Abstract
Autoimmune encephalitis and neurodegenerative disorders share several clinical features, including behavioural and psychiatric manifestations, cognitive impairment, sleep and movement disorders. Therefore, it is not surprising that autoimmune encephalitis is one of the main differential diagnoses of rapidly progressive dementia. However, more chronic presentations of autoimmune disorders have been reported and can lead to the misdiagnosis of a neurodegenerative disease. On the other hand, antibodies against neuronal proteins, such as those directed against NMDAR, can occur during established neurogenerative disorders, and their role in this context is still unclear. They might be simple bystanders or modify the disease course and phenotype. Indeed, autoimmune encephalitis can leave long-term cognitive sequelae and specific antibodies to neuronal surface antigens are associated with clinical and pathological neurodegenerative features. Here we review the link between these antibodies and neurodegeneration. In particular we discuss: (a) the possibility that autoimmune encephalitis presents as a neurodegenerative disease, identifying the red flags that can help in the differential diagnosis between antibody-mediated and neurodegenerative disorders; (b) the occurrence of antibodies against neuronal surface antigens in patients with neurodegenerative disorders and their possible role in the disease course; and (c) the long-term cognitive and neuroradiological changes associated with autoimmune encephalitis, as well as the biomarkers that can help to predict the cognitive outcome. Finally, we review the clinical and pathological features of IgLON5 antibodies-related encephalitis, a unique model of the relationship between antibodies and neurodegeneration.
1. Introduction
Autoimmune encephalitis (AE) represents a group of brain disorders characterised by the subacute onset of diverse neurological and neuropsychiatric symptoms due to a pathologic immune response directed against self-antigens expressed in the central nervous system. AE is often associated with antibodies against neuronal surface antigens (NSA-Ab) [1,2]. Due to the wide and different expression of the various antigenic targets, clinical manifestations are variable and include diverse combinations of cognitive dysfunction, psychiatric and behavioural disturbances, movement disorders, seizures, sleep disorders and dysautonomia, among others (Table 1) [2].
Many NSA-Ab-related syndromes are associated with cognitive impairment and sometimes this is the first and prevalent manifestation, challenging the diagnosis. This is particularly true in the elderly, as encephalitic signs can be absent [3], and the disease can progress slowly, mimicking a primary neurodegenerative disorder.
On the other hand, the idea that autoantibodies could be involved in dementia is old [4,5,6] and several targets have been investigated over time including neuronal receptors and glial protein (for review see [7]). Antibodies against several neuronal proteins involved in different neurodegenerative diseases (i.e., amyloid β (Aβ), tau and alpha-synuclein) occur in both healthy subjects and in pathological conditions and they could have either a physiological role in preventing the accumulation of pathological proteins or exert additional damage [8]. Antibodies against neuronal proteins might therefore be part of the homeostatic immunity, emerging for example secondarily to the physiological turnover of neurons outside the CNS, such as in the enteric nervous system [9]. After the discovery of NMDAR-Ab in patients with NMNDAR encephalitis [10], the presence of these antibodies was investigated in a few cohorts of patients with neurodegenerative disorders [11,12,13]. Oppositely to what is generally observed in patients with NSA-Ab-related AE, these antibodies are often found only in the serum in patients without encephalitic signs and often belong to the IgA/IgM subclasses (instead of IgG) [11,12,13,14,15], and therefore their role is not yet clear. In this context, NSA-Ab could be bystanders of a degenerative process or play a pathogenic function, if not primary at least secondary, acting as modifiers of the clinical phenotype and of the disease course. Indeed, NSA-Ab have the potential to cause secondary neurodegeneration even in typical AE, with mechanisms that have not yet been fully elucidated.

Table 1.
The main clinical features and pathogenic mechanisms of NSA-Ab.
Table 1.
The main clinical features and pathogenic mechanisms of NSA-Ab.
| Antibody Target | Age/ Sex Prevalence | Clinical Syndromes/Symptoms | Acute Phase Cognitive Features | Ancillary Test | Antibody Pathogenicity and Mechanism of Cognitive Impairment |
|---|---|---|---|---|---|
| NMDAR [10,16,17,18] | Young/F | Encephalitis with psychiatric symptoms, seizures, movement disorders, autonomic instability, hypoventilation, coma | Severe cognitive dysfunction involving all domains (memory, executive function, attention, language, visuospatial processing and social cognition) | MRI: mainly normal (70–80%) or unspecific. CSF: 80% abnormal (pleocytosis, oligoclonal bands); EEG: usually abnormal (90%) with slow wave or epileptic changes and rare delta brush | NMDAR-Ab cause cross-linking and internalization with reduced NMDARs density and consequent reduced NMDAR-dependent LTP leading to memory dysfunction |
| LGI1 [19,20,21,22,23] | Elderly/M | Limbic encephalitis with memory impairment and seizures. FBDS can precede the onset of cognitive impairment | Disorientation, confusion and autobiographical memory impairment | MRI: abnormal (75%), mostly showing increased medial temporal lobe and hippocampus signal; CSF: abnormal 25% (pleocytosis and increased proteins); EEG: 50% abnormal (epileptiform activity or slowing) | Disruption of the interaction with ADAM22 and reduction of AMPAR; possible complement activation. Antibodies abrogate LTP induction at CA3-CA1 hippocampal synapses in animal model leading to memory impairment |
| CASPR2 [19,24,25,26,27] | Elderly/M | Limbic encephalitis, neuromyotonia and Morvan’s syndrome, neuropathic pain | Common cognitive dysfunction, anterograde and episodic memory impairment | MRI: more often normal (30% abnormal signals in the hippocampus); CSF: often normal (30% pleocytosis, increased proteins or oligoclonal bands); EEG: 70% abnormal (epileptic changes or slowing) | CAPSR2 internalization and inhibition of CASPR2/TAG1 interaction. Possible reduction of Kv1 and AMPARs leading to neuronal hyperexcitability and ineffective recruitment of post-synaptic AMPARs leading to memory impairment |
| GABAB receptor [28,29,30,31] | Elderly/M | Limbic encephalitis, seizures | Cognitive/behavioural dysfunction in most patients with memory impairment and confusion | MRI: abnormal signals in the temporal lobe and hippocampus (70%); CSF: frequently altered (80%); EEG: epileptic changes (80%) | Antibodies can prevent the activation of the GABABR and block its function; however, how this mechanism can impact memory has not been investigated in vivo |
| AMPA receptor [32,33,34,35,36] | Middle aged/F | Limbic encephalitis, seizures | Prominent memory impairment: amnesia could be isolated at onset | MRI: often bilateral medial temporal lobes and insular cortex involvement; CSF: 70% abnormal; EEG: abnormal in about half cases | Internalisation of GluA2 containing AMPARs with reduction of extrasynaptic AMPAR leading to LTP changes and consequent memory impairment |
| GABAA receptor [37,38] | Broad age range/no sex predominance | Encephalitis and refractory seizures or status epilepticus | Cognitive impairment in two thirds of patients | MRI: multifocal cortical and subcortical FLAIR signal abnormalities: CSF. Abnormal in about 50% of cases; EEG: usually abnormal | Antibodies selectively remove GABAAR from synapses, downregulation of the GABAAR function; however how this mechanism can impact cognition has not been investigated in vivo |
| DPPX [39,40] | Middle aged/M | Weight loss/diarrhoea, cognitive/behavioural changes, CNS hyperexcitability (myoclonus, tremors and hyperekplexia) | Memory loss, confusion | MRI: normal or unspecific; CSF: often normal; EEG: 70% abnormal | DPPX and Kv4.2 membrane expression is reduced by patient antibodies, whilst enteric neuron activity is increased; how this mechanism can impact cognition has not been investigated in vivo |
| Glycine receptor [41,42,43,44] | Broad age range/M | Progressive encephalopathy with rigidity and myoclonus (PERM), epilepsy, SPS | Cognitive deficits, encephalopathy | MRI: often normal or non-specific; CSF: abnormal in less than half cases; EEG: often abnormal (70%), with focal/diffuse slowing or rarer epileptic changes | GlyR internalisation; how this mechanism can impact cognition has not been investigated in vivo |
| IgLON5 [45,46,47] | Elderly/no sex predominance | Sleep disorder, bulbar symptoms, gait abnormality followed by cognitive dysfunction | Sleep disorder, bulbar symptoms and gait abnormality followed by cognitive dysfunction | MRI: often normal or non-specific; CSF: 60% pleocytosis | Internalisation with reduction of IgLon5 expression (irreversible in the long term after the removal of antibodies); memory impairment due to gamma-oscillation dysfunction, neurodegeneration and synaptic dysfunction |
CSF: cerebrospinal fluid; EEG: electroencephalogram; F: female; M: male; MRI: magnetic resonance imaging.
Finally, in the last years, the borders between autoimmunity and neurodegeneration have been further thinned by the discovery of IgLON5 antibodies [45], which has shed new light on the possible role of autoimmunity in neurodegenerative disorders.
Others have reviewed the role of antibodies directed against neuronal proteins involved in different neurodegenerative diseases (e.g., amyloid β (Aβ)) in the pathogenesis of these disorders [8]. Here, we focus specifically on antibodies against neuronal surface antigens primarily involved in AE, reviewing the different interplay between neuronal surface antibody-mediated autoimmunity and neurodegeneration (Figure 1). We first discuss the presentation of AE as a neurodegenerative-like disorder and the red flags that could help in the differential diagnosis between these conditions. Secondly, we present the current evidence of the occurrence and possible role of NSA-Ab in neurodegenerative diseases. We further highlight the long-term neuropsychological, neuroimaging and biomarker changes associated with NSA-Ab in AE patients and discuss the possible underlying mechanisms leading to neurodegeneration in this context. Finally, we present the current knowledge on IgLON5 disease as a new model of interaction between autoimmunity and neurodegeneration.
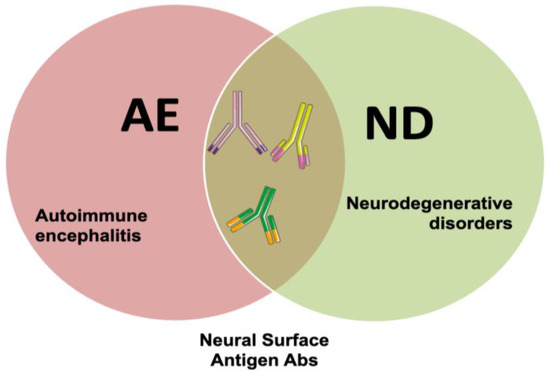
Figure 1.
Interplay between AE and neurodegeneration mediated by NSA-Ab. AE can present with neurodegenerative-like phenotype and NSA-Ab can cause neurodegeneration in patients with AE. On the other hand, NSA-Ab might be found in patients with neurodegenerative disorders, although their significance is still unclear.
2. Neuronal Surface Antibody-Mediated Encephalitis Presenting as a Neurodegenerative Disorder
AE and neurodegenerative disorders share several features including behavioural and psychiatric disorders, cognitive impairment, movement and sleep disorders.
Patients with AE usually present a combination of clinical features associated with variable brain MRI and CSF inflammatory changes (Table 1). However, cognitive impairment and/or movement and sleep disorders can be the main clinical feature misleading the diagnosis towards a neurodegenerative disorder. It is not surprising that AEs are increasingly recognised as one of the most common causes of treatable rapid progressive dementias, with frequencies progressively increasing over time thanks also to the expanding list of NSA-Ab [48,49,50,51,52].
