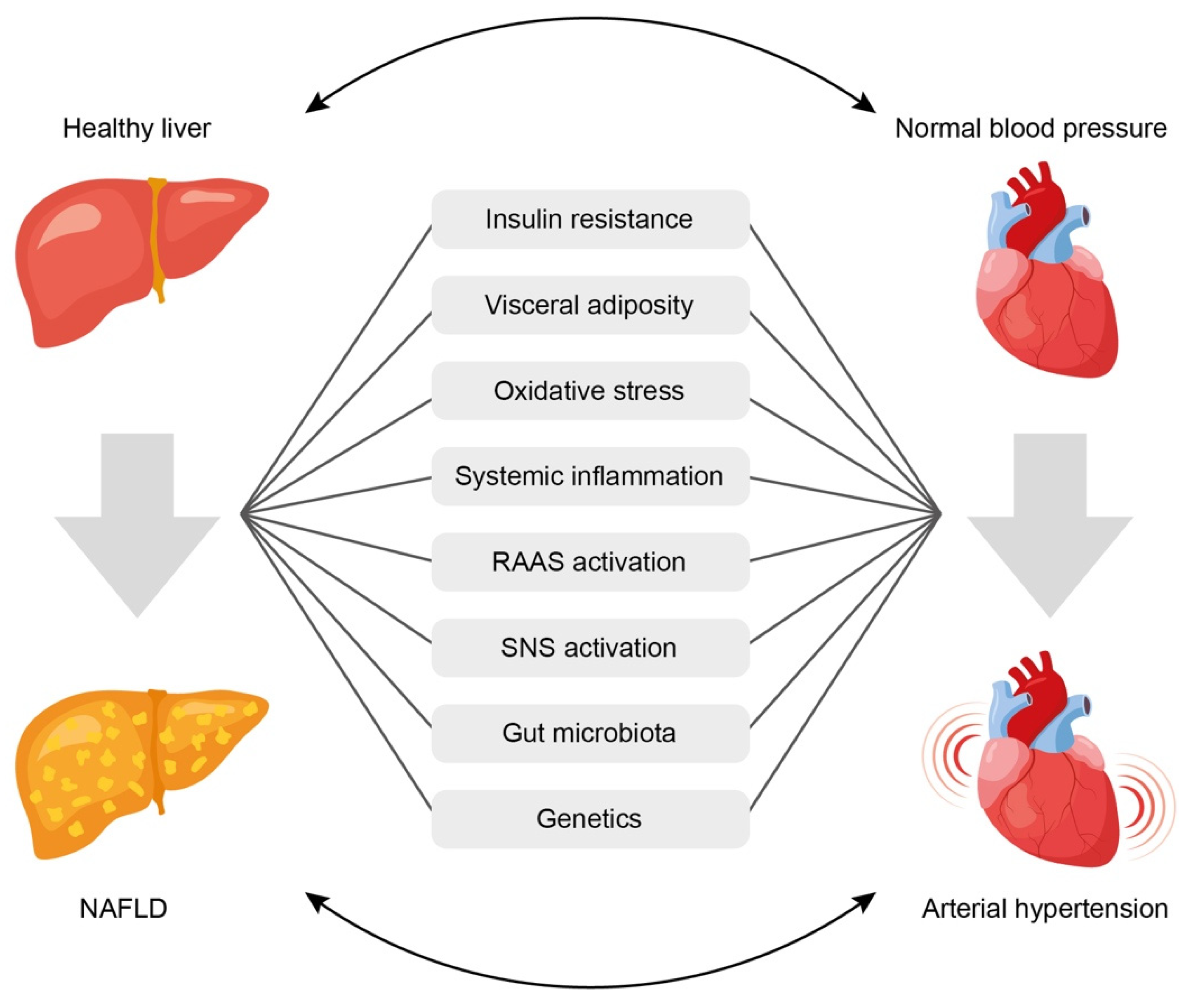
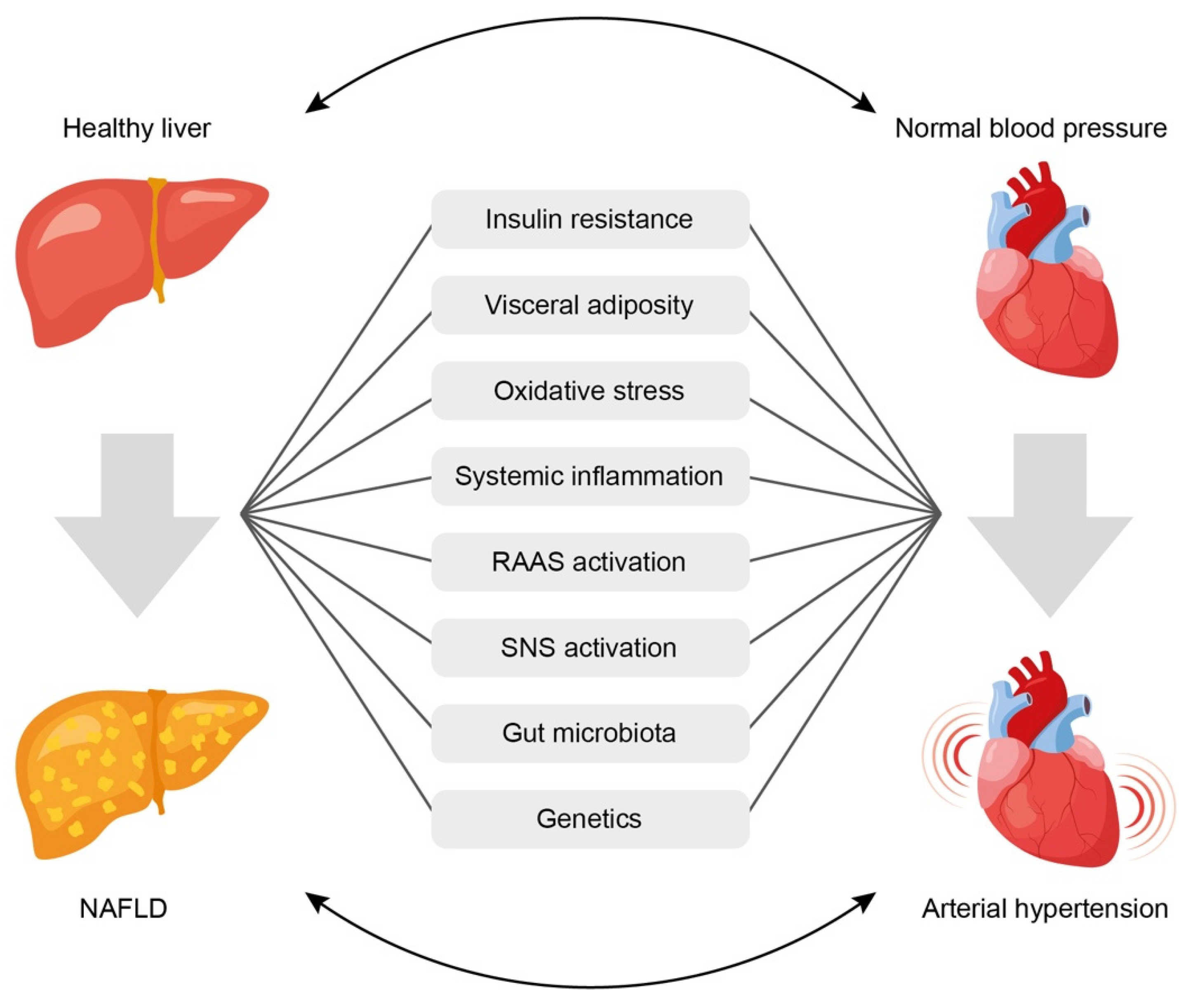
4. Mechanisms of the Relationship between NAFLD and AH
- (1)
- Insulin resistance,
- (2)
- Systemic inflammation,
- (3)
- Activation of the renin-angiotensin-aldosterone system (RAAS),
- (4)
- Oxidative stress,
- (5)
- Activation of the sympathetic nervous system,
- (6)
- Gut microbiota disbalance,
- (7)
- Genetic factors.