In a setting when an autoimmune aetiology for the dementia is strongly suspected, Flanagan et al. [53] reported the presence of autoantibodies targeting the VGKC complex in 15% of cases. In the same cohort, 64% of cases responded to immunotherapy. A shorter delay from symptom onset to initiation of therapy for autoimmune dementia increased the likelihood of response [53]. However, 41% of immunotherapy-responsive dementia patients had normal brain MRIs, and many patients showed normal CSF and EEG; almost 9% of the disorders had been initially diagnosed as Creutzfeldt-Jakob disease (CJD) [53]. Several other reports described patients with rapidly progressive dementia and antibody mediated encephalitis [28,54,55].
However, not all patients with antibody-mediated dementia present with a rapidly evolving clinical picture. Autoimmune-mediated cognitive decline can progress slowly over many months, and therefore may be mistaken for a primary neurodegenerative disorder such as Alzheimer’s disease (AD) or frontotemporal dementia (FTD) [48,56]. NMDAR encephalitis might present with atypical features and prominent cognitive disturbances, including memory loss, cognitive fluctuations, visual hallucinations and sleep disorder, reminiscent of Lewy Body Dementia (LBD) [57]. Similarly, due to the prominent memory impairment, often associated with confusion and disorientation, LGI1 encephalitis could be misdiagnosed as neurodegenerative dementia including AD and LBD [58,59,60]. In a recent retrospective study, exploring the frequency of dementia diagnosis in patients over the age of 45 with LGI, NMDAR, CASPR2 and GABABR encephalitis, 67/175 (38%) were found to satisfy the 2011 NINCDS-ADRDA criteria for dementia and in 33 (52%) a neurodegenerative disorder was suspected [61]. Interestingly, imaging or CSF inflammatory signs were absent in 25% of patients and in 61% of 44 patients in whom neurodegenerative CSF biomarkers were assessed, at least one was found altered [61], underlying the difficulties of differential diagnosis between AE and dementia in certain clinical contexts.
However, not only dementias but also other neurodegenerative diseases, mainly presenting with movement disorders, can be associated with the presence of NSA-Ab. Patients with LGI1, IgLON5, DPPX and GABABR antibodies have been misdiagnosed with Parkinson’s disease (PD), progressive supranuclear palsy (PSP), cortico-basal syndrome (CBS) or multisystem atrophy (MSA) [45,62,63,64,65]. REM sleep behaviour disorder (RBD), which can anticipate by decades the onset of a neurodegenerative disease, in particular MSA and PD [66], has been reported in patients with CASPR2 and LGI1-antibody-associated limbic encephalitis [19,67,68] and with IgLON5-antibody linked neurodegeneration [45]. Finally, status dissociatus, a complete breakdown of the boundaries of the different states of being, which are wakefulness, REM sleep, and non-REM sleep, with motor hyperactivity, which may be observed in the final phase of several different neurodegenerative disorders, has also been reported in patients with CASPR2-Abs, and less commonly, LGI1-Abs or NMDAR-Abs [69,70], widening the overlap between neurodegenerative and autoimmune conditions.
We explored the frequency of NSA-Ab in patients who had received a diagnosis of a wide range of neurodegenerative disorders. We observed NSA-Ab in 13.8% (13/93) of patients vs. 2% of controls, with no difference between patients presenting with prominent dementia or parkinsonism [71]. When we retrospectively applied the AE criteria [1] in the seropositive cases without including the antibody results, five patients did not meet the criteria for possible AE because they had a chronic course, whereas in three cases it was not possible to exclude other diagnoses. Of the remaining five patients, who met criteria for possible AE, only two showed inflammatory CSF changes and none had AE-like MRI abnormalities [71]. Altogether these observations underline on one hand, the difficulty of differentiating AE from neurodegenerative disorders in the setting of a slowly evolving clinical picture, on the other hand they support the possibility that secondary NSA-Ab might develop during the course of neurodegenerative disorders, as discussed below.
Diagnostic Clues to the Autoimmune Aetiology
Given the fact that AE and “autoimmune dementia” are potentially treatable disorders, it is important to achieve an early diagnosis. On the other hand, the indiscriminate search of NSA-Ab could lead to misdiagnosis and overtreatment along with over-utilization of resources. The need to identify patients with AE early led to the creation of consensus criteria [1], mainly based on the clinical features (i.e., subacute onset of cognitive impairment, behavioural and psychiatric changes and focal CNS signs) and brain MRI (i.e., hyperintense T2/FLAIR signal involving one or both medial temporal lobes, or in multifocal areas involving grey matter, white matter, or both) and CSF findings suggestive of neuroinflammation (i.e., pleocytosis and oligoclonal bands) or epileptic or slow-wave activity involving the temporal lobes on the EEG [1]. In this setting, the differential diagnosis of AE from even rapidly progressive dementia should be relatively easy. Moreover, patients with neurodegenerative dementia should have additional supporting findings such as specific changes in CSF neurodegeneration markers (i.e., altered Aβ42, total tau and phosphorylated tau values and ratios) [72] or molecular imaging specific alterations that should be absent in patients with AE [73]. However, in clinical practice this distinction is not always straightforward. Indeed, antibody-mediated cognitive deficits can evolve slowly over time, and inflammatory changes can be absent, particularly in the elderly population and in association with certain antibodies [3]. On the other hand, a tumultuous evolution of cognitive deficits and a frankly inflammatory CSF can be observed also in patients with primary degenerative disorders [74,75].
To help clinicians to promptly recognize patients with potentially treatable disorders, several studies tried to identify the main features associated with an “autoimmune dementia”. These have been classically defined as: (1) subacute onset with a rapidly progressive course; (2) symptoms fluctuation; (3) presence of tremor/myoclonus; (3) coexisting organ-specific autoimmunity; (4) inflammatory CSF; (5) presence of MRI changes suggestive of an inflammatory process; (6) risk factors or recent history of tumour [48,53,56,76]. A more recent study suggested that the presence of subtle seizures could be a red flag for AE in patients with dementia [61]. Nevertheless, seizures can occur also in neurodegenerative disorders [77], even early in the disease course [78], therefore the results of other investigations including imaging features and pathological biomarkers should be considered in the differential diagnosis. Nevertheless, in the same study, as already mentioned, 14 out of 44 patients tested had a CSF biomarker profile suggestive of AD or CJD, challenging the differential diagnosis [61]. In these cases, more specific tests such as RT-QuIC can help to exclude CJD, whereas an increased IgG index might suggest an autoimmune aetiology. We found that an irregular progression and the lack of a diagnosis for a specific type of dementia or parkinsonism associate with the presence of NSA-Ab [71]. However, the utility of these suggested red-flags in guiding the antibody testing has not been systematically assessed in prospective studies. Dubey and colleagues recently evaluated the diagnostic performance of the antibody-prevalence-in-epilepsy and encephalopathy (APE2) and responsive-to-immunotherapy-in-epilepsy and encephalopathy (RITE2) scores in the evaluation and management of patients with encephalopathy and cognitive impairment [79]. They observed that an APE2 score ≥ 4 was 99% sensitive and 93% specific for neural-specific-antibodies and that a RITE2 score ≥ 7 had 96% sensitivity and 86% specificity for favourable initial immunotherapy response supporting the utility of this approach in selecting patients candidate to antibody screening. However, these results warrant replication in prospective cohorts.
3. NSA-Ab in Patients with Neurodegenerative Disorders
Despite the clear existence of cases of immunotherapy-responsive dementia associated with NSA-Ab, the presence of several neuronal antibodies has been reported in patients with ascertained prion disease [80,81,82,83,84,85], challenging the significance of this finding and further complicating this diagnostic dilemma in cases with more chronic presentations. However, it is likely that these findings reflect the inclusion of patients with other diagnoses or a secondary phenomenon. Indeed, the frequency of NSA-Ab appears rare in patients with pathologically confirmed CJD [84,86].
To explore the prevalence of NSA-Ab in patients with defined primary dementias, Çoban A et al. [87] investigated 50 patients, finding NMDAR antibodies in one case presenting with LBD phenotype. Even though this patient had some features suggestive of an autoimmune aetiology, the presence of “atypical features” in the remaining cases failed to predict the presence of an NSA-Ab, highlighting the necessity to identify other markers predictive of an autoimmune dementia. However, the concept that “atypical” features associate more frequently with the presence of NSA-Ab recur in different studies (for a review see [88]).
Indeed, Prüss et al. [11] described the presence of NMDAR-Ab of the IgA isotype in a small cohort of patients with atypical dementia. A subgroup of positive patients partially responded to immunotherapy [11]. Purified IgA containing NMDAR IgA antibodies caused substantial loss of NMDARs and further synaptic proteins in primary hippocampal cultures, resulting in marked changes of NMDAR-mediated currents [11]. These results were further explored in a large series of 660 cases including different neurological disorders and controls. Serum NMDAR-Ab of IgM, IgA, or IgG subtypes were detected in 16.1% of 286 dementia patients and in 2.8% of 217 cognitively healthy controls. Higher prevalence of serum antibodies was detected in patients with “unclassified dementia” followed by PSP, CBD, PD-related dementia (PDD), and primary progressive aphasia (PPA). Among the unclassified dementia group, 60% of 20 patients had NMDAR-Ab, accompanied by higher frequency of CSF abnormalities, and subacute or fluctuating disease progression. Immunotherapy in selected prospective cases resulted in improvement of clinical and functional imaging parameters, as well as antibody disappearance. Epitope mapping showed varied determinants in patients with NMDAR IgA-associated cognitive decline. However, antibodies were rarely found in CSF [12], and therefore their role in the clinical manifestations is unclear. Another study found increased frequency of IgA/IgM serum NMDAR antibodies in patients with atypical AD (i.e., with minimal or no hippocampal atrophy or normal CSF Tau and amyloid β) [13].
In PD, an initial study involving 258 PD patients and 1730 controls did not find an association with the presence of NSA-Ab, although detailed clinical information was not available [89]. In the already discussed Doss et al., study [12], however, NMDAR-Ab, although not overall more frequent in patients with PD, appeared to be more common in PDD than PD without dementia patients [12]. Nevertheless, in a further cohort of PD patients with and without cognitive impairment (n = 296) and controls (n = 295), the association between NMDAR IgA/IgM antibodies and the presence and severity of cognitive impairment in PD was not confirmed. Moreover, unexpectedly, the frequency of NMDAR IgA/IgM antibodies was lower in PD (13%) than in controls (22%), and positively correlated with age in the latter [90].
Although the appearance of antibodies in neurodegenerative disorders is likely to be secondary, a role in modulating the phenotype has not been systemically investigated. Doss et al. [12] found no association between NMDAR-Abs and neuropsychiatric features in patients with dementia, whereas these antibodies have been associated with minimal symptomatic response to cholinesterase inhibitor treatment, more pronounced cognitive decline and overall poorer prognosis in patients with “non-classical” AD [13].