4.1. Insulin Resistance
4.2. Visceral Obesity
4.3. Oxidative Stress and Biologically Active Substances
4.4. Gut Microbiota
4.4.1. Short-Chain Fatty Acids
4.4.2. Trimethylamine and Trimethylamine N-oxide
4.4.3. Increased Intestinal Permeability
4.5. Genetic Factors
4.6. Influence of Gender on the Development of AH and NAFLD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE | angiotensin-converting enzyme |
| ADIPOQ | adiponectin |
| AGTR1 | angiotensin II receptor type 1 |
| AH | arterial hypertension |
| AHR | aryl hydrocarbon receptor |
| Akt | protein kinase B |
| BMI | body mass index |
| BP | blood pressure |
| CI | confidence interval |
| CVD | cardiovascular disease |
| DBP | diastolic blood pressure |
| ET-1 | endothelin-1 |
| FFAs | free fatty acids |
| FGF-21 | fibroblast growth factor 21 |
| FIB-4 | fibrosis-4 index |
| FLI | fatty liver index |
| GPR | G protein-coupled receptor |
| HDL | high-density lipoproteins |
| HFF | hepatic fat fraction |
| HOMA-IR | homeostasis model assessment of insulin resistance |
| HR | hazard ratio |
| hsCRP | high-sensitivity C-reactive protein |
| IL | interleukin |
| IR | insulin resistance |
| LDL | low-density lipoproteins |
| LEP | leptin |
| MAPK | mitogen-activated protein kinase |
| MRI | magnetic resonance imagining |
| N/A | not applicable |
| NAFLD | non-alcoholic fatty liver disease |
| NFS | NAFLD fibrosis score |
| NO | nitric oxide |
| Olfr | olfactory receptor |
| OR | odds ratio |
| PCOS | polycystic ovary syndrome |
| PERK | protein kinase RNA- like endoplasmic reticulum kinase |
| PI3K | phosphatidylinositol 3-kinase |
| RAAS | renin-angiotensin-aldosterone system |
| RR | risk ratio |
| SBP | systolic blood pressure |
| SCFAs | short-chain fatty acids |
| SD | standard deviation |
| SUA | serum uric acid |
| T2DM | type 2 diabetes mellites |
| TGFB1 | transforming growth factor beta-1 |
| TMA | trimethylamine |
| TMAO | trimethylamine N-oxide |
| TNF-α | tumor necrosis factor-α |
| US | ultrasound |
| VLDL | very-low-density lipoproteins |
References
- Wu, S.; Wu, F.; Ding, Y.; Hou, J.; Bi, J.; Zhang, Z. Association of non-alcoholic fatty liver disease with major adverse cardiovascular events: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 33386. [Google Scholar] [CrossRef]
- Kuang, M.; Lu, S.; Xie, Q.; Peng, N.; He, S.; Yu, C.; Qiu, J.; Sheng, G.; Zou, Y. Abdominal obesity phenotypes are associated with the risk of developing non-alcoholic fatty liver disease: Insights from the general population. BMC Gastroenterol. 2022, 1, 311. [Google Scholar] [CrossRef] [PubMed]
- Huh, Y.; Cho, Y.J.; Nam, G.E. Recent Epidemiology and Risk Factors of Nonalcoholic Fatty Liver Disease. J. Obes. Metab. Syndr. 2022, 1, 17–27. [Google Scholar] [CrossRef]
- Zarghamravanbakhsh, P.; Frenkel, M.; Poretsky, L. Metabolic causes and consequences of nonalcoholic fatty liver disease (NAFLD). Metabol. Open. 2021, 12, 100149, Erratum in Metabol. Open. 2023, 17, 100231. [Google Scholar] [CrossRef] [PubMed]
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2021, 7, 921–937. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kang, K.; Sheol, H.; Shin, J.; Sim, Y.; Yang, T.; Hwang, J.; Lee, J.-M. The Association between Serum Uric Acid Levels and 10-Year Cardiovascular Disease Risk in Non-Alcoholic Fatty Liver Disease Patients. Int. J. Environ. Res. Public Health 2022, 19, 1042. [Google Scholar] [CrossRef]
- Duan, Y.; Pan, X.; Luo, J.; Xiao, X.; Li, J.; Bestman, P.L.; Luo, M. Association of Inflammatory Cytokines with Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2022, 13, 880298. [Google Scholar] [CrossRef]
- Iglesias Morcillo, M.; Freuer, D.; Peters, A.; Heier, M.; Teupser, D.; Meisinger, C.; Linseisen, J. Association between fatty liver index and blood coagulation markers: A population-based study. Lipids Health Dis. 2023, 1, 83. [Google Scholar] [CrossRef]
- Emamat, H.; Tangestani, H.; Behrad Nasab, M.; Ghalandari, H.; Hekmatdoost, A. The association between epicardial adipose tissue and non-alcoholic fatty liver disease: A systematic review of existing human studies. EXCLI J. 2021, 20, 1096–1105. [Google Scholar] [CrossRef]
- Vu, H.; Khanh Tuong, T.T.; Hoang Lan, N.; Quoc Thang, T.; Bilgin, K.; Hoa, T.; Minh Duc, N.; The Dung, B. Association between nonalcoholic fatty liver disease and carotid intima-media thickness. Clin. Ter. 2023, 174, 42–47. [Google Scholar] [CrossRef]
- Huang, P.L. A comprehensive definition for metabolic syndrome. Dis. Model. Mech. 2009, 2, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, F.D.; Whelton, P.K. High Blood Pressure and Cardiovascular Disease. Hypertension 2020, 75, 285–292. [Google Scholar] [CrossRef]
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002, 25, 3143–3421. [Google Scholar] [CrossRef]
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C.; et al. Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005, 17, 2735–2752. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Corey, K.E.; Byrne, C.D. NAFLD, and cardiovascular and cardiac diseases: Factors influencing risk, prediction and treatment. Diabetes Metab. 2021, 47, 101215. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, Y.; Kim, S.U.; Kim, H.C. Metabolic dysfunction-associated fatty liver disease and incident cardiovascular disease risk: A nationwide cohort study. Clin. Gastroenterol. Hepatol. 2021, 19, 2138–2147. [Google Scholar] [CrossRef]
- Cattazzo, F.; Lombardi, R.; Mantovani, A.; Bevilacqua, M.; Zoncapè, M.; Iogna Prat, L.; Roccarina, D.; Fortuna, L.; Cespiati, A.; Sacerdoti, D.; et al. Subclinical and clinical atherosclerosis in non-alcoholic fatty liver disease is associated with the presence of hypertension. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 2839–2847. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef]
- Ng, C.H.; Wong, Z.Y.; Chew, N.W.S.; Chan, K.E.; Xiao, J.; Sayed, N.; Lim, W.H.; Tan, D.J.H.; Loke, R.W.K.; Tay, P.W.L.; et al. Hypertension is prevalent in non-alcoholic fatty liver disease and increases all-cause and cardiovascular mortality. Front. Cardiovasc. Med. 2022, 9, 942753. [Google Scholar] [CrossRef]
- Song, Q.; Liu, S.; Ling, Q.H.; Gao, Q.N.; Yang, R.X.; Chen, S.H.; Wu, S.; Chen, M.L.; Cai, J. Severity of nonalcoholic fatty liver disease is associated with cardiovascular outcomes in patients with prehypertension or hypertension: A community–based cohort study. Front. Endocrinol. 2022, 13, 942647. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Li, L.; Dai, J.; Natarajan, Y.; Yu, X.; Asch, S.M.; El-Serag, H.B. Effect of metabolic traits on the risk of cirrhosis and hepatocellular cancer in nonalcoholic fatty liver disease. Hepatology 2020, 71, 808–819. [Google Scholar] [CrossRef] [PubMed]
- Welzel, T.M.; Graubard, B.I.; Zeuzem, S.; El-Serag, H.B.; Davila, J.A.; McGlynn, K.A. Metabolic syndrome increases the risk of primary liver cancer in the United States: A study in the SEER-Medicare database. Hepatology 2011, 2, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.J.; Kim, W.; Kim, D.; Yoon, J.H.; Lee, K.; Kim, J.H.; Cho, E.J.; Lee, J.H.; Kim, H.Y.; Kim, Y.J.; et al. Visceral obesity predicts significant fibrosis in patients with nonalcoholic fatty liver disease. Medicine 2015, 94, e2159. [Google Scholar] [CrossRef]
- Feng, R.N.; Du, S.S.; Wang, C.; Li, Y.C.; Liu, L.Y.; Guo, F.C.; Sun, C.H. Lean-non-alcoholic fatty liver disease increases risk for metabolic disorders in a normal weight Chinese population. World. J. Gastroenterol. 2014, 20, 17932–17940. [Google Scholar] [CrossRef]
- Yang, D.; Lan, J.; Cen, J.; Han, Y.; Hu, H. Association between hypertension and new-onset non-alcoholic fatty liver disease in chinese non-obese people: A longitudinal cohort study. Diabetes. Metab. Syndr. Obes. 2023, 16, 345–363. [Google Scholar] [CrossRef] [PubMed]
- WHO. Report of a WHO consultation. Obesity: Preventing and managing the global epidemic. World Health Organ. Tech. Rep. Ser. 2000, 894, 1–253. [Google Scholar]
- WHO Expert Consultation. Appropriate body-mass index for Asian populations and its implications for policy and intervention strategies. Lancet 2004, 9403, 157–163. [Google Scholar] [CrossRef]
- Ryoo, J.H.; Suh, Y.J.; Shin, H.C.; Cho, Y.K.; Choi, J.M.; Park, S.K. Clinical association between non-alcoholic fatty liver disease and the development of hypertension. J. Gastroenterol. Hepatol. 2014, 29, 1926–1931. [Google Scholar] [CrossRef]
- Sung, K.C.; Wild, S.H.; Byrne, C.D. Development of new fatty liver, or resolution of existing fatty liver, over five years of follow-up, and risk of incident hypertension. J. Hepatol. 2014, 60, 1040–1045. [Google Scholar] [CrossRef]
- Vasunta, R.L.; Kesäniemi, Y.A.; Ylitalo, A.S.; Ukkola, O.H. High ambulatory blood pressure values associated with non-alcoholic fatty liver in middle-aged adults. J. Hypertens. 2012, 30, 2015–2019. [Google Scholar] [CrossRef]
- Huh, J.H.; Ahn, S.V.; Koh, S.B.; Choi, E.; Kim, J.Y.; Sung, K.C.; Kim, E.J.; Park, J.B. A prospective study of fatty liver index and incident hypertension: The KoGES-ARIRANG study. PLoS ONE 2015, 10, e0143560. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, F.; Gastaldelli, A.; Pihan-Le Bars, F.; Natali, A.; Roussel, R.; Petrie, J.; Tichet, J.; Marre, M.; Fromenty, B.; Balkau, B. Gamma-glutamyltransferase, fatty liver index and hepatic insulin resistance are associated with incident hypertension in two longitudinal studies. J. Hypertens. 2017, 35, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Lorbeer, R.; Bayerl, C.; Auweter, S.; Rospleszcz, S.; Lieb, W.; Meisinger, C.; Heier, M.; Peters, A.; Bamberg, F.; Hetterich, H. Association between MRI-derived hepatic fat fraction and blood pressure in participants without history of cardiovascular disease. J. Hypertens. 2017, 35, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Tang, Y.; Guo, X.; Zhu, X.; He, M.; Yuan, J.; Wang, Y.; Wei, S.; Chen, W.; Zhang, X.; et al. Bidirectional association between nonalcoholic fatty liver disease and hypertension from the Dongfeng-Tongji cohort study. J. Am. Soc. Hypertens. 2018, 12, 660–670. [Google Scholar] [CrossRef]
- Roh, J.H.; Park, J.H.; Lee, H.; Yoon, Y.H.; Kim, M.; Kim, Y.G.; Park, G.M.; Lee, J.H.; Seong, I.W. A close relationship between nonalcoholic fatty liver disease marker and new-onset hypertension in healthy Korean adults. Korean Circ. J. 2020, 50, 695–705. [Google Scholar] [CrossRef]
- Higashiura, Y.; Furuhashi, M.; Tanaka, M.; Takahashi, S.; Mori, K.; Miyamori, D.; Koyama, M.; Ohnishi, H.; Moniwa, N.; Numata, K.; et al. Elevated fatty liver index is independently associated with new onset of hypertension during a 10-year period in both male and female subjects. J. Am. Heart Assoc. 2021, 10, e021430. [Google Scholar] [CrossRef] [PubMed]
- Ciardullo, S.; Grassi, G.; Mancia, G.; Perseghin, G. Nonalcoholic fatty liver disease and risk of incident hypertension: A systematic review and meta-analysis. Eur. J. Gastroenterol. Hepatol. 2022, 34, 365–371. [Google Scholar] [CrossRef]
- Siafi, E.; Andrikou, I.; Thomopoulos, C.; Konstantinidis, D.; Kakouri, N.; Tatakis, F.; Kariori, M.; Filippou, C.; Zamanis, I.; Manta, E.; et al. Fatty liver index and cardiovascular outcomes in never-treated hypertensive patients: A prospective cohort. Hypertens. Res. 2023, 46, 119–127. [Google Scholar] [CrossRef]
- Yadav, D.; Choi, E.; Ahn, S.V.; Koh, S.B.; Sung, K.C.; Kim, J.Y.; Huh, J.H. Fatty liver index as a simple predictor of incident diabetes from the KoGES-ARIRANG study. Medicine 2016, 31, e4447. [Google Scholar] [CrossRef]
- Seo, I.H.; Lee, H.S.; Lee, Y.J. Fatty liver index as a predictor for incident type 2 diabetes in community-dwelling adults: Longitudinal findings over 12 years. Cardiovasc. Diabetol. 2022, 1, 209. [Google Scholar] [CrossRef]
- García-Escobar, E.; Valdés, S.; Soriguer, F.; Vendrell, J.; Urrutia-Etxebarria, I.M.; Maldonado-Araque, C.; Ortega, E.; Ocón, P.; Montanya, E.; Menéndez, E.; et al. Fatty liver index as a predictor for type 2 diabetes in subjects with normoglycemia in a nationwide cohort study. Sci. Rep. 2021, 1, 16453. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, D.J.; Koskinen, J.; Brown, E.; Magnussen, C.G.; Hutri-Kähönen, N.; Sabin, M.; Tossavainen, P.; Jokinen, E.; Laitinen, T.; Viikari, J.; et al. Fatty liver index predicts incident risk of prediabetes, type 2 diabetes and non-alcoholic fatty liver disease (NAFLD). Ann. Med. 2021, 1, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, Y.; Lin, C.; Chen, Z. Hypertension and non-alcoholic fatty liver disease proven by transient elastography. Hepatol. Res. 2016, 46, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Yu, H.; Zhong, X.; Tian, Y.; Cui, Z.; Quan, Z. Association between hypertension and nonalcoholic fatty liver disease: A cross-sectional and meta-analysis study. J. Hum. Hypertens. 2023, 37, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Aneni, E.C.; Oni, E.T.; Martin, S.S.; Blaha, M.J.; Agatston, A.S.; Feldman, T.; Veledar, E.; Conçeicao, R.D.; Carvalho, J.A.M.; Santos, R.D.; et al. Blood pressure is associated with the presence and severity of nonalcoholic fatty liver disease across the spectrum of cardiometabolic risk. J. Hypertens. 2015, 33, 1207–1214. [Google Scholar] [CrossRef]
- Tsuneto, A.; Hida, A.; Sera, N.; Imaizumi, M.; Ichimaru, S.; Nakashima, E.; Seto, S.; Maemura, K.; Akahoshi, M. Fatty liver incidence and predictive variables. Hypertens. Res. 2010, 33, 638–643. [Google Scholar] [CrossRef]
- Sorrentino, P.; Terracciano, L.; D’Angelo, S.; Ferbo, U.; Bracigliano, A.; Vecchione, R. Predicting fibrosis worsening in obese patients with NASH through parenchymal fibronectin, HOMA-IR, and hypertension. Am. J. Gastroenterol. 2010, 105, 336–344. [Google Scholar] [CrossRef]
- Gawrieh, S.; Wilson, L.A.; Cummings, O.W.; Clark, J.M.; Loomba, R.; Hameed, B.; Abdelmalek, M.F.; Dasarathy, S.; Neuschwander-Tetri, B.A.; Kowdley, K.; et al. Histologic findings of advanced fibrosis and cirrhosis in patients with nonalcoholic fatty liver disease who have normal aminotransferase levels. Am. J. Gastroenterol. 2019, 114, 1626–1635. [Google Scholar] [CrossRef]
- Labenz, C.; Huber, Y.; Kalliga, E.; Nagel, M.; Ruckes, C.; Straub, B.K.; Galle, P.R.; Wörns, M.A.; Anstee, Q.M.; Schuppan, D.; et al. Predictors of advanced fibrosis in non-cirrhotic non-alcoholic fatty liver disease in Germany. Aliment. Pharmacol. Ther. 2018, 48, 1109–1116. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654. [Google Scholar] [CrossRef]
- Ma, J.; Hwang, S.J.; Pedley, A.; Massaro, J.M.; Hoffmann, U.; Chung, R.T.; Benjamin, E.J.; Levy, D.; Fox, C.S.; Long, M.T. Bi-directional analysis between fatty liver and cardiovascular disease risk factors. J. Hepatol. 2017, 66, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Peng, Y.; Chen, Z.; Li, H.; Liu, D.; Ye, X. Bidirectional association between hypertension and NAFLD: A systematic review and meta-analysis of observational studies. Int. J. Endocrinol. 2022, 2022, 8463640. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Ye, J.; Sun, Y.; Feng, S.; Chen, Y.; Zhong, B. The Additive Values of the Classification of Higher Serum Uric Acid Levels as a Diagnostic Criteria for Metabolic-Associated Fatty Liver Disease. Nutrients 2022, 14, 3587. [Google Scholar] [CrossRef]
- Wei, F.; Li, J.; Chen, C.; Zhang, K.; Cao, L.; Wang, X.; Ma, J.; Feng, S.; Li, W.D. Higher Serum Uric Acid Level Predicts Non-alcoholic Fatty Liver Disease: A 4-Year Prospective Cohort Study. Front. Endocrinol. 2020, 11, 179. [Google Scholar] [CrossRef]
- Paschos, P.; Athyros, V.G.; Tsimperidis, A.; Katsoula, A.; Grammatikos, N.; Giouleme, O. Can Serum Uric Acid Lowering Therapy Contribute to the Prevention or Treatment of Nonalcoholic Fatty Liver Disease? Curr. Vasc. Pharmacol. 2018, 3, 269–275. [Google Scholar] [CrossRef]