Antibodies against brain proteins were found in healthy individuals [89,91] as well as in patients with several neurological or psychiatric disorders (reviewed in [92,93]. Whereas the physiological role of some natural occurring antibodies is partially understood (i.e., cellular debris removal) [94], for antibodies against specific neuronal antigens this is unknown, nor are the possible circumstances and mechanisms that may turn these potentially ‘harmless’ antibodies into disease-relevant ones. Dahm L et al. [89] found no difference in immunoglobulin classes distribution (IgM, IgG and IgA) or antibody titres between patients and controls. Therefore, the role of the blood-brain barrier (BBB) in defining the disease status was investigated. It was found that schizophrenic individuals with a past or present history of BBB disturbance were more likely to have more pronounced neurological symptoms if NMDAR antibody seropositive [95].
An interesting possibility is that the difference between health- or disease-status could be related to different functionality or epitope specificity of the antibodies. Castillo-Gómez E et al. [96] showed that all NMDAR-Ab positive sera, derived from randomly selected individuals, were able to induce NMDAR internalisation in inducible pluripotent stem cell (IPSC)-derived human cortical neurons. Several different epitopes recognised by NMDAR-Abs were identified, without any consistent functional or epitope pattern related to health/disease state. However, these findings were contradicted by a subsequent study showing different functional effects of NMDAR-Abs from schizophrenic patients versus those found in healthy individuals [97]. NMDAR-Ab from patients, but not from healthy subjects, were shown to alter the surface dynamics and nanoscale organization of synaptic NMDAR and its anchoring partner the ephrin-B2 receptor in heterologous cells, cultured neurons and in mouse brain and prevent long-term potentiation at glutamatergic synapses, while leaving NMDAR-mediated calcium influx intact.
It is clear that more studies are needed to clarify the role of the antibodies in health and disease, mainly to identify the factors associated with the disease status, looking not only at the antibodies per se but also at all those factors that could influence their ability to reach their targets (i.e., BBB integrity) and those which could modulate the immune response (i.e., HLA status). The implications of these findings could help support the use of immunotherapy in patients that otherwise would have remained untreated and avoid ineffective and potentially harmful treatment in those cases which would not benefit.
4. Secondary Neurodegeneration in Patients with Autoimmune Encephalitis
NSA-Abs disrupt the function of their target in different ways, including internalization, complement activation and through the loss of interaction with other proteins (see Figure 2) [98].
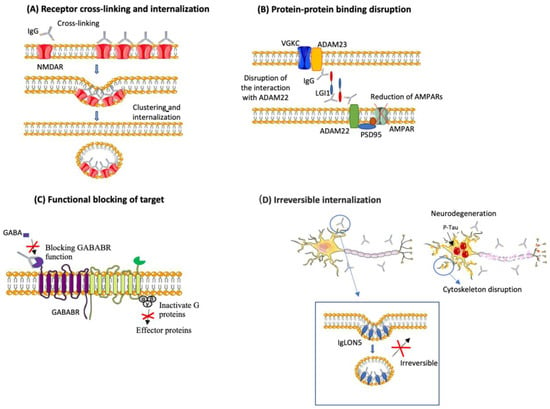
Figure 2.
Pathogenic mechanisms of selected NSA-Ab. NSA-Ab alter their target through different mechanisms. (A) Some, such as NMDAR-Ab and AMPAR-Ab cause internalization of their targets. (B) others (i.e., LGI1-Ab) alter the interaction between their target and partners proteins; (C) others (i.e., GABABR-Ab) block their targets. These effects are usually reversible upon removal of the antibody. (D) IgLON5 antibodies cause irreversible internalization of IgLON5, causing alterations of the neuronal cytoskeleton, tau accumulation and neurodegeneration.
In most cases, both animal and in vitro studies demonstrated that the antibody effects are reversible upon antibody removal and indeed patients with AE mediated by NSA-Ab significantly improve with immunotherapy. Nonetheless, is becoming increasingly evident that despite the clinical improvement, a variable proportion of patients present long-term sequelae and do not go back to their pre-morbid condition in terms of cognitive status, employment, and quality of life [21,99]. These observations are supported by changes in long-term cognitive function and brain anatomy as well as changes of neurodegeneration biomarkers, as discussed below.
4.1. Long-Term Cognitive Outcome
Long-term cognitive impairment is an increasingly recognized sequela of AE associated with NSA-Ab (Table 2).
Table 2.
Representative studies investigating long-term cognitive outcome in patients with AE.
In NMDAR encephalitis, cognitive dysfunction is prominent in the acute phase, affecting all domains [107,108]. Memory deficits are the most encountered, followed by executive dysfunction, language impairment and visuospatial difficulties [107]. In the post-acute phase, many patients with NMDAR encephalitis report self-observed deficits including memory problems, concentration difficulties, and social withdrawal [100,102,109]. Several studies have consistently shown persistent cognitive deficits in patients with previous NMDAR encephalitis. A recent systematic review outlined that more than 75% of patients have neuropsychological deficits at any point after treatment, affecting mainly memory and executive function [108]. In most cases, the dysfunction remains stable or improves, although up to 25% of patients may fluctuate or deteriorate over time [108]. As for the general clinical outcome [10,16,110], a worst long-term cognitive function is related to a delay in treatment [100]. Indeed, patients treated more than 3 months after onset have almost eight times the odds of an adverse neuropsychological outcome, compared to those treated earlier [108]. These data were confirmed in a recent study [111] showing persistent cognitive deficits in more than 80% of patients, which was severe in 50%, 2.3 years after onset. At subsequent follow up, 4.9 years after onset, most patients had improved but 30% continued to have severe deficits, mainly involving memory and executive function and few patients had deteriorated, despite the absence of relapse. A poor cognitive long-term outcome was predicted by delayed treatment, older age, and higher disease severity, and longer overall disease duration. The cognitive deficits were not mirrored by functional assessment as measured by the modified Rankin scale (mRS) which showed no deficit in 47.1% of cases, pointing out the importance of neuropsychological assessment in the evaluation of the outcome in these patients. Importantly, this study showed that although cognitive improvement occurs mostly in the immediate post-acute phase, it continues over time, even years after onset [103].
In LGI1 encephalitis, cognitive impairment or encephalopathy are frequent in the acute phase and can persist over time [21,112]. Van Sonderen and colleagues reported persistent amnesia for the disease period and retrograde amnesia in 86% of 21 patients observed over a median follow up period of 42 months [22]. In another study, 29% of patients had moderate to severe cognitive impairment at 24 months follow up, but only 35% were able to return to work or to all premorbid activities [21]. A more in-depth characterization of the associated cognitive impairment showed episodic verbal and visuospatial memory deficits, impaired working memory, impairment of executive function and attention as well as impaired semantic and phonemic fluency [104,113]. As in NMDAR encephalitis, higher disease severity, delays to immunotherapy or longer immunotherapy courses were associated with more severe cognitive dysfunction [104].
Data on long-term cognitive outcome are very scarce in patients with CASPR2 encephalitis. In a study pooling LGI1- and CASPR2-Ab positive cases, some form of cognitive impairment was detected in 28% of patients, with 9% showing a severe dementia at last follow-up using the clinical dementia rating scale (CDR ≥ 0.5) [60]. In another study, including patients with CASPR2-Abs in the CSF, 19% exhibited persistent amnesia at 2 years follow-up [24], although formal neuropsychological testing were not performed. In a small study, assessing memory performance in four patients with CASPR2-Abs at an average of 4.2 (range 3–5) months after immunoadsorption, no memory recovery was observed, despite an overall clinical improvement [114]. However, the observation time was too short and the sample too small to achieve definitive conclusions. Therefore, more data are needed to establish the long-term cognitive outcome of CASPR2 encephalitis.
Similarly, few data are available for other, rarer, forms of AE. In a retrospective study on 22 patients with paraneoplastic GABABR encephalitis, severe anterograde amnesia was observed in all surviving patients at 2-year follow up [105]. In a more recent prospective study, cognitive deficits were detected in 57.1% of patients by CRS and 50% by Montreal cognitive assessment (MoCA) 24 months after onset. Neuropsychological testing revealed deficits in working memory, visual memory, executive function and nonverbal reasoning [106]. Only an older age at onset (≥45 years) was predictive of poor cognitive outcome in this study [106]. In AMPAR encephalitis [33] and DPPX encephalitis [39] 57% and 22% of patients, respectively, had persistent cognitive impairment, although no specific testing was performed. Although cognitive impairment is often overlooked in patients with GlyR-Abs, a recent study found evidence of mild cognitive impairment or dementia in five patients, with prevalent memory impairment, [44] suggesting the need to better explore the cognitive function in patients with GlyR-Ab. No study to date assessed the long-term outcome of GABAAR encephalitis, although in a case series most patients showed a good recovery despite the frequent presence of cognitive impairment at onset [37].
4.2. Brain MRI Changes
The long-term cognitive alterations observed in patients with AE are mirrored by permanent structural brain changes. Finke and colleagues [101] first described functional and microstructural alterations in patients with previous NMDAR encephalitis. Using resting state fMRI, they showed reduced bilateral functional connectivity between the hippocampus and the anterior default mode network (aDMN). These alterations correlated with memory performance. Diffusion tensor imaging (DTI) analysis showed widespread white matter changes, which correlated with mRS [101]. Patients with persistent cognitive impairment showed extensive damage of the superficial white matter (WM) including U-fibres and intracortical myelin [115]. On the contrary, grey matter (GM) and routine clinical MRI were normal, despite a tendency towards smaller hippocampal volumes in patients compared to controls [101]. Indeed, in a subsequent study, the same group, using a multimodal imaging approach that combined analysis of hippocampal subfield volumes with DTI derived assessment of hippocampal microstructural integrity, observed hippocampal subfield atrophy and impaired microstructural integrity of the hippocampus in patients recovering from NMDAR encephalitis [116]. Hippocampal volume and microstructural changes correlated with memory performance and disease severity and duration [116]. However, brain structural and functional abnormalities in NMDAR encephalitis seems to extend beyond hippocampus. A number of recent works using different MR techniques as DTI, diffusion kurtosis imaging, voxel-based morphometry (VBM) and surface-based morphometry disclosed diffuse structural changes in hemispheric WM and cortical and subcortical GM, with correlation with cognitive scores (Table 3) [117,118,119,120,121]. Accordingly, network analysis of resting-state fMRI data in patients with post-acute NMDAR encephalitis revealed alterations in whole-brain functional connectivity including impairment of the connectivity between the hippocampus and the medial temporal lobe (MTL) and the DMN, and significant reductions in intranetwork connectivity within the MTL, sensorimotor, lateral-temporal, and visual networks [122]. The disruption of the hippocampal and medial-temporal-lobe network connectivity correlated with memory impairment, whereas schizophrenia-like symptoms were associated with functional connectivity changes in functional connectivity of the frontoparietal control network and the ventral attention network [122].
Table 3.
Representative studies investigating long-term brain imaging changes and their correlation with cognitive outcome in patients with AE.
Long term structural brain changes have also been described in patients with encephalitis previously attributed to VGKC antibodies. In LGI1 encephalitis, follow-up routine MRI showed hippocampal atrophy on visual inspection in most patients, ranging from 77% to 96% of cases [104,113,127]. Volumetric analysis group revealed reductions of all hippocampal subfield volumes except CA1 and mean diffusivity (MD) studies showed altered microstructural integrity. These alterations correlated with memory deficits [104]. Further studies have reported rather conflicting results. Miller and colleagues [124] reported a hippocampal volume loss confined to bilateral CA3, and associated with severe episodic but not semantic amnesia. On the contrary, Hanert and colleagues [113] disclosed a global hippocampal volume reduction in patients with a median time of 3.5 years from the onset of LGI1 encephalitis, associated with deficits in pattern separation performance best predicted by the volume of the hippocampal dentate gyrus, whereas CA1 volume was highly predictive of recognition memory deficits [113]. Regarding extra-hippocampal involvement, Szots and colleagues [128] studied a small sample of patients and reported widespread DTI changes in the cerebral and cerebellar WM, most prominent in the anterior parts of the corona radiata, capsula interna and corpus callosum. Furthermore, MR spectroscopy revealed lower glutamine/glutamate at WM levels and volumetric analysis significantly smaller volume in several brain areas (i.e., cerebellum, brainstem, accumbens, hippocampus, corpus callosum, cerebral WM) [128]. These widespread alterations were not confirmed on a larger sample in which VBM analysis revealed no GM volume change other than in hippocampus and there was no evidence of structural WM damage as assessed using DTI [126]. Notwithstanding, functional connectivity alterations in several large-scale networks, including the DMN which showed an aberrant structure-function relationship with the damaged hippocampus, were evident. In addition, connectivity in the sensorimotor, salience and higher visual networks was impaired independent of hippocampal damage [126].
For encephalitis associated with other type of NSA-Ab, as CASPR2, GABAAR, GABABR and AMPAR, imaging studies focused on follow-up findings are not available except on very small groups or case reports [38,123,129,130,131], generally reporting bilateral mediotemporal abnormalities with possible and variable extra hippocampal involvement, but more in-depth studies are needed for a better characterization.
4.3. Biomarkers and Predictors of Neurodegeneration
Fluid biomarkers of neurodegeneration in AE have been the subject of several studies, especially in the last few years (Table 4). The large majority refers to the CSF and the two most extensively investigated analytes are neurofilament light chain (NFL) and total tau (T-tau). NFL is a component of the cytoskeletal scaffold of large myelinated axons and its CSF concentrations are increased as a consequence of neuroaxonal damage [132]; tau is likewise a mainly axonal protein and is considered a marker of neuronal injury [133].
Table 4.
Representative studies investigating neurochemical biomarkers of neurodegeneration in autoimmune encephalitides.
Increased CSF levels of T-tau and NFL have been reported in AE [135,136] and specifically both in NMDAR [135] and in LGI1 encephalitides [140]. Indeed, the two CSF biomarkers correlate with each other in AE [143]. On the other hand, while NFL levels are similar to those observed in AD [140], T-tau in AE is generally lower than in AD and the more AD-specific biomarker phosphorylated tau (P-tau181) is in most cases not increased [140,144]. Actually, in the large study of Day et al., on 45 cases of NSA-Ab positive AE (NSA-AE; NMDAR, LGI1 and CASPR2), after correction for age CSF levels of T-tau did not significantly differ from those of neurologically healthy controls (HC) [137]. NFL has also been evaluated in serum, where its levels moderately correlate with those in the CSF [145]. Increased levels of NFL in both serum and plasma have been reported in AEs, including NSA-AEs and specifically NMDAR encephalitis compared to HC [138,145,146]. Serum NFL levels in NMDAR encephalitis, however, are lower compared to herpes simplex encephalitis (HSE) [138].
CSF levels of NFL have been reported to be lower in NSA-AEs compared to AEs with antibodies against intracellular antigens (ICAs), while for CSF T-tau the difference was not statistically significant [143]. Conflicting data exist, however, regarding specific forms of NSA-AE, as, for example, some investigations found lower CSF and/or serum levels of NFL in NMDAR encephalitis [142,145], whereas another group observed increased CSF NFL levels in this form but not in LGI1/CASPR2 encephalitis [137]. Several investigations (actually including both NSA-AEs and AEs with antibodies against ICAs) have found higher CSF levels of NFL, but not of T-tau, in patients with neoplasia compared to those without [136,139], although the study of Day et al., on NSA-AEs reported lower CSF NFL levels in the presence of a tumour [137]. While in NMDAR encephalitis an association between CSF NFL and the presence of ovarian teratoma has not been established [147], forms of NMDAR encephalitis developing after HSE or in association with other tumours or demyelinating diseases seem to display higher CSF NFL levels compared to idiopathic or teratoma-associated cases [142].
Several associations between biomarker levels and clinical phenotype have been proposed in AE, albeit not by all investigators [145]. In a cohort including both NSA-AEs and AEs with antibodies against ICAs, CSF NFL correlated negatively with scores on both mini mental state examination (MMSE) and frontal assessment battery (FAB) [143]. In LGI1 encephalitis, CSF levels of NFL seem to be higher in patients with seizures (either clinical or electrographic), but they are not associated with facio-brachial dystonic seizures (FBDSs) specifically [140]. In NMDAR encephalitis, higher CSF levels of NFL have been reported to be associated with extreme delta brush (EDB) on EEG [147], involuntary movements [142], occurrence of prodromal symptoms, seizures or status epilepticus (SE), admission to intensive care unit (ICU), absence of immunotherapy in the first 4 weeks of disease, and subsequent need of second-line immunosuppressive treatment, while for serum NFL levels the only significant associations were those with ICU admission and initial absence of immunotherapy [138].
NFL might correlate with disability, especially longitudinally. In two studies including both NSA-AEs and other AEs, initial CSF levels of NFL, but not of T-tau, correlated with disability (expressed as score on the modified Rankin Scale, mRS) at last follow-up (namely, after 12 months) [135,136]. Similar findings were confirmed for NMDAR and LGI1 encephalitides [142,148], although not in all cohorts [138,140], as well as for serum NFL in NMDAR encephalitis, with the biomarker discriminating between patients with mRS score above vs. below 2 at 1-year follow-up with an area under the curve (AUC) of 0.697 [141]. In agreement with this, serum NFL is lower in NMDAR encephalitis patients with good treatment response compared to those with poor response [141].
Associations between biomarkers and neuroimaging features have been described. In NSA-AEs, Körtvelyessy et al. [139] found higher CSF levels of T-tau and NFL in association with temporal FLAIR hyperintensities on MRI followed, in a later phase, by hippocampal sclerosis. All patients developing hippocampal sclerosis had increased levels of at least one of the two biomarkers, with T-tau seeming to be more predictive of hippocampal sclerosis [139]. In NMDAR and LGI1 encephalitides, higher CSF NFL levels have been reported in patients with abnormal MRI [142], while an association between increased CSF NFL and hippocampal atrophy has been shown in NMDAR encephalitis [147]. Other investigations, however, did not found significant associations with neuroimaging [140,145]. In some studies, biomarkers have been reported to be associated with other laboratory data. In NMDAR encephalitis, CSF levels have been shown to be positively associated with the intensity of antibody positivity in serum [142] and to be higher in patients with CSF pleocytosis [138]. In LGI1 encephalitis, CSF NFL levels are higher in the presence of abnormal CSF and hyponatremia [142].
Regarding potential diagnostic utility of T-tau and NFL in AEs, the most relevant aspects pertain to the differentiation from CJD and psychiatric conditions. CSF levels of T-tau are lower in encephalitis with antibodies against voltage-gated potassium channels (VGKC), LGI1 and other NSAs compared to CJD and discriminate well between NSA-AEs and CJD [134,140,149]. CSF levels of NFL and P-tau181 are lower in LGI1 encephalitis compared to CJD as well [140].
Constantinescu et al. evaluated longitudinal kinetics of CSF NFL and T-tau in a group of 25 patients with AEs (including, but not limited to, NSA-AEs). While NFL levels were in most cases still high at 3-month follow-up and were normalized in one-third of cases at 1-year follow-up, T-tau exhibited faster kinetics, displaying lower levels at 3-month follow-up compared to presentation and being normalized in almost all cases at 1-year follow-up [135]. While in NMDAR encephalitis CSF levels of NFL have been shown to decrease over the disease course in the majority of cases, in LGI1 encephalitis more heterogeneity has been observed, including relatively stable levels [140,142]. Pertaining to T-tau and P-tau181, the absence of significant longitudinal modifications has been reported for CSF levels in LGI1 encephalitis [140]. An investigation performed on patients with NMDAR encephalitis with at least three CSF samplings over the disease course indicated that CSF levels of NFL peaked at a median time of 2.5 months, namely relatively late compared to CSF antibody peak, clinical nadir, and reduction of CSF inflammatory indices (cell count and intrathecal IgG synthesis) [147]. Interestingly, in NMDAR encephalitis, CSF levels of NFL at follow-up have been reported to correlate with mRS scores from the same timepoint, and the same applies to the corresponding differentials of the two measures relative to clinical onset [148]. Moreover, in NSA-AEs the evolution of serum NFL levels seems to mirror the clinical course, with stable levels in clinically stable patients, increasing levels in cases with clinical worsening, and decreasing levels in patients with clinical improvement [145]. Another small study on AEs, not limited to NSA-AEs, reported longitudinally decreasing plasma levels of NFL; accordingly, patients with chronic AE (i.e., evaluated at least 6 months after symptom onset or last clinical deterioration) had lower plasma NFL levels, albeit non-significantly, compared to those with active AE [146]. Although in a study on LGI1 encephalitis no significant difference was observed between levels of T-tau, P-tau181 and NFL from CSF samples taken before or after initiation of immunosuppressive treatment [140], in NSA-AEs reduced CSF levels of T-tau have been reported following immunosuppressive treatment, while the normalization of CSF NFL levels paralleled the resolution of MRI abnormalities [139]. In NMDAR and LGI1 encephalitides, plasma exchange alone or in combination with intravenous immune globulin (IVIG) has been shown to reduce CSF levels of NFL to a greater extent than IVIG alone [142].
As in other neurological diseases, also in AE the phosphorylated form of neurofilament heavy chain (pNFH) has been less extensively studied compared to NFL. In a cohort of AEs, including several cases without antibodies against NSAs, CSF levels of pNFH were higher compared to neurologically healthy controls, were higher in patients who died, but did not differ between cases with vs. without paraneoplastic aetiology and with vs. without antibodies [150]. In NMDAR encephalitis, CSF levels of pNFH have been reported to be lower compared to non-inflammatory neurological controls, without, however, correlating with disability [148]. Similarly, few studies have investigated CSF levels of amyloid β (Aβ) in AE. Although one study on AE patients (of whom not all had NSA-Ab) found a median CSF level of Aβ42 below the cut-off for AD [134], another investigation on AEs with antibodies (including some against ICAs) demonstrated lower CSF Aβ42 and lower CSF Aβ42/Aβ40 ratio compared to AD patients [144]. In a study on LGI1 encephalitis, almost one-third of patients had CSF levels of Aβ42 below the cut-off for AD; however, median CSF Aβ42 was higher compared to AD and similar to CJD and psychiatric patients, and no patient with LGI1 encephalitis had a true CSF AD profile (i.e., reduced Aβ42 along with increased T-tau and P-tau181) [140]. In the same study, baseline CSF levels of Aβ42 correlated positively with MMSE score at 6- and 12-month follow-up, while CSF Aβ42 was longitudinally stable over the disease course [140].