- Sanchez-Lozada, L.G.; Rodriguez-Iturbe, B.; Kelley, E.E.; Nakagawa, T.; Madero, M.; Feig, D.I.; Borghi, C.; Piani, F.; Cara-Fuentes, G.; Bjornstad, P.; et al. Uric Acid and Hypertension: An Update With Recommendations. Am. J. Hypertens. 2020, 7, 583–594. [Google Scholar] [CrossRef]
- Cicero, A.F.G.; Fogacci, F.; Giovannini, M.; Grandi, E.; D’Addato, S.; Borghi, C.; Brisighella Heart Study Group. Interaction between low-density lipoprotein-cholesterolaemia, serum uric level and incident hypertension: Data from the Brisighella Heart Study. J. Hypertens. 2019, 4, 728–731. [Google Scholar] [CrossRef]
- Piani, F.; Cicero, A.F.G.; Borghi, C. Uric Acid and Hypertension: Prognostic Role and Guide for Treatment. J. Clin. Med. 2021, 10, 448. [Google Scholar] [CrossRef]
- Kunutsor, S.K.; Bakker, S.J.; Gansevoort, R.T.; Chowdhury, R.; Dullaart, R.P. Circulating total bilirubin and risk of incident cardiovascular disease in the general population. Arterioscler. Thromb. Vasc. Biol. 2015, 3, 716–724. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, X.; Li, Z.; Li, L. Relationship between Serum Bilirubin and Left Ventricular Hypertrophy in Patients with Essential Hypertension. PLoS ONE 2015, 4, e0125275. [Google Scholar] [CrossRef]
- Bakirci, E.M.; Degirmenci, H.; Hamur, H.; Cosgun, M.S.; Coskun, R.; Gunduz, T.; Tan, M.; Dogan, M.O.; Tanriseven, H.I.; Cakir, M.; et al. Predictors of left atrial remodeling in newly diagnosed hypertensive patients: A speckle-tracking echocardiographic study. Int. J. Cardiovasc. Imaging 2021, 10, 2881–2889. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zou, L.; He, Y.; Luo, L.; Dong, W.; Zhang, Y.; Lei, X. Associations between neonatal serum bilirubin and childhood hypertension. PLoS ONE 2019, 7, e0219942. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Takeda, Y.; Tomimoto, S.; Tani, T.; Narita, H.; Kimura, G. Bilirubin as a prognostic marker in patients with pulmonary arterial hypertension. BMC Pulm. Med. 2010, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Jiang, H.; Zhao, B.; Lin, Y.; Lin, S.; Chen, T.; Su, Y.; Zhang, Y.; Zhou, L.; Li, L.; et al. The association between bilirubin and hypertension among a Chinese ageing cohort: A prospective follow-up study. J. Transl. Med. 2022, 1, 108. [Google Scholar] [CrossRef]
- Lanone, S.; Bloc, S.; Foresti, R.; Almolki, A.; Taillé, C.; Callebert, J.; Conti, M.; Goven, D.; Aubier, M.; Dureuil, B.; et al. Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: Implications for protection against endotoxic shock in rats. FASEB J. 2005, 13, 1890–1892. [Google Scholar] [CrossRef]
- Vera, T.; Stec, D.E. Moderate hyperbilirubinemia improves renal hemodynamics in ANG II-dependent hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 4, R1044–R1049. [Google Scholar] [CrossRef]
- Stec, D.E.; Hosick, P.A.; Granger, J.P. Bilirubin, renal hemodynamics, and blood pressure. Front. Pharmacol. 2012, 3, 18. [Google Scholar] [CrossRef]
- Bulmer, A.C.; Bakrania, B.; Du Toit, E.F.; Boon, A.C.; Clark, P.J.; Powell, L.W.; Wagner, K.H.; Headrick, J.P. Bilirubin acts as a multipotent guardian of cardiovascular integrity: More than just a radical idea. Am. J. Physiol. Heart Circ. Physiol. 2018, 3, H429–H447. [Google Scholar] [CrossRef]
- Ilan, Y. Analogy between non-alcoholic steatohepatitis (NASH) and hypertension: A stepwise patient-tailored approach for NASH treatment. Ann. Gastroenterol. 2018, 3, 1296–1304. [Google Scholar] [CrossRef]
- Zhao, Y.C.; Zhao, G.J.; Chen, Z.; She, Z.G.; Cai, J.; Li, H. Nonalcoholic fatty liver disease: An emerging driver of hypertension. Hypertension 2020, 75, 275–284. [Google Scholar] [CrossRef]
- Muniyappa, R.; Chen, H.; Montagnani, M.; Sherman, A.; Quon, M.J. Endothelial dysfunction due to selective insulin resistance in vascular endothelium: Insights from mechanistic modeling. Am. J. Physiol. Endocrinol. Metab. 2020, 3, E629–E646. [Google Scholar] [CrossRef] [PubMed]
- Artunc, F.; Schleicher, E.; Weigert, C.; Fritsche, A.; Stefan, N.; Häring, H.U. The impact of insulin resistance on the kidney and vasculature. Nat. Rev. Nephrol. 2016, 12, 721–737. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.A.; do Carmo, J.M.; Li, X.; Wang, Z.; Mouton, A.J.; Hall, J.E. Role of hyperinsulinemia and insulin resistance in hypertension: Metabolic syndrome revisited. Can. J. Cardiol. 2020, 36, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, D.; Georgiopoulos, G.; Katsi, V.; Kourek, C.; Tsioufis, C.; Alexopoulou, A.; Koutli, E.; Tousoulis, D. Non-alcoholic fatty liver disease and hypertension: Coprevalent or correlated? Eur. J. Gastroenterol. Hepatol. 2018, 30, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Yu, Y.; Zhang, Z.; Guo, F.; Beltz, T.G.; Thunhorst, R.L.; Felder, R.B.; Johnson, A.K. Leptin mediates high-fat diet sensitization of angiotensin II-elicited hypertension by upregulating the brain renin-angiotensin system and inflammation. Hypertension 2016, 67, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Catena, C.; Bernardi, S.; Sabato, N.; Grillo, A.; Ermani, M.; Sechi, L.A.; Fabris, B.; Carretta, R.; Fallo, F. Ambulatory arterial stiffness indices and non-alcoholic fatty liver disease in essential hypertension. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 389–393. [Google Scholar] [CrossRef]
- D’Elia, L.; Giaquinto, A.; Iacone, R.; Russo, O.; Strazzullo, P.; Galletti, F. Serum leptin is associated with increased pulse pressure and the development of arterial stiffening in adult men: Results of an eight-year follow-up study. Hypertens. Res. 2021, 11, 1444–1450. [Google Scholar] [CrossRef]
- Ferrara, D.; Montecucco, F.; Dallegri, F.; Carbone, F. Impact of different ectopic fat depots on cardiovascular and metabolic diseases. J. Cell Physiol. 2019, 12, 21630–21641. [Google Scholar] [CrossRef]
- Poetsch, M.S.; Strano, A.; Guan, K. Role of Leptin in Cardiovascular Diseases. Front. Endocrinol. 2020, 11, 354. [Google Scholar] [CrossRef]
- D’Elia, L.; Strazzullo, P. Excess Body Weight, Insulin Resistance and Isolated Systolic Hypertension: Potential Pathophysiological Links. High Blood Press. Cardiovasc. Prev. 2018, 1, 17–23. [Google Scholar] [CrossRef]
- Brandes, R.P. Endothelial dysfunction and hypertension. Hypertension 2014, 64, 924–928. [Google Scholar] [CrossRef]
- Meex, R.C.R.; Watt, M.J. Hepatokines: Linking nonalcoholic fatty liver disease and insulin resistance. Nat. Rev. Endocrinol. 2017, 13, 509–520. [Google Scholar] [CrossRef]
- Guzik, T.J.; Touyz, R.M. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Statsenko, M.E.; Streltsova, A.M.; Turovets, M.I. Effect of non-alcoholic fatty liver disease on arterial stiffness and the risk of cardiovascular complications in patients with arterial hypertension. Russ. Arch. Intern. Med. 2020, 10, 296–304. [Google Scholar] [CrossRef]
- Jung, T.W.; Youn, B.S.; Choi, H.Y.; Lee, S.Y.; Hong, H.C.; Yang, S.J.; Yoo, H.J.; Kim, B.H.; Baik, S.H.; Choi, K.M. Salsalate and adiponectin ameliorate hepatic steatosis by inhibition of the hepatokine fetuin-A. Biochem. Pharmacol. 2013, 7, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.M.; Han, K.A.; Ahn, H.J.; Lee, S.Y.; Hwang, S.Y.; Kim, B.H.; Hong, H.C.; Choi, H.Y.; Yang, S.J.; Yoo, H.J.; et al. The effects of caloric restriction on fetuin-A and cardiovascular risk factors in rats and humans: A randomized controlled trial. Clin. Endocrinol. 2013, 3, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Hong, H.C.; Choi, H.Y.; Yoo, H.J.; Cho, G.J.; Hwang, T.G.; Baik, S.H.; Choi, D.S.; Kim, S.M.; Choi, K.M. Effects of a three-month combined exercise programme on fibroblast growth factor 21 and fetuin-A levels and arterial stiffness in obese women. Clin. Endocrinol. 2011, 4, 464–469. [Google Scholar] [CrossRef]