Glial fibrillary acidic protein (GFAP) is a biomarker of astrocytosis and has been investigated by few studies in AEs up to now. In a cohort composed of paraneoplastic neurological syndromes (most of which with brain involvement) and non-paraneoplastic AEs, CSF levels of GFAP were not increased and did not correlate with disability at last follow-up [136]. In a longitudinal study on a similar cohort, GFAP in the CSF tended to increase initially and then to decrease, but the differences between the three timepoints were not statistically significant; equally non-significant was the correlation between CSF levels of GFAP and disability at the time of CSF sampling after correction for age [135]. On the other hand, CSF levels of the microglial (but also astrocytic) biomarker YKL-40 were found to be increased in NSA-AEs compared to neurologically healthy controls [137] as well as in NMDAR encephalitis compared to patients with viral meningitis [151]. In NSA-AEs, CSF levels of YKL-40 correlated with mRS score at 12-month follow-up [137], while in NMDAR encephalitis its levels were diminished at 3-month follow-up, with the extent of decrease correlating with that of mRS score decrease over the same interval [151].
Day et al. also evaluated the less investigated visinin-like protein 1 (VILIP-1, a biomarker of neuronal injury, as the protein is mainly localized in the neuronal cell body) and synaptic biomarkers synaptosome-associated protein of 25 kDa (SNAP-25) and neurogranin in a cohort of 45 NSA-AE cases (NMDAR, LGI1, CASPR2) [137]. Interestingly, all three biomarkers were reduced in AEs compared to neurologically healthy controls, with VILIP-1 also being lower in NMDAR encephalitis than in LGI1/CASPR2 encephalitis. Both VILIP-1 and SNAP-25 were inversely associated with worse mRS scores, ICU admission and presence of a tumour (with the latter two conditions being more frequently observed in NMDAR encephalitis); on the other hand, both SNAP-25 and neurogranin were positively associated with mRS score at 12-month follow-up. Pertaining to the relationships of the synaptic biomarkers, the authors interpreted the early inverse associations with clinical severity as expression of antibody-induced internalization of synapses, and the late direct associations as expression, on the contrary, of loss of synapse integrity [137].
Finally, a recent German multicentre study investigated neurochemical biomarkers in a large cohort of patients with IgLON5 disease (N = 53) [46]. Only 1/25, 3/20, 2/27, and 1/16 patients had high CSF levels of T-tau, high levels of P-tau181, low levels of Aβ42, and low values of the Aβ42/Aβ40 ratio, respectively, with no patients displaying a typical CSF AD profile (i.e., both increased P-tau181 and decreased Aβ42). In 27 patients who had not yet undergone immunosuppressive treatment, serum NFL and GFAP correlated moderately with each other and negatively with CSF cell count. Serum levels of NFL also correlated moderately with CSF T-tau, while serum GFAP was lower in patients with subacute symptom onset and paucisymptomatic phenotype. Moreover, low pre-treatment serum NFL levels independently predicted response to long-term immunotherapy [46].
4.4. Mechanisms of Neurodegeneration in AE
Although most of the mechanisms underlying NSA-Ab pathogenicity appear to have been elucidated [98], less is known about the mechanisms leading to neurodegeneration in patients with AE (Figure 3). On the contrary, the data available so far from in vitro and in vivo studies seems to be at least partially in contrast to what is observed in patients.
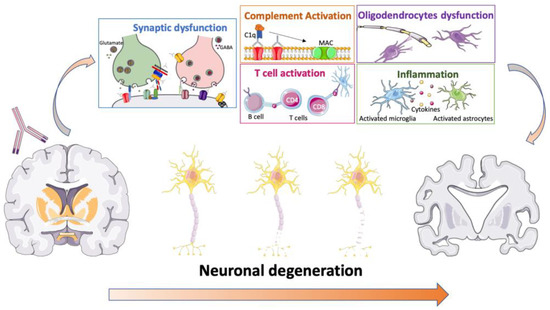
Figure 3.
Possible mechanisms of NSA-Ab-mediated neurodegeneration. NSA-Ab can induce synaptic dysfunction, complement activation, T cell, microglia and astrocyte activation and neuroinflammation as well as oligodendrocyte dysfunction. Overall, these alterations might lead to neuroaxonal or neuronal damage with progressive neuronal degeneration and loss.
NMDAR antibodies significantly decrease the NMDAR surface and total cluster densities in a titre-dependent fashion with consequent reduction of synaptic NMDAR currents but without disruption of the neuronal structure. The reduction of NMDAR expression is induced by cross-linking and internalization, without complement deposition [17]. In parallel, the antibodies alter the NMDARs trafficking inducing dispersal of GluN2A-NMDAR through the blockade of the interaction between the extracellular domains of GluN1/2 subunits and Ephrin-B2 receptors (EPHB2R) [152]. Altogether these changes affect long term potentiation, which is the cellular substrate of learning and memory, explaining the cognitive symptoms observed in patients. Indeed, the acute administration of NMDAR antagonists, such as ketamine and phencyclidine, induces memory impairment, as well as hallucinations, delusions, agitation and dissociation [153], overall partially reminiscent of NMDAR encephalitis. Similarly, endogenous NMDAR antagonists, including kynurenic acid, endocannabinoids and zinc, among others, have been implicated in the pathogenesis of several neuropsychiatric disorders [154], and particularly schizophrenia [155]. In both in vitro and in vivo models, the effects of NMDAR antibodies on the receptor are reversible. Mice infused with pooled CSFs from individuals with anti-NMDAR encephalitis over 14 days showed memory deficits, anhedonia and depression-like behaviour associated with IgG deposition and a decrease in NMDAR clusters in the hippocampi, which resolved within days after discontinuing the infusion [156]. However, the demonstration of long-term cognitive deficit and structural hippocampal damage suggests that while the acute effects of NMDAR-Abs are functional and reversible, their persistence can impact on neuronal function in a way that goes beyond the established mechanisms and not yet understood. NMDAR is not expressed only on neurons but also on oligodendrocytes where they control the supply of energy substrates to support the axonal functioning via GLUT1 translocation to the oligodendrocyte membrane [157]. Patients’ CSF antibodies reduce the levels of oligodendroglial GLUT1 [158] providing a link with the white matter alterations described in patients with NMDAR encephalitis. It has been shown that the prolonged loss of NMDAR function in oligodendroglia leads to axonal pathology [157] explaining the correlation between disease duration and white matter damage and, indeed patients not recovering from NMDAR encephalitis showed extensive superficial white matter damage [115]. Therefore, it is possible that while neurons recover, the damage to the oligodendrocyte might become permanent, affecting whole brain function. The effects of the antibodies depend on the duration of the exposure. Indeed, in experimental animal studies, NMDAR-antagonists are able to produce reversible or irreversible effects depending on the dosage and duration of the administration [153]. Another, not mutually exclusive possibility, is that the long-term damage is related to the accumulation of excitatory mediators such as glutamate at synaptic levels. Indeed, patients’ CSF and purified IgGs resulted in an acute increase of the glutamate levels of the motor cortex in a rat model [18]. Moreover, the involvement of T cell mediated mechanisms have not been investigated and cannot be derived by in vitro or passive transfer animal models but their possible role has been recently pointed out in an active immunization model [159]. In the few available neuropathological studies, T cell infiltrates have been reported although they usually do not display a cytotoxic phenotype and neuronal degeneration is variable whilst co-occurring pathologies might affect the neuropathological features [160].
Oppositely to NMDAR-Ab, LGI1 antibodies are predominantly IgG4. These kind of IgGs are less effective than IgG1 in crosslinking and internalization and are thought to affect their targets by interference with protein-protein interactions. LGI1-Abs disrupt the ligand-receptor interaction of LGI1 with ADAM22, resulting in reversible reduction in synaptic AMPARs, Kv1.1 and VGKC [23,161]. Nevertheless, LGI1-Abs have been detected in the sera of cats with a spontaneous form of autoimmune encephalitis with complex partial seizures with orofacial involvement. Post-mortem analysis in these cases showed complement deposition, a feature also shared by post-mortem examination of the small number of available brains from patients with LGI1-related encephalitis [162]. The finding of complement deposition suggests that LGI1-Abs can induce neuronal death through the activation of the complement cascade which may explain the persistence of long-term cognitive deficits as well as structural brain changes in affected patients. The post-mortem findings reflect the relatively small, but more pathogenic, IgG1 antibodies that are present in some patients with LGI1 encephalitis and occur more frequently in patients with cognitive impairment [19,20]. The same might be true for CASPR2-Abs. Indeed, CASPR2-Abs are mainly IgG4, and therefore the most likely common mechanism of their action is a disruption of the interaction of CASPR2 with its associated molecules rather than internalization and complement activation. Accordingly, CASPR2-Abs did not reduce CASPR2 expression on the surface of cultured hippocampal neurons, but they appeared to act by altering CASPR2 interaction with contactin-2 [163,164]. However, these findings contrast with the results of others who found CASPR2-Abs mediated internalization of their target [25,26,27] as well as with neuropathology showing reduced CASPR2 expression in the brain [165] neuronal loss and complement deposition [166,167]. These discrepancies might be related to different proportion of IgG1 and IgG4 in the samples used in various studies which might affect the prevalent observed mechanism. Moreover, similarly to LGI1-Abs, CASPR2-Abs may also alter AMPAR synaptic traffic affecting cognitive function at least in the acute phase [26,27].
AMPAR [35,36] and GABABR [31] antibodies act causing internalization and block of the receptor, respectively. However, neuropathological evidence of T cell activation and inflammation [32,168] together with the evidence of hippocampal sclerosis and evolution in brain atrophy suggest the presence of additional mechanisms promoting neurodegeneration.
5. IgLON5 Disease: The Interface between Antibody-Mediated Immunity and Neurodegeneration
The question of the relation between antibodies and neurodegeneration is made more complex by the description of IgLON5 antibodies. These were firstly described in a small group of patients with prominent sleep-related movement disorders, mainly characterised by a non-REM sleep parasomnia with simple or finalistic movements, resembling daytime activities such as eating, drinking or manipulating objects. Other sleep abnormalities included RBD and periodic limb movements during sleep. Breathing disorders, including obstructive sleep apnoea (OSAS) and stridor, both in sleep and wakefulness, are almost a constant feature and are often associated with respiratory failure. Other clinical manifestations include ataxia, bulbar signs, abnormal eye movements and dysautonomia [28,45,167,169]. Over time, other clinical manifestations were reported including dementia and chorea [170,171,172], motor neuron-like disease and seizures [147,173]. In the first case series, all eight patients reported received immunotherapy, but six of them died during the follow-up period [45]. To date more than 100 patients have been reported and response to immunotherapy seems to occur in certain cases and to some extent [46,170,171,172,173,174,175]. Initiation of first-line immune treatment within 6 weeks, or the initiation of long-term treatment within 1 year of onset, seems to predict clinical improvement; moreover, a low serum level of neurofilament light chain at onset correlated with a better prognosis [46]. However, mortality remains high [46,175,176]. Neuropathology, intriguingly, show the presence of hyperphosphorylated three- and four-repeat tau aggregates in neurons, and neuronal loss predominantly in the hypothalamus and the brainstem tegmentum, and absence of inflammatory infiltrates [45,177]. However, not all patients show these neuropathological features. Some cases showed no p-tau deposition [173,178] or no deposition in the brainstem [179,180], together with evidence of neuroinflammation, including lymphocyte infiltration and microglia activation [173,178,179,180]. These findings, together with the finding of increased B cell numbers in the CSF [181], suggest that neurodegeneration might be a secondary disease mechanism, that set after an initial immune response, which would fit with the observed response to immunotherapy in patients treated close to disease onset. Moreover, all genotyped patients had HLA-DQB1*0501 and HLA-DRB1*1001 alleles, suggesting a genetic susceptibility for autoimmunity [45].