- Kikuchi, N.; Satoh, K.; Kurosawa, R.; Yaoita, N.; Elias-Al-Mamun, M.; Siddique, M.A.H.; Omura, J.; Satoh, T.; Nogi, M.; Sunamura, S.; et al. Selenoprotein P Promotes the Development of Pulmonary Arterial Hypertension: Possible Novel Therapeutic Target. Circulation 2018, 6, 600–623. [Google Scholar] [CrossRef]
- Sun, Q.; Hackler, J.; Hilger, J.; Gluschke, H.; Muric, A.; Simmons, S.; Schomburg, L.; Siegert, E. Selenium and Copper as Biomarkers for Pulmonary Arterial Hypertension in Systemic Sclerosis. Nutrients 2020, 12, 1894. [Google Scholar] [CrossRef]
- Nosalski, R.; McGinnigle, E.; Siedlinski, M.; Guzik, T.J. Novel immune mechanisms in hypertension and cardiovascular risk. Curr. Cardiovasc. Risk Rep. 2017, 11, 12. [Google Scholar] [CrossRef]
- Simons, N.; Bijnen, M.; Wouters, K.A.M.; Rensen, S.S.; Beulens, J.W.J.; van Greevenbroek, M.M.J.; ’t Hart, L.M.; Greve, J.W.M.; van der Kallen, C.J.H.; Schaper, N.C.; et al. The endothelial function biomarker soluble E-selectin is associated with nonalcoholic fatty liver disease. Liver Int. 2020, 40, 1079–1088. [Google Scholar] [CrossRef]
- Satou, R.; Penrose, H.; Navar, L.G. Inflammation as a regulator of the renin-angiotensin system and blood pressure. Curr. Hypertens. Rep. 2018, 20, 100. [Google Scholar] [CrossRef]
- Houghton, D.; Zalewski, P.; Hallsworth, K.; Cassidy, S.; Thoma, C.; Avery, L.; Slomko, J.; Hardy, T.; Burt, A.D.; Tiniakos, D.; et al. The degree of hepatic steatosis associates with impaired cardiac and autonomic function. J. Hepatol. 2019, 70, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Sharpton, S.R.; Ajmera, V.; Loomba, R. Emerging role of the gut microbiome in nonalcoholic fatty liver disease: From composition to function. Clin. Gastroenterol. Hepatol. 2019, 17, 296–306. [Google Scholar] [CrossRef]
- Tokarek, J.; Budny, E.; Saar, M.; Kućmierz, J.; Młynarska, E.; Rysz, J.; Franczyk, B. Does the Composition of Gut Microbiota Affect Hypertension? Molecular Mechanisms Involved in Increasing Blood Pressure. Int. J. Mol. Sci. 2023, 24, 1377. [Google Scholar] [CrossRef] [PubMed]
- Verhaar, B.J.H.; Prodan, A.; Nieuwdorp, M.; Muller, M. Gut Microbiota in Hypertension and Atherosclerosis: A Review. Nutrients 2020, 12, 2982. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, N.; Hori, D.; Flavahan, S.; Steppan, J.; Flavahan, N.A.; Berkowitz, D.E.; Pluznick, J.L. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol. Genom. 2016, 48, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, J.; Kasubuchi, M.; Nakajima, A.; Irie, J.; Itoh, H.; Kimura, I. The role of short-chain fatty acid on blood pressure regulation. Curr. Opin. Nephrol. Hypertens. 2016, 25, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, H.; Tu, X.; Gao, Z. The Role of Short-Chain Fatty Acids of Gut Microbiota Origin in Hypertension. Front. Microbiol. 2021, 12, 730809. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, Q.; Lu, A.; Liu, X.; Zhang, L.; Xu, C.; Liu, X.; Li, H.; Yang, T. Sodium butyrate suppresses angiotensin II-induced hypertension by inhibition of renal (pro)renin receptor and intrarenal renin-angiotensin system. J. Hypertens. 2017, 9, 1899–1908. [Google Scholar] [CrossRef]
- Kim, S.; Goel, R.; Kumar, A.; Qi, Y.; Lobaton, G.; Hosaka, K.; Mohammed, M.; Handberg, E.M.; Richards, E.M.; Pepine, C.J.; et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin. Sci. 2018, 6, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Chun, P. Therapeutic effects of histone deacetylase inhibitors on heart disease. Arch. Pharm. Res. 2020, 12, 1276–1296. [Google Scholar] [CrossRef]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fu, C.; Li, F. Acetate Affects the Process of Lipid Metabolism in Rabbit Liver, Skeletal Muscle and Adipose Tissue. Animals 2019, 9, 799. [Google Scholar] [CrossRef]
- Xie, L.; Alam, M.J.; Marques, F.Z.; Mackay, C.R. A major mechanism for immunomodulation: Dietary fibres and acid metabolites. Semin. Immunol. 2023, 66, 101737. [Google Scholar] [CrossRef]
- Mishima, Y.; Ishihara, S. Enteric Microbiota-Mediated Serotonergic Signaling in Pathogenesis of Irritable Bowel Syndrome. Int. J. Mol. Sci. 2021, 22, 10235. [Google Scholar] [CrossRef]
- Skelly, A.N.; Sato, Y.; Kearney, S.; Honda, K. Mining the microbiota for microbial and metabolite-based immunotherapies. Nat. Rev. Immunol. 2019, 19, 305–323. [Google Scholar] [CrossRef]
- Chen, M.; Hui, S.; Lang, H.; Zhou, M.; Zhang, Y.; Kang, C.; Zeng, X.; Zhang, Q.; Yi, L.; Mi, M. SIRT3 Deficiency Promotes High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease in Correlation with Impaired Intestinal Permeability through Gut Microbial Dysbiosis. Mol. Nutr. Food Res. 2019, 63, 1800612. [Google Scholar] [CrossRef]
- Barrea, L.; Annunziata, G.; Muscogiuri, G.; Di Somma, C.; Laudisio, D.; Maisto, M.; de Alteriis, G.; Tenore, G.C.; Colao, A.; Savastano, S. Trimethylamine-N-oxide (TMAO) as Novel Potential Biomarker of Early Predictors of Metabolic Syndrome. Nutrients 2018, 10, 1971. [Google Scholar] [CrossRef] [PubMed]
- Aragonès, G.; Colom-Pellicer, M.; Aguilar, C.; Guiu-Jurado, E.; Martínez, S.; Sabench, F.; Antonio Porras, J.; Riesco, D.; Del Castillo, D.; Richart, C.; et al. Circulating microbiota-derived metabolites: A “liquid biopsy?”. Int. J. Obes. 2020, 44, 875–885. [Google Scholar] [CrossRef]
- Cordeiro, A.; Costa, R.; Andrade, N.; Silva, C.; Canabrava, N.; Pena, M.J.; Rodrigues, I.; Andrade, S.; Ramalho, A. Does adipose tissue inflammation drive the development of non-alcoholic fatty liver disease in obesity? Clin. Res. Hepatol. Gastroenterol. 2020, 44, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Liu, Y.; Long, J.; Chen, S.; Liao, G.; Wu, S.; Li, C.; Wang, L.; Ling, W.; Zhu, H. Trimethylamine N-Oxide Aggravates Liver Steatosis through Modulation of Bile Acid Metabolism and Inhibition of Farnesoid X Receptor Signaling in Nonalcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, 1900257. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.H.; Xin, F.Z.; Zhou, D.; Xue, Y.Q.; Liu, X.L.; Yang, R.X.; Pan, Q.; Fan, J.G. Trimethylamine N-oxide attenuates high-fat high-cholesterol diet-induced steatohepatitis by reducing hepatic cholesterol overload in rats. World. J. Gastroenterol. 2019, 25, 2450–2462. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 7341, 57–63. [Google Scholar] [CrossRef]
- Vu, V.; Kim, Y.; Cho, M. Effects of SCFAs and TMAO on non-alcoholic fatty liver disease indicating the therapeutic benefits of plant-based diet, and supplemental prebiotics, probiotics and synbiotics. Appl. Biol. Chem. 2023, 66, 11. [Google Scholar] [CrossRef]
- Maksymiuk, K.M.; Szudzik, M.; Gawryś-Kopczyńska, M.; Onyszkiewicz, M.; Samborowska, E.; Mogilnicka, I.; Ufnal, M. Trimethylamine, a gut bacteria metabolite and air pollutant, increases blood pressure and markers of kidney damage including proteinuria and KIM-1 in rats. J. Transl. Med. 2022, 20, 470. [Google Scholar] [CrossRef]
- Brunt, V.E.; Casso, A.G.; Gioscia-Ryan, R.A.; Sapinsley, Z.J.; Ziemba, B.P.; Clayton, Z.S.; Bazzoni, A.E.; VanDongen, N.S.; Richey, J.J.; Hutton, D.A.; et al. Gut Microbiome-Derived Metabolite Trimethylamine N-Oxide Induces Aortic Stiffening and Increases Systolic Blood Pressure With Aging in Mice and Humans. Hypertension 2021, 78, 499–511. [Google Scholar] [CrossRef]
- Huc, T.; Drapala, A.; Gawrys, M.; Konop, M.; Bielinska, K.; Zaorska, E.; Samborowska, E.; Wyczalkowska-Tomasik, A.; Pączek, L.; Dadlez, M.; et al. Chronic, low-dose TMAO treatment reduces diastolic dysfunction and heart fibrosis in hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, 1805–1820. [Google Scholar] [CrossRef]
- Ge, X.; Zheng, L.; Zhuang, R.; Yu, P.; Xu, Z.; Liu, G.; Xi, X.; Zhou, X.; Fan, H. The Gut Microbial Metabolite Trimethylamine N-Oxide and Hypertension Risk: A Systematic Review and Dose-Response Meta-analysis. Adv. Nutr. 2020, 1, 66–76. [Google Scholar] [CrossRef]
- Mutengo, K.H.; Masenga, S.K.; Mweemba, A.; Mutale, W.; Kirabo, A. Gut microbiota dependant trimethylamine N-oxide and hypertension. Front. Physiol. 2023, 14, 1075641. [Google Scholar] [CrossRef]
- Jiang, S.; Shui, Y.; Cui, Y.; Tang, C.; Wang, X.; Qiu, X.; Hu, W.; Fei, L.; Li, Y.; Zhang, S.; et al. Gut microbiota dependent trimethylamine N-oxide aggravates angiotensin II-induced hypertension. Redox Biol. 2021, 46, 102115. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, M. Trimethylamine N-Oxide Generated by the Gut Microbiota Is Associated with Vascular Inflammation: New Insights into Atherosclerosis. Mediat. Inflamm. 2020, 2020, 4634172. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Qiu, J.; Lian, J.; Yang, X.; Zhou, J. Gut Metabolite Trimethylamine-N-Oxide in Atherosclerosis: From Mechanism to Therapy. Front. Cardiovasc. Med. 2021, 8, 723886. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lin, F.; Tang, R.; Bao, C.; Zhou, Q.; Ye, K.; Shen, Y.; Liu, C.; Hong, C.; Yang, K.; et al. Gut Microbial Metabolite Trimethylamine N-Oxide Aggravates Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2022, 4, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, D.; Li, B.; Li, X.; Lai, X.; Lei, S.; Li, N.; Zhang, X. Relationship between Plasma Trimethylamine N-Oxide Levels and Renal Dysfunction in Patients with Hypertension. Kidney Blood Press. Res. 2021, 4, 421–432. [Google Scholar] [CrossRef]
- Naghipour, S.; Cox, A.J.; Peart, J.N.; Du Toit, E.F.; Headrick, J.P. Trimethylamine N-oxide: Heart of the microbiota-CVD nexus? Nutr. Res. Rev. 2021, 1, 125–146. [Google Scholar] [CrossRef]
- Li, Z.; Wu, Z.; Yan, J.; Liu, H.; Liu, Q.; Deng, Y.; Ou, C.; Chen, M. Gut microbe-derived metabolite trimethylamine N-oxide induces cardiac hypertrophy and fibrosis. Lab. Investig. 2019, 3, 346–357. [Google Scholar] [CrossRef]
- Ma, J.; Li, H. The role of gut microbiota in atherosclerosis and hypertension. Front. Pharmacol. 2018, 9, 1082. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef]
- Li, F.; Ye, J.; Shao, C.; Zhong, B. Compositional alterations of gut microbiota in nonalcoholic fatty liver disease patients: A systematic review and meta-analysis. Lipids Health Dis. 2021, 20, 22. [Google Scholar] [CrossRef] [PubMed]
- Ntlahla, E.E.; Mfengu, M.M.; Engwa, G.A.; Nkeh-Chungag, B.N.; Sewani-Rusike, C.R. Gut permeability is associated with hypertension and measures of obesity but not with Endothelial Dysfunction in South African youth. Afr. Health Sci. 2021, 3, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Sturgeon, C.; Fasano, A. Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases. Tissue Barriers 2016, 4, e1251384. [Google Scholar] [CrossRef] [PubMed]
- Santisteban, M.M.; Qi, Y.; Zubcevic, J.; Kim, S.; Yang, T.; Shenoy, V.; Cole-Jeffrey, C.T.; Lobaton, G.O.; Stewart, D.C.; Rubiano, A.; et al. Hypertension-linked pathophysiological alterations in the gut. Circ. Res. 2017, 120, 312–323. [Google Scholar] [CrossRef]
- Li, C.; Xiao, P.; Lin, D.; Zhong, H.J.; Zhang, R.; Zhao, Z.G.; He, X.X. Risk Factors for Intestinal Barrier Impairment in Patients With Essential Hypertension. Front. Med. 2021, 7, 543698. [Google Scholar] [CrossRef]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef]
- De Munck, T.J.I.; Xu, P.; Verwijs, H.J.A.; Masclee, A.A.M.; Jonkers, D.; Verbeek, J.; Koek, G.H. Intestinal permeability in human nonalcoholic fatty liver disease: A systematic review and meta-analysis. Liver Int. 2020, 40, 2906–2916. [Google Scholar] [CrossRef]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Muñoz, L.; Caparrós, E.; Albillos, A.; Francés, R. The shaping of gut immunity in cirrhosis. Front. Immunol. 2023, 14, 1139554. [Google Scholar] [CrossRef]
- Forlano, R.; Mullish, B.H.; Roberts, L.A.; Thursz, M.R.; Manousou, P. The Intestinal Barrier and Its Dysfunction in Patients with Metabolic Diseases and Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 662. [Google Scholar] [CrossRef]
- Ma, C.; Yan, K.; Wang, Z.; Zhang, Q.; Gao, L.; Xu, T.; Sai, J.; Cheng, F.; Du, Y. The association between hypertension and nonalcoholic fatty liver disease (NAFLD): Literature evidence and systems biology analysis. Bioengineered 2021, 12, 2187–2202. [Google Scholar] [CrossRef] [PubMed]
- Roderburg, C.; Krieg, S.; Krieg, A.; Demir, M.; Luedde, T.; Kostev, K.; Loosen, S.H. Non-alcoholic fatty liver disease (NAFLD) is associated with an increased incidence of chronic kidney disease (CKD). Eur. J. Med. Res. 2023, 1, 153. [Google Scholar] [CrossRef] [PubMed]
- Hartl, L.; Rumpf, B.; Domenig, O.; Simbrunner, B.; Paternostro, R.; Jachs, M.; Poglitsch, M.; Marculescu, R.; Trauner, M.; Reindl-Schwaighofer, R.; et al. The systemic and hepatic alternative renin-angiotensin system is activated in liver cirrhosis, linked to endothelial dysfunction and inflammation. Sci. Rep. 2023, 1, 953. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Kirkwood, J.; Won, K.J.; Tjota, N.; Jeong, H.; Isoherranen, N. Characterization of Vitamin A Metabolome in Human Livers With and Without Nonalcoholic Fatty Liver Disease. J. Pharmacol. Exp. Ther. 2019, 1, 92–103. [Google Scholar] [CrossRef]
- Yang, C.K.; Wang, X.K.; Liao, X.W.; Han, C.Y.; Yu, T.D.; Qin, W.; Zhu, G.Z.; Su, H.; Yu, L.; Liu, X.G.; et al. Aldehyde dehydrogenase 1 (ALDH1) isoform expression and potential clinical implications in hepatocellular carcinoma. PLoS ONE 2017, 8, e0182208. [Google Scholar] [CrossRef]
- Makia, N.L.; Bojang, P.; Falkner, K.C.; Conklin, D.J.; Prough, R.A. Murine hepatic aldehyde dehydrogenase 1a1 is a major contributor to oxidation of aldehydes formed by lipid peroxidation. Chem. Biol. Interact. 2011, 1–3, 278–287. [Google Scholar] [CrossRef]
- Saeed, A.; Dullaart, R.P.F.; Schreuder, T.C.M.A.; Blokzijl, H.; Faber, K.N. Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2017, 1, 29. [Google Scholar] [CrossRef]
- Musso, G.; Saba, F.; Cassader, M.; Paschetta, E.; De Michieli, F.; Pinach, S.; Framarin, L.; Berrutti, M.; Leone, N.; Parente, R.; et al. Angiotensin II type 1 receptor rs5186 gene variant predicts incident NAFLD and associated hypertension: Role of dietary fat-induced pro-inflammatory cell activation. Am. J. Gastroenterol. 2019, 114, 607–619. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104, Erratum in Eur. Heart J. 2019, 40, 475. [Google Scholar] [CrossRef]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018, 71, 13–115, Erratum in Hypertension 2018, 71, 140–144. [Google Scholar] [CrossRef]
- Connelly, P.J.; Currie, G.; Delles, C. Sex Differences in the Prevalence, Outcomes and Management of Hypertension. Curr. Hypertens. Rep. 2022, 6, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Meinert, F.; Thomopoulos, C.; Kreutz, R. Sex and gender in hypertension guidelines. J. Hum. Hypertens. 2023, 8, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, K.; Ji, H. Sex differences in primary hypertension. Biol. Sex. Differ. 2012, 3, 7. [Google Scholar] [CrossRef]
- Khalid, F.; Siddique, A.; Siddiqui, J.A.; Panhwar, G.; Singh, S.; Anwar, A.; Hashmi, A.A. Correlation Between Body Mass Index and Blood Pressure Levels Among Hypertensive Patients: A Gender-Based Comparison. Cureus 2020, 12, e10974. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A. Associations between smoking and alcohol consumption with blood pressure in a middle-aged population. Tob. Induc. Dis. 2023, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.M. Physical Activity and Hypertension: Knowing Is Not Enough; We Must Apply. Willing Is Not Enough; We Must Do-von Goethe. Hypertension 2017, 69, 404–406. [Google Scholar] [CrossRef]
- Kojima, S.; Watanabe, N.; Numata, M.; Ogawa, T.; Matsuzaki, S. Increase in the prevalence of fatty liver in Japan over the past 12 years: Analysis of clinical background. J. Gastroenterol. 2003, 38, 954–961. [Google Scholar] [CrossRef]