The IgLON5 antibodies target the extracellular domain of the protein and are predominantly of the IgG4 subtype, and to a lesser extent IgG1. The latter are responsible for the irreversible internalisation of IgLON5 in rat neuronal cultures [182], whereas this effect was not seen with IgG4 antibodies, which are likely to act in a different way. Another study, demonstrated that IgLON5 antibodies disrupt the cytoskeleton in rat hippocampal neurons, resulting in dystrophic axons and axon swelling after 3 weeks (Figure 2D). These alterations might explain the raised concentration of NFL in patients’ CSF [183]. Ryding et al. [184], showed progressive p-tau accumulation, synaptic disruption, neuronal death as well as reduction in IgLON5 concentrations, in differentiated human neural stem cells incubated with IgLON5 antibodies [184]. These pathogenetic effects of IgLON5 antibodies have been confirmed in vivo [47,185]. Mice infused with these antibodies showed p-tau deposition in the hippocampal CA4 region, mossy fibres, and posterior periependymal areas [185]. In another study, mice exposed to IgLON5 antibodies showed neuronal loss, microglial and astrocytic activation as well as an increase in the relative mRNA expression levels of several inflammatory factors, including TGF-β, CCL5, and CXCL13 [47]. Altogether, these observations suggest that IgLON5 antibodies are directly pathogenic and can initiate an inflammatory and neurodegenerative process. However, the differential role of IgG1 and IgG4 in these process remains to be clarified, as well as the contribution of adaptive immunity and neuroinflammation in the progression of the disease.
6. Conclusions
Several NSA-Ab can present with cognitive impairment, phenocopying different neurodegenerative disorders. These antibodies are pathogenic in this context and the unusual presentation, often associated with the lack of typical inflammatory CSF and imaging findings, might lead to misdiagnosis, and delay the immune treatment. Although few reports have suggested clinical red-flags for this autoimmune dementia, their real utility in predicting this diagnosis needs to be investigated in prospective studies. On the other hand, NSA-Ab have been detected in patients with established neurogenerative disorders; in these non-encephalitis situations their pathogenic potential is debated and to date there is no evidence on the possible utility of immunotherapy.
In addition to NSA-Ab, several antibodies have been described in patients with different neurodegenerative disorders [8,186,187] and they might have a role in modulating the neurodegenerative process as well as the clinical phenotype. We suggest that a similar role might be also played by NSA-Ab outside of encephalitis. Indeed, these neuronal antibodies have the potential to induce neurodegeneration with permanent cognitive and brain structural changes in patients with AE. Moreover, IgLON5 antibodies suggest a continuum between autoimmunity and neurodegeneration. Indeed, it is possible that multiple antibodies against neuronal proteins either with detrimental or neuroprotective function occur together contributing to the individual risk of developing a neurodegenerative disorder [188]. Therefore, future studies should investigate the pathogenic potential of NSA-Ab in neurodegenerative disorders. A better understanding of the interaction between humoral immunity and neurodegeneration might pose the base for novel therapeutic options in patients with neurodegenerative disorders.
Author Contributions
Conceptualization: M.P.G. and R.L.; data curation: M.P.G., F.V., L.M., G.R. and F.R.; supervision: M.P.G. and R.L.; visualization: M.P.G., L.M. and F.R.; writing: M.P.G., F.V. and G.R.; review and editing: M.P.G., F.V., L.M., G.R., F.R. and R.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data have been included in the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, R.C.; Gelfand, J.M.; Geschwind, M.; et al. A Clinical Approach to Diagnosis of Autoimmune Encephalitis. Lancet Neurol. 2016, 15, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Giannoccaro, M.P.; Crisp, S.J.; Vincent, A. Antibody-Mediated Central Nervous System Diseases. Brain Neurosci. Adv. 2018, 2, 239821281881749. [Google Scholar] [CrossRef] [PubMed]
- Escudero, D.; Guasp, M.; Ariño, H.; Gaig, C.; Martínez-Hernández, E.; Dalmau, J.; Graus, F. Antibody-Associated CNS Syndromes without Signs of Inflammation in the Elderly. Neurology 2017, 89, 1471–1475. [Google Scholar] [CrossRef] [PubMed]
- Ingram, C.R.; Phegan, K.J.; Blumenthal, H.T. Significance of An Aging-Linked Neuron Binding Gamma Globulin Fraction of Human Sera. J. Gerontol. 1974, 29, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Mayer, P.P.; Chughtai, M.A.; Cape, R.D.T. An immunological approach to dementia in the elderly. Age Ageing 1976, 5, 164–170. [Google Scholar] [CrossRef]
- McRae-Degueurce, A.; Booj, S.; Haglid, K.; Rosengren, L.; Karlsson, J.E.; Karlsson, I.; Wallin, A.; Svennerholm, L.; Gottfries, C.G.; Dahlstrom, A. Antibodies in Cerebrospinal Fluid of Some Alzheimer Disease Patients Recognize Cholinergic Neurons in the Rat Central Nervous System. Proc. Natl. Acad. Sci. USA 1987, 84, 9214–9218. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, L. Autoantibodies in Alzheimer’s Disease: Potential Biomarkers, Pathogenic Roles, and Therapeutic Implications. J. Biomed. Res. 2016, 30, 361–372. [Google Scholar] [CrossRef]
- Kocurova, G.; Ricny, J.; Ovsepian, S.V. Autoantibodies Targeting Neuronal Proteins as Biomarkers for Neurodegenerative Diseases. Theranostics 2022, 12, 3045–3056. [Google Scholar] [CrossRef]
- Ovsepian, S.V.; O’Leary, V.B. Adult Neurogenesis in the Gut, Homeostatic Autoimmunity and Neurodegenerative Disease Biomarkers. Neuroscience 2022, 504, 75–78. [Google Scholar] [CrossRef]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, J.E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-Receptor Encephalitis: Case Series and Analysis of the Effects of Antibodies. Lancet Neurol. 2008, 7, 1091–1098. [Google Scholar] [CrossRef]
- Pruss, H.; Holtje, M.; Maier, N.; Gomez, A.; Buchert, R.; Harms, L.; Ahnert-Hilger, G.; Schmitz, D.; Terborg, C.; Kopp, U.; et al. IgA NMDA Receptor Antibodies Are Markers of Synaptic Immunity in Slow Cognitive Impairment. Neurology 2012, 78, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Doss, S.; Wandinger, K.-P.; Hyman, B.T.; Panzer, J.A.; Synofzik, M.; Dickerson, B.; Mollenhauer, B.; Scherzer, C.R.; Ivinson, A.J.; Finke, C.; et al. High Prevalence of NMDA Receptor IgA/IgM Antibodies in Different Dementia Types. Ann. Clin. Transl. Neurol. 2014, 1, 822–832. [Google Scholar] [CrossRef]
- Busse, M.; Kunschmann, R.; Dobrowolny, H.; Hoffmann, J.; Bogerts, B.; Steiner, J.; Frodl, T.; Busse, S. Dysfunction of the Blood-Cerebrospinal Fluid-Barrier and N-Methyl-d-Aspartate Glutamate Receptor Antibodies in Dementias. Eur. Arch. Psychiatry Clin. Neurosci. 2018, 268, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Busse, S.; Busse, M.; Brix, B.; Probst, C.; Genz, A.; Bogerts, B.; Stoecker, W.; Steiner, J. Seroprevalence of N-Methyl-d-Aspartate Glutamate Receptor (NMDA-R) Autoantibodies in Aging Subjects without Neuropsychiatric Disorders and in Dementia Patients. Eur. Arch. Psychiatry Clin. Neurosci. 2014, 264, 545–550. [Google Scholar] [CrossRef]
- Ehrenreich, H. Autoantibodies against N-Methyl-d-Aspartate Receptor 1 in Health and Disease. Curr. Opin. Neurol. 2018, 31, 306–312. [Google Scholar] [CrossRef]
- Titulaer, M.J.; McCracken, L.; Gabilondo, I.; Armangué, T.; Glaser, C.; Iizuka, T.; Honig, L.S.; Benseler, S.M.; Kawachi, I.; Martinez-Hernandez, E.; et al. Treatment and Prognostic Factors for Long-Term Outcome in Patients with Anti-NMDA Receptor Encephalitis: An Observational Cohort Study. Lancet Neurol. 2013, 12, 157–165. [Google Scholar] [CrossRef]
- Hughes, E.G.; Peng, X.; Gleichman, A.J.; Lai, M.; Zhou, L.; Tsou, R.; Parsons, T.D.; Lynch, D.R.; Dalmau, J.; Balice-Gordon, R.J. Cellular and Synaptic Mechanisms of Anti-NMDA Receptor Encephalitis. J. Neurosci. 2010, 30, 5866–5875. [Google Scholar] [CrossRef]
- Manto, M.; Dalmau, J.; Didelot, A.; Rogemond, V.; Honnorat, J. In Vivo Effects of Antibodies from Patients with Anti-NMDA Receptor Encephalitis: Further Evidence of Synaptic Glutamatergic Dysfunction. Orphanet J. Rare Dis. 2010, 5, 31. [Google Scholar] [CrossRef]
- Irani, S.R.; Alexander, S.; Waters, P.; Kleopa, K.A.; Pettingill, P.; Zuliani, L.; Peles, E.; Buckley, C.; Lang, B.; Vincent, A. Antibodies to Kv1 Potassium Channel-Complex Proteins Leucine-Rich, Glioma Inactivated 1 Protein and Contactin-Associated Protein-2 in Limbic Encephalitis, Morvan’s Syndrome and Acquired Neuromyotonia. Brain 2010, 133, 2734–2748. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of Multiple Sclerosis: 2017 Revisions of the McDonald Criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Ariño, H.; Armangué, T.; Petit-Pedrol, M.; Sabater, L.; Martinez-Hernandez, E.; Hara, M.; Lancaster, E.; Saiz, A.; Dalmau, J.; Graus, F. Anti-LGI1–Associated Cognitive Impairment. Neurology 2016, 87, 759–765. [Google Scholar] [CrossRef] [PubMed]