- Nagral, A.; Bangar, M.; Menezes, S.; Bhatia, S.; Butt, N.; Ghosh, J.; Manchanayake, J.H.; Mahtab, M.A.; Singh, S.P. Gender Differences in Nonalcoholic Fatty Liver Disease. Euroasian J. Hepatogastroenterol. 2022, 12 (Suppl. S1), S19–S25. [Google Scholar] [CrossRef]
- Martin-Grau, M.; Monleon, D. Sex dimorphism and metabolic profiles in management of metabolic-associated fatty liver disease. World J. Clin. Cases. 2023, 11, 1236–1244. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Li, Y.Y.; Nie, Y.Q.; Ma, J.X.; Lu, L.G.; Shi, S.L.; Chen, M.H.; Hu, P.J. Prevalence of fatty liver disease and its risk factors in the population of South China. World J. Gastroenterol. 2007, 13, 6419–6424. [Google Scholar] [CrossRef]
- Gariani, K.; Jornayvaz, F.R. Pathophysiology of NASH in endocrine diseases. Endocr. Connect. 2021, 10, R52–R65. [Google Scholar] [CrossRef] [PubMed]
- Singeap, A.M.; Stanciu, C.; Huiban, L.; Muzica, C.M.; Cuciureanu, T.; Girleanu, I.; Chiriac, S.; Zenovia, S.; Nastasa, R.; Sfarti, C.; et al. Association between Nonalcoholic Fatty Liver Disease and Endocrinopathies: Clinical Implications. Can. J. Gastroenterol. Hepatol. 2021, 2021, 6678142. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Ho, H.N. Hepatic manifestations of women with polycystic ovary syndrome. Best. Pract. Res. Clin. Obstet. Gynaecol. 2016, 37, 119–128. [Google Scholar] [CrossRef]
- Jaruvongvanich, V.; Sanguankeo, A.; Riangwiwat, T.; Upala, S. Testosterone, Sex Hormone-Binding Globulin and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Ann. Hepatol. 2017, 16, 382–394. [Google Scholar] [CrossRef]
- Yang, M.; Ma, F.; Guan, M. Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 320. [Google Scholar] [CrossRef] [PubMed]
- Della Torre, S. Beyond the X Factor: Relevance of Sex Hormones in NAFLD Pathophysiology. Cells 2021, 10, 2502. [Google Scholar] [CrossRef]
- Varlamov, O.; Bethea, C.L.; Roberts, C.T., Jr. Sex-specific differences in lipid and glucose metabolism. Front. Endocrinol. 2015, 5, 241. [Google Scholar] [CrossRef]
- Ceccarelli, I.; Bioletti, L.; Peparini, S.; Solomita, E.; Ricci, C.; Casini, I.; Miceli, E.; Aloisi, A.M. Estrogens and phytoestrogens in body functions. Neurosci. Biobehav. Rev. 2022, 132, 648–663. [Google Scholar] [CrossRef]
- Von-Hafe, M.; Borges-Canha, M.; Vale, C.; Leite, A.R.; Sérgio Neves, J.; Carvalho, D.; Leite-Moreira, A. Nonalcoholic Fatty Liver Disease and Endocrine Axes-A Scoping Review. Metabolites 2022, 12, 298. [Google Scholar] [CrossRef]
- Link, J.C.; Chen, X.; Arnold, A.P.; Reue, K. Metabolic impact of sex chromosomes. Adipocyte 2013, 2, 74–79. [Google Scholar] [CrossRef]
- Chen, X.; McClusky, R.; Chen, J.; Beaven, S.W.; Tontonoz, P.; Arnold, A.P.; Reue, K. The number of x chromosomes causes sex differences in adiposity in mice. PLoS Genet. 2012, 8, e1002709. [Google Scholar] [CrossRef]
- Michopoulos, S.; Chouzouri, V.I.; Manios, E.D.; Grapsa, E.; Antoniou, Z.; Papadimitriou, C.A.; Zakopoulos, N.; Dimopoulos, A.M. Untreated newly diagnosed essential hypertension is associated with nonalcoholic fatty liver disease in a population of a hypertensive center. Clin. Exp. Gastroenterol. 2016, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, M.; Patel, P.; Dunn-Valadez, S.; Dao, C.; Khan, V.; Ali, H.; El-Serag, L.; Hernaez, R.; Sisson, A.; Thrift, A.P.; et al. Women Have a Lower Risk of Nonalcoholic Fatty Liver Disease but a Higher Risk of Progression vs Men: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2021, 19, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Villela-Nogueira, C.A.; Leite, N.C.; Cardoso, C.R.; Salles, G.F. NAFLD and Increased Aortic Stiffness: Parallel or Common Physiopathological Mechanisms? Int. J. Mol. Sci. 2016, 17, 460. [Google Scholar] [CrossRef] [PubMed]
- Cadeddu, C.; Franconi, F.; Cassisa, L.; Campesi, I.; Pepe, A.; Cugusi, L.; Maffei, S.; Gallina, S.; Sciomer, S.; Mercuro, G.; et al. Arterial hypertension in the female world: Pathophysiology and therapy. J. Cardiovasc. Med. 2016, 17, 229–236. [Google Scholar] [CrossRef]
- Sevre, K.; Lefrandt, J.D.; Nordby, G.; Os, I.; Mulder, M.; Gans, R.O.; Rostrup, M.; Smit, A.J. Autonomic function in hypertensive and normotensive subjects: The importance of gender. Hypertension 2001, 37, 1351–1356. [Google Scholar] [CrossRef]
- Narkiewicz, K.; Phillips, B.G.; Kato, M.; Hering, D.; Bieniaszewski, L.; Somers, V.K. Gender-selective interaction between aging, blood pressure, and sympathetic nerve activity. Hypertension 2005, 45, 522–525. [Google Scholar] [CrossRef]
- Matsukawa, T.; Sugiyama, Y.; Watanabe, T.; Kobayashi, F.; Mano, T. Gender difference in age-related changes in muscle sympathetic nerve activity in healthy subjects. Am. J. Physiol. 1998, 275, R1600–R1604. [Google Scholar] [CrossRef]
- Meloni, A.; Cadeddu, C.; Cugusi, L.; Donataccio, M.P.; Deidda, M.; Sciomer, S.; Gallina, S.; Vassalle, C.; Moscucci, F.; Mercuro, G.; et al. Gender Differences and Cardiometabolic Risk: The Importance of the Risk Factors. Int. J. Mol. Sci. 2023, 24, 1588. [Google Scholar] [CrossRef]
- Reckelhoff, J.F. Gender differences in the regulation of blood pressure. Hypertension 2001, 37, 1199–1208. [Google Scholar] [CrossRef]
- Oparil, S.; Miller, A.P. Gender and blood pressure. J. Clin. Hypertens. 2005, 7, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Barris, C.T.; Faulkner, J.L.; Belin de Chantemèle, E.J. Salt Sensitivity of Blood Pressure in Women. Hypertension 2023, 80, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Mercuro, G.; Podda, A.; Pitzalis, L.; Zoncu, S.; Mascia, M.; Melis, G.B.; Rosano, G.M. Evidence of a role of endogenous estrogen in the modulation of autonomic nervous system. Am. J. Cardiol. 2000, 85, 787–789. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Reference | Study Design | Study Population | NAFLD Diagnosis | Follow-Up | Key Results |
|---|---|---|---|---|---|
| R.L. Vasunta et al., 2012 [30] | Analysis of the dataset from the population-based epidemiological case–control OPERA (The Oulu Project Elucidating Risk of Atherosclerosis) study. Cross-sectional design | 890 hypertensive (n = 433) and normotensive (n = 457) individuals (mean age ± SD 51.0 ± 6.0 years; 49% were males) | Abdominal ultrasound | N/A | After adjusting for BMI, gender and age, individuals with NAFLD had significantly higher ambulatory daytime (p < 0.02) and nighttime (p < 0.05) SBP values, compared with individuals without fatty liver disease |
| J.H. Ryoo et al., 2014 [28] | Prospective cohort study | 22,090 Korean men without AH (mean age ± SD: 42.1 ± 6.8 years). NAFLD was diagnosed in 7561 patients (34.2%) | Abdominal ultrasound, exclusion of liver disease of other etiology | 5 years | The incidence of AH was higher in patients with NAFLD vs. those without it (30.1% vs. 14.4%, p < 0.001). After adjusting for multiple covariates, the OR for developing AH were higher in mild NAFLD patients (1.07; 95% CI 1.00–1.15) and moderate to severe NAFLD patients (1.14; 95% CI 1.00–1.30), compared with the control group (p < 0.001) |
| K.C. Sung et al., 2014 [29] | Retrospective cohort study | 11,448 patients without AH at baseline. Of them, 911 patients developed AH (mean age ± SD: 42.28 ± 6.45 years; 84.4% were males), and 1418 patients developed fatty liver (mean age ± SD: 40.84 ± 5.58 years; 83.9% were males) during follow-up | Abdominal ultrasound, exclusion of liver disease of other etiology | 5 years | The development of fatty liver was associated with the incidence of AH even after adjusting for multiple confounding factors (adjusted OR 1.60; 95% CI 1.30, 1.96; p < 0.001). |
| J.H. Huh et al., 2015 [31] | Prospective cohort study (analysis of the data from the Korean Genome and Epidemiology Study on Atherosclerosis Risk of Rural Areas in the Korean General Population [KoGES-ARIRANG]) | 1521 adults (31.8% were males) aged 40 to 70 years without AH at baseline | FLI | Mean follow-up period was 2.6 years | During the follow-up period, 153 subjects (10.06%) developed AH. The OR values (95% CI) for developing AH in the 30 ≤ FLI ≤ 59 group and FLI ≥ 60 group were 1.87 (1.2–2.91) and 2.22 (1.16–4.25), respectively, compared with values in the FLI < 30 group, after adjustment for confounding factors (e.g., liver enzyme activity, adiponectin, IR, and etc.) |
| F. Bonnet et al., 2017 [32] |
Analysis of data from two longitudinal studies in subjects without AH:
| Men and women aged 30–65 years; n = 2565 (D.E.S.I.R. cohort) and n = 321 (RISC cohort). Of them, 1021 subjects (39.8%) from the D.E.S.I.R. cohort (mean age ± SD: 48 ± 9 years; 54% were males) and 63 (19.6%) subjects from the RISC cohort (mean age ± SD: 47.6 ± 7.7 years; 62% were males) developed AH during follow-up | FLI |
| After adjusting for confounding factors, only GGT was significantly associated with the onset of AH (standardized OR, 1.21; 95% CI, 1.10–1.34; p = 0.0001). FLI as a continuous value or ≥60 at baseline was predictive of the onset incidence of AH in the multivariate model |
| R. Lorbeer et al., 2017 [33] | Cross-sectional data from the MRI subset of the KORA FF4 study | 384 participants (58.1% males) aged 39–73 years | HFF measured in the left and right lobe of the liver using single voxel multi-echo 1H-spectroscopy and at the level of the portal vein using a multi-echo Dixon-sequence (MRI) | N/A | The best prediction of AH among all HFF variables was observed for the left lobe HFF threshold (3.57%; OR = 2.62, p = 0.003). Alcohol consumption appeared to be an effect modifier for the association between HFF and AH (non-drinkers: OR = 3.76, p = 0.025; alcohol consumers: OR = 1.59, p = 0.165). |
| P. Liu et al., 2018 [34] | Analysis of the dataset from the Dongfeng–Tongji cohort study | 6704 eligible hypertension-free subjects and 9328 NAFLD-free subjects at baseline (mean age 59.8 ± 7.6 years; 36.3% were males) | Abdominal ultrasound, exclusion of liver disease of other etiology | N/A | Incidental NAFLD, as well as persistent NAFLD, were significantly associated with an increased OR for incidental AH (OR 1.49, 95% CI 1.26–1.76, p < 0.0001 and OR 1.50, 95% CI 1.27–1.78, p < 0.0001, respectively). At the same time, incidental AH was associated with risk of incidental NAFLD (OR 1.45, 95% CI 1.23–1.71, p < 0.0001). Similar data were obtained for persistent AH (OR 1.61, 95% CI 1.35–1.92, p < 0.0001) |
| J.H. Roh et al., 2020 [35] | Retrospective analysis of the data set from the National Health Insurance Service–National Sample Cohort 2.0 (NHIS-NSC 2.0) | 334,280 healthy Korean people with no known comorbidities aged ≥20 years. Of these, 24,678 subjects (7.4%) developed AH during follow-up (60.6% aged ≥ 50 years, 51.1% men) | FLI The following quartiles of the FLI values were suggested: Q1, 0–4.9; Q2, 5.0–12.5; Q3, 12.6–31.0; Q4, >31.0. | The median follow-up was 5.2 years (interquartile range: 3.5–6.3) | The highest FLI values were associated with an increased risk of new onset AH (adjusted OR between Q4 and Q1 FLI values 2.330; 95% CI 2.218–2.448; p < 0.001). |
| Y. Higashiura et al., 2021 [36] | Retrospective cohort study | 15,965 subjects (9566 males, mean age ± SD: 45 ± 11 years; 6499 females, mean age ± SD 45 ± 11 years) | FLI | The mean follow-up period was 6.0 years (range: 1–10 years) | Over a 10-year period, 2,304 men (24.3%) and 745 women (11.5%) developed AH. The combination of FLI with traditional risk factors significantly improved the discriminatory power of the AH risk stratification model for both men and women (p < 0.001) |
| S. Ciardullo et al., 2022 [37] | A systematic review and meta-analysis of 11 observational studies | 390,348 middle-aged individuals (52% men) | Abdominal ultrasound in 6 studies (n = 45,924) Computed tomography in 1 study (n = 1051) FLI in 4 studies (n = 343,373) | Mean follow-up period was 5.7 years (range, 2.6–9 years) | NAFLD was associated with a 1.6-fold increase in the risk of developing AH |
| E. Siafi et al., 2023 [38] | Prospective cohort study | 903 hypertensive patients without a history of cardiovascular disease (mean age ± SD: 52.7 ± 11.4 years; 55% were males) | FLI | Mean follow-up period was 5.2 ± 3.2 years | Patients with FLI < 60 (n = 625) had better BP control vs. their counterparts with FLI ≥ 60 (n = 278) at follow-up (43% vs 33%, p = 0.02). |
| Reference | Study Design | Study Population | NAFLD Diagnosis | Follow-Up | Key Results |
|---|---|---|---|---|---|
| A. Tsuneto et al., 2010 [46] | Prospective cohort study | 1635 Nagasaki atomic bomb survivors (606 men) without fatty liver at baseline (mean age ± SD: 63.1 ± 8.9 years; 37.1% were males) | Abdominal ultrasound, exclusion of liver disease of other etiology | The mean follow-up duration was 11.6 years (SD, 4.6; median, 14.0; range, 1.3–17.1) | AH was an independent predictor of NAFLD (relative risk 1.63; 95%CI, 1.30–2.04; p < 0.001 after adjusting for age, gender, smoking and drinking habits; risk ratio 1.31; 95% CI, 1.01–1.71; p = 0.046) |
| P. Sorrentino et al., 2010 [47] | Prospective cohort study | 271 obese patients with NAFLD and abnormal liver enzymes. 132 patients were included in the final analysis (mean age ± SD: 41.5 ± 9.2 years; 40.15% were males) | Liver biopsy | The median follow-up was 6.4 years (range: 5–8.3 years). | The presence of AH at baseline was independently associated with aggravation of hepatic fibrosis during the follow-up (OR 4.8; 95% CI, 2.7–18.2; p = 0.028) |
| E.C. Aneni et al., 2015 [45] | Cross-sectional study | A Brazilian cohort of 5362 healthy middle-aged men and women (without a history of cardiovascular or liver disease) Mean age ± SD: 43.5 ± 9.3 years; 77% were males) | Abdominal ultrasound, exclusion of liver disease of other etiology, FIB-4 | N/A | Multivariate analysis demonstrated that AH was associated with increased risk of NAFLD (adjusted OR 1.8; 95% CI 1.4–2.3). |
| Y. Wang et al., 2016 [43] | Retrospective cohort study | 836 subjects (mean age ± SD: 53.6 ± 13.5 years), of them, 333 (39.83%) had NAFLD |
| N/A | In patients with AH, NAFLD was independently associated with AH and BP category (OR for NAFLD was 1.476; 95% CI 1.166–2.551 when comparing patients with SBP ≥ 180 mmHg and/or DBP ≥ 110 mmHg and patients with normal BP) |
| Q. Huang et al., 2023 [44] | Meta-analysis (11 cross-sectional studies) | 2049 adults aged ≥20 years (42.5% were males). Of them, 804 (39.2%) had NAFLD at baseline (mean age ± SD: 48.1 ± 13.4; 57.7% were males) | Abdominal ultrasound, exclusion of liver disease of other etiology. Additionally:
| N/A | In the fixed effects model, it was shown that AH was a risk factor for NAFLD (Z = 13.46, p < 0.001) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golubeva, J.A.; Sheptulina, A.F.; Elkina, A.Y.; Liusina, E.O.; Kiselev, A.R.; Drapkina, O.M. Which Comes First, Nonalcoholic Fatty Liver Disease or Arterial Hypertension? Biomedicines 2023, 11, 2465. https://doi.org/10.3390/biomedicines11092465
Golubeva JA, Sheptulina AF, Elkina AY, Liusina EO, Kiselev AR, Drapkina OM. Which Comes First, Nonalcoholic Fatty Liver Disease or Arterial Hypertension? Biomedicines. 2023; 11(9):2465. https://doi.org/10.3390/biomedicines11092465
Chicago/Turabian StyleGolubeva, Julia A., Anna F. Sheptulina, Anastasia Yu. Elkina, Ekaterina O. Liusina, Anton R. Kiselev, and Oxana M. Drapkina. 2023. "Which Comes First, Nonalcoholic Fatty Liver Disease or Arterial Hypertension?" Biomedicines 11, no. 9: 2465. https://doi.org/10.3390/biomedicines11092465
APA StyleGolubeva, J. A., Sheptulina, A. F., Elkina, A. Y., Liusina, E. O., Kiselev, A. R., & Drapkina, O. M. (2023). Which Comes First, Nonalcoholic Fatty Liver Disease or Arterial Hypertension? Biomedicines, 11(9), 2465. https://doi.org/10.3390/biomedicines11092465