- van Sonderen, A.; Thijs, R.D.; Coenders, E.C.; Jiskoot, L.C.; Sanchez, E.; de Bruijn, M.A.A.M.; van Coevorden-Hameete, M.H.; Wirtz, P.W.; Schreurs, M.W.J.; Sillevis Smitt, P.A.E.; et al. Anti-LGI1 Encephalitis: Clinical Syndrome and Long-Term Follow-Up. Neurology 2016, 87, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Petit-Pedrol, M.; Sell, J.; Planagumà, J.; Mannara, F.; Radosevic, M.; Haselmann, H.; Ceanga, M.; Sabater, L.; Spatola, M.; Soto, D.; et al. LGI1 Antibodies Alter Kv1.1 and AMPA Receptors Changing Synaptic Excitability, Plasticity and Memory. Brain 2018, 141, 3144–3159. [Google Scholar] [CrossRef] [PubMed]
- Joubert, B.; Saint-Martin, M.; Noraz, N.; Picard, G.; Rogemond, V.; Ducray, F.; Desestret, V.; Psimaras, D.; Delattre, J.-Y.; Antoine, J.-C.; et al. Characterization of a Subtype of Autoimmune Encephalitis with Anti–Contactin-Associated Protein-like 2 Antibodies in the Cerebrospinal Fluid, Prominent Limbic Symptoms, and Seizures. JAMA Neurol. 2016, 73, 1115. [Google Scholar] [CrossRef]
- Giannoccaro, M.P.; Menassa, D.A.; Jacobson, L.; Coutinho, E.; Prota, G.; Lang, B.; Leite, M.I.; Cerundolo, V.; Liguori, R.; Vincent, A. Behaviour and Neuropathology in Mice Injected with Human Contactin-Associated Protein 2 Antibodies. Brain 2019, 142, 2000–2012. [Google Scholar] [CrossRef]
- Joubert, B.; Petit-Pedrol, M.; Planagumà, J.; Mannara, F.; Radosevic, M.; Marsal, M.; Maudes, E.; García-Serra, A.; Aguilar, E.; Andrés-Bilbé, A.; et al. Human CASPR2 Antibodies Reversibly Alter Memory and the CASPR2 Protein Complex. Ann. Neurol. 2022, 91, 801–813. [Google Scholar] [CrossRef]
- Fernandes, D.; Santos, S.D.; Coutinho, E.; Whitt, J.L.; Beltrão, N.; Rondão, T.; Leite, M.I.; Buckley, C.; Lee, H.-K.; Carvalho, A.L. Disrupted AMPA Receptor Function upon Genetic- or Antibody-Mediated Loss of Autism-Associated CASPR2. Cereb. Cortex 2019, 29, 4919–4931. [Google Scholar] [CrossRef]
- Van Coevorden-Hameete, M.H.; de Bruijn, M.A.A.M.; de Graaff, E.; Bastiaansen, D.A.E.M.; Schreurs, M.W.J.; Demmers, J.A.A.; Ramberger, M.; Hulsenboom, E.S.P.; Nagtzaam, M.M.P.; Boukhrissi, S.; et al. The Expanded Clinical Spectrum of Anti-GABABR Encephalitis and Added Value of KCTD16 Autoantibodies. Brain 2019, 142, 1631–1643. [Google Scholar] [CrossRef]
- Hoftberger, R.; Titulaer, M.J.; Sabater, L.; Dome, B.; Rozsas, A.; Hegedus, B.; Hoda, M.A.; Laszlo, V.; Ankersmit, H.J.; Harms, L.; et al. Encephalitis and GABAB Receptor Antibodies: Novel Findings in a New Case Series of 20 Patients. Neurology 2013, 81, 1500–1506. [Google Scholar] [CrossRef]
- Lancaster, E.; Lai, M.; Peng, X.; Hughes, E.; Constantinescu, R.; Raizer, J.; Friedman, D.; Skeen, M.B.; Grisold, W.; Kimura, A.; et al. Antibodies to the GABAB Receptor in Limbic Encephalitis with Seizures: Case Series and Characterisation of the Antigen. Lancet Neurol. 2010, 9, 67–76. [Google Scholar] [CrossRef]
- Nibber, A.; Mann, E.O.; Pettingill, P.; Waters, P.; Irani, S.R.; Kullmann, D.M.; Vincent, A.; Lang, B. Pathogenic Potential of Antibodies to the GABAB Receptor. Epilepsia Open 2017, 2, 355–359. [Google Scholar] [CrossRef]
- Ricken, G.; Zrzavy, T.; Macher, S.; Altmann, P.; Troger, J.; Falk, K.K.; Kiefer, A.; Fichtenbaum, A.; Mitulovic, G.; Kubista, H.; et al. Autoimmune Global Amnesia as Manifestation of AMPAR Encephalitis and Neuropathologic Findings. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1019. [Google Scholar] [CrossRef]
- Joubert, B.; Kerschen, P.; Zekeridou, A.; Desestret, V.; Rogemond, V.; Chaffois, M.-O.; Ducray, F.; Larrue, V.; Daubail, B.; Idbaih, A.; et al. Clinical Spectrum of Encephalitis Associated with Antibodies Against the α-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid Receptor: Case Series and Review of the Literature. JAMA Neurol. 2015, 72, 1163. [Google Scholar] [CrossRef]
- Hoftberger, R.; van Sonderen, A.; Leypoldt, F.; Houghton, D.; Geschwind, M.; Gelfand, J.; Paredes, M.; Sabater, L.; Saiz, A.; Titulaer, M.J.; et al. Encephalitis and AMPA Receptor Antibodies: Novel Findings in a Case Series of 22 Patients. Neurology 2015, 84, 2403–2412. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Hughes, E.G.; Peng, X.; Zhou, L.; Gleichman, A.J.; Shu, H.; Matà, S.; Kremens, D.; Vitaliani, R.; Geschwind, M.D.; et al. AMPA Receptor Antibodies in Limbic Encephalitis Alter Synaptic Receptor Location. Ann. Neurol. 2009, 65, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Haselmann, H.; Mannara, F.; Werner, C.; Planagumà, J.; Miguez-Cabello, F.; Schmidl, L.; Grünewald, B.; Petit-Pedrol, M.; Kirmse, K.; Classen, J.; et al. Human Autoantibodies against the AMPA Receptor Subunit GluA2 Induce Receptor Reorganization and Memory Dysfunction. Neuron 2018, 100, 91–105.e9. [Google Scholar] [CrossRef] [PubMed]
- Spatola, M.; Petit-Pedrol, M.; Simabukuro, M.M.; Armangue, T.; Castro, F.J.; Barcelo Artigues, M.I.; Julià Benique, M.R.; Benson, L.; Gorman, M.; Felipe, A.; et al. Investigations in GABA A Receptor Antibody-Associated Encephalitis. Neurology 2017, 88, 1012–1020. [Google Scholar] [CrossRef]
- Pettingill, P.; Kramer, H.B.; Coebergh, J.A.; Pettingill, R.; Maxwell, S.; Nibber, A.; Malaspina, A.; Jacob, A.; Irani, S.R.; Buckley, C.; et al. Antibodies to GABAA receptor α1 and γ2 subunits: Clinical and serologic characterization. Neurology 2015, 84, 1233–1241. [Google Scholar] [CrossRef]
- Hara, M.; Ariño, H.; Petit-Pedrol, M.; Sabater, L.; Titulaer, M.J.; Martinez-Hernandez, E.; Schreurs, M.W.J.; Rosenfeld, M.R.; Graus, F.; Dalmau, J. DPPX Antibody–Associated Encephalitis: Main Syndrome and Antibody Effects. Neurology 2017, 88, 1340–1348. [Google Scholar] [CrossRef]
- Piepgras, J.; Höltje, M.; Michel, K.; Li, Q.; Otto, C.; Drenckhahn, C.; Probst, C.; Schemann, M.; Jarius, S.; Stöcker, W.; et al. Anti-DPPX Encephalitis: Pathogenic Effects of Antibodies on Gut and Brain Neurons. Neurology 2015, 85, 890–897. [Google Scholar] [CrossRef]
- Carvajal-González, A.; Leite, M.I.; Waters, P.; Woodhall, M.; Coutinho, E.; Balint, B.; Lang, B.; Pettingill, P.; Carr, A.; Sheerin, U.-M.; et al. Glycine Receptor Antibodies in PERM and Related Syndromes: Characteristics, Clinical Features and Outcomes. Brain 2014, 137, 2178–2192. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-González, A.; Jacobson, L.; Clover, L.; Wickremaratchi, M.; Shields, S.; Lang, B.; Vincent, A. Systemic Delivery of Human GlyR IgG Antibody Induces GlyR Internalization into Motor Neurons of Brainstem and Spinal Cord with Motor Dysfunction in Mice. Neuropathol. Appl. Neurobiol. 2021, 47, 316–327. [Google Scholar] [CrossRef]
- Rauschenberger, V.; von Wardenburg, N.; Schaefer, N.; Ogino, K.; Hirata, H.; Lillesaar, C.; Kluck, C.J.; Meinck, H.; Borrmann, M.; Weishaupt, A.; et al. Glycine Receptor Autoantibodies Impair Receptor Function and Induce Motor Dysfunction. Ann. Neurol. 2020, 88, 544–561. [Google Scholar] [CrossRef]
- Hansen, N.; Bartels, C.; Stöcker, W.; Wiltfang, J.; Fitzner, D. Impaired Verbal Memory Recall in Patients with Axonal Degeneration and Serum Glycine-Receptor Autoantibodies—Case Series. Front. Psychiatry 2022, 12, 778684. [Google Scholar] [CrossRef]
- Sabater, L.; Gaig, C.; Gelpi, E.; Bataller, L.; Lewerenz, J.; Torres-Vega, E.; Contreras, A.; Giometto, B.; Compta, Y.; Embid, C.; et al. A Novel Non-Rapid-Eye Movement and Rapid-Eye-Movement Parasomnia with Sleep Breathing Disorder Associated with Antibodies to IgLON5: A Case Series, Characterisation of the Antigen, and Post-Mortem Study. Lancet Neurol. 2014, 13, 575–586. [Google Scholar] [CrossRef]
- Grüter, T.; Möllers, F.E.; Tietz, A.; Dargvainiene, J.; Melzer, N.; Heidbreder, A.; Strippel, C.; Kraft, A.; Höftberger, R.; Schöberl, F.; et al. Clinical, Serological and Genetic Predictors of Response to Immunotherapy in Anti-IgLON5 Disease. Brain 2022, 146, 600–611. [Google Scholar] [CrossRef]
- Ni, Y.; Feng, Y.; Shen, D.; Chen, M.; Zhu, X.; Zhou, Q.; Gao, Y.; Liu, J.; Zhang, Q.; Shen, Y.; et al. Anti-IgLON5 Antibodies Cause Progressive Behavioral and Neuropathological Changes in Mice. J. Neuroinflamm. 2022, 19, 140. [Google Scholar] [CrossRef]
- Geschwind, M.D.; Shu, H.; Haman, A.; Sejvar, J.J.; Miller, B.L. Rapidly Progressive Dementia. Ann. Neurol. 2008, 64, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Chitravas, N.; Jung, R.S.; Kofskey, D.M.; Blevins, J.E.; Gambetti, P.; Leigh, R.J.; Cohen, M.L. Treatable Neurological Disorders Misdiagnosed as Creutzfeldt-Jakob Disease. Ann. Neurol. 2011, 70, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Sala, I.; Marquié, M.; Sánchez-Saudinós, M.B.; Sánchez-Valle, R.; Alcolea, D.; Gómez-Ansón, B.; Gómez-Isla, T.; Blesa, R.; Lleó, A. Rapidly Progressive Dementia: Experience in a Tertiary Care Medical Center. Alzheimer Dis. Assoc. Disord. 2012, 26, 267–271. [Google Scholar] [CrossRef]
- Papageorgiou, S.G.; Kontaxis, T.; Bonakis, A.; Karahalios, G.; Kalfakis, N.; Vassilopoulos, D. Rapidly Progressive Dementia: Causes Found in a Greek Tertiary Referral Center in Athens. Alzheimer Dis. Assoc. Disord. 2009, 23, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Anuja, P.; Venugopalan, V.; Darakhshan, N.; Awadh, P.; Wilson, V.; Manoj, G.; Manish, M.; Vivek, L. Rapidly Progressive Dementia: An Eight Year (2008–2016) Retrospective Study. PLoS ONE 2018, 13, e0189832. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, E.P.; McKeon, A.; Lennon, V.A.; Boeve, B.F.; Trenerry, M.R.; Tan, K.M.; Drubach, D.A.; Josephs, K.A.; Britton, J.W.; Mandrekar, J.N.; et al. Autoimmune Dementia: Clinical Course and Predictors of Immunotherapy Response. Mayo Clin. Proc. 2010, 85, 881–897. [Google Scholar] [CrossRef]
- Li, X.; Yuan, J.; Liu, L.; Hu, W. Antibody-LGI 1 Autoimmune Encephalitis Manifesting as Rapidly Progressive Dementia and Hyponatremia: A Case Report and Literature Review. BMC Neurol. 2019, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Sun, L.; Cao, J.; Liu, C. Creutzfeldt-Jakob Disease versus Anti-LGI1 Limbic Encephalitis in a Patient with Progressive Cognitive Dysfunction, Psychiatric Symptoms, Involuntary Facio-Brachio-Crural Movement, and an Abnormal Electroencephalogram: A Case Report. Neuropsychiatr. Dis. Treat. 2015, 11, 1427–1430. [Google Scholar] [CrossRef]
- McKeon, A.; Marnane, M.; O’Connell, M.; Stack, J.P.; Kelly, P.J.; Lynch, T. Potassium Channel Antibody–Associated Encephalopathy Presenting with a Frontotemporal Dementia–like Syndrome. Arch. Neurol. 2007, 64, 1528. [Google Scholar] [CrossRef]
- Abe, K.; Chiba, Y. A Case of Treatable Dementia with Lewy Bodies Remarkably Improved by Immunotherapy. J. Neuroimmunol. 2019, 330, 35–37. [Google Scholar] [CrossRef]
- Marquetand, J.; van Lessen, M.; Bender, B.; Reimold, M.; Elsen, G.; Stoecker, W.; Synofzik, M. Slowly Progressive LGI1 Encephalitis with Isolated Late-Onset Cognitive Dysfunction: A Treatable Mimic of Alzheimer’s Disease. Eur. J. Neurol. 2016, 23, e28–e29. [Google Scholar] [CrossRef]
- Arakawa, I.; Oguri, T.; Nakamura, T.; Sakurai, K.; Yuasa, H. Anti-voltage-gated potassium channel (VGKC) complex/leucine-rich glioma-inactivated protein 1 (LGI1) antibody-associated limbic encephalitis mimicking dementia with Lewy bodies in a patient with subacute REM sleep behavior disorder. Rinsho Shinkeigaku 2022, 62, 22–26. [Google Scholar] [CrossRef]
- Gadoth, A.; Pittock, S.J.; Dubey, D.; McKeon, A.; Britton, J.W.; Schmeling, J.E.; Smith, A.; Kotsenas, A.L.; Watson, R.E.; Lachance, D.H.; et al. Expanded Phenotypes and Outcomes among 256 LGI1/CASPR2-IgG-Positive Patients: LGI1/CASPR2-IgG + Patients. Ann. Neurol. 2017, 82, 79–92. [Google Scholar] [CrossRef]
- Bastiaansen, A.E.M.; van Steenhoven, R.W.; de Bruijn, M.A.A.M.; Crijnen, Y.S.; van Sonderen, A.; van Coevorden-Hameete, M.H.; Nühn, M.M.; Verbeek, M.M.; Schreurs, M.W.J.; Smitt, P.A.E.S.; et al. Autoimmune Encephalitis Resembling Dementia Syndromes. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1039. [Google Scholar] [CrossRef] [PubMed]
- Tobin, W.O.; Lennon, V.A.; Komorowski, L.; Probst, C.; Clardy, S.L.; Aksamit, A.J.; Appendino, J.P.; Lucchinetti, C.F.; Matsumoto, J.Y.; Pittock, S.J.; et al. DPPX Potassium Channel Antibody: Frequency, Clinical Accompaniments, and Outcomes in 20 Patients. Neurology 2014, 83, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Kurtis, M.M.; Toledano, R.; García-Morales, I.; Gil-Nagel, A. Immunomodulated Parkinsonism as a Presenting Symptom of LGI1 Antibody Encephalitis. Park. Relat. Disord. 2015, 21, 1286–1287. [Google Scholar] [CrossRef] [PubMed]
- Kannoth, S.; Anandakkuttan, A.; Mathai, A.; Sasikumar, A.N.; Nambiar, V. Autoimmune Atypical Parkinsonism—A Group of Treatable Parkinsonism. J. Neurol. Sci. 2016, 362, 40–46. [Google Scholar] [CrossRef]
- Abdullah, N.S.; Jan, T.H.; Remli, R.; Mukari, S.A.M.; Ibrahim, N.M. Anti-GABAB Receptor Encephalitis Presenting with Atypical Corticobasal Syndrome in a Patient with Parkinson’s Disease. J. Mov. Disord. 2020, 13, 235–237. [Google Scholar] [CrossRef]
- Iranzo, A.; Molinuevo, J.L.; Santamaría, J.; Serradell, M.; Martí, M.J.; Valldeoriola, F.; Tolosa, E. Rapid-Eye-Movement Sleep Behaviour Disorder as an Early Marker for a Neurodegenerative Disorder: A Descriptive Study. Lancet Neurol. 2006, 5, 572–577. [Google Scholar] [CrossRef]
- Iranzo, A.; Graus, F.; Clover, L.; Morera, J.; Bruna, J.; Vilar, C.; Martínez-Rodriguez, J.E.; Vincent, A.; Santamaría, J. Rapid Eye Movement Sleep Behavior Disorder and Potassium Channel Antibody-Associated Limbic Encephalitis. Ann. Neurol. 2006, 59, 178–181. [Google Scholar] [CrossRef]
- Cornelius, J.R.; Pittock, S.J.; McKeon, A.; Lennon, V.A.; Aston, P.A.; Josephs, K.A.; Tippmann-Peikert, M.; Silber, M.H. Sleep Manifestations of Voltage-Gated Potassium Channel Complex Autoimmunity. Arch. Neurol. 2011, 68, 733–738. [Google Scholar] [CrossRef]
- Stamelou, M.; Plazzi, G.; Lugaresi, E.; Edwards, M.J.; Bhatia, K.P. The Distinct Movement Disorder in Anti-NMDA Receptor Encephalitis May Be Related to Status Dissociatus: A Hypothesis. Mov. Disord. 2012, 27, 1360–1363. [Google Scholar] [CrossRef]
- Abgrall, G.; Demeret, S.; Rohaut, B.; Leu-Semenescu, S.; Arnulf, I. Status Dissociatus and Disturbed Dreaming in a Patient with Morvan Syndrome plus Myasthenia Gravis. Sleep Med. 2015, 16, 894–896. [Google Scholar] [CrossRef]
- Giannoccaro, M.P.; Gastaldi, M.; Rizzo, G.; Jacobson, L.; Vacchiano, V.; Perini, G.; Capellari, S.; Franciotta, D.; Costa, A.; Liguori, R.; et al. Antibodies to Neuronal Surface Antigens in Patients with a Clinical Diagnosis of Neurodegenerative Disorder. Brain Behav. Immun. 2021, 96, 106–112. [Google Scholar] [CrossRef]
- Hazan, J.; Wing, M.; Liu, K.Y.; Reeves, S.; Howard, R. Clinical Utility of Cerebrospinal Fluid Biomarkers in the Evaluation of Cognitive Impairment: A Systematic Review and Meta-Analysis. J. Neurol. Neurosurg. Psychiatry 2023, 94, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Barthel, H.; Villemagne, V.L.; Drzezga, A. Future Directions in Molecular Imaging of Neurodegenerative Disorders. J. Nucl. Med. 2022, 63, 68S–74S. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, M.D.; Murray, K. Differential Diagnosis with Other Rapid Progressive Dementias. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 153, pp. 371–397. ISBN 978-0-444-63945-5. [Google Scholar]
- Janssen, J.C.; Godbolt, A.K.; Ioannidis, P.; Thompson, E.J.; Rossor, M.N. The Prevalence of Oligoclonal Bands in the CSF of Patients with Primary Neurodegenerative Dementia. J. Neurol. 2004, 251, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, E.P.; Drubach, D.A.; Boeve, B.F. Autoimmune Dementia and Encephalopathy. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 133, pp. 247–267. ISBN 978-0-444-63432-0. [Google Scholar]
- Beagle, A.J.; Darwish, S.M.; Ranasinghe, K.G.; La, A.L.; Karageorgiou, E.; Vossel, K.A. Relative Incidence of Seizures and Myoclonus in Alzheimer’s Disease, Dementia with Lewy Bodies, and Frontotemporal Dementia. J. Alzheimer’s Dis. 2017, 60, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Lozsadi, D.A.; Larner, A.J. Prevalence and Causes of Seizures at the Time of Diagnosis of Probable Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2006, 22, 121–124. [Google Scholar] [CrossRef]
- Dubey, D.; Kothapalli, N.; McKeon, A.; Flanagan, E.P.; Lennon, V.A.; Klein, C.J.; Britton, J.W.; So, E.; Boeve, B.F.; Tillema, J.-M.; et al. Predictors of Neural-Specific Autoantibodies and Immunotherapy Response in Patients with Cognitive Dysfunction. J. Neuroimmunol. 2018, 323, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Mackay, G.; Ahmad, K.; Stone, J.; Sudlow, C.; Summers, D.; Knight, R.; Will, R.; Irani, S.R.; Vincent, A.; Maddison, P. NMDA Receptor Autoantibodies in Sporadic Creutzfeldt-Jakob Disease. J. Neurol. 2012, 259, 1979–1981. [Google Scholar] [CrossRef]
- Fujita, K.; Yuasa, T.; Takahashi, Y.; Tanaka, K.; Sako, W.; Koizumi, H.; Iwasaki, Y.; Yoshida, M.; Izumi, Y.; Kaji, R. Antibodies to N-Methyl-D-Aspartate Glutamate Receptors in Creutzfeldt–Jakob Disease Patients. J. Neuroimmunol. 2012, 251, 90–93. [Google Scholar] [CrossRef]
- Fujita, K.; Yuasa, T.; Watanabe, O.; Takahashi, Y.; Hashiguchi, S.; Adachi, K.; Izumi, Y.; Kaji, R. Voltage-Gated Potassium Channel Complex Antibodies in Creutzfeldt-Jakob Disease. J. Neurol. 2012, 259, 2249–2250. [Google Scholar] [CrossRef]
- Angus-Leppan, H.; Rudge, P.; Mead, S.; Collinge, J.; Vincent, A. Autoantibodies in Sporadic Creutzfeldt-Jakob Disease. JAMA Neurol. 2013, 70, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Mead, S.; Collinge, J.; Rudge, P.; Vincent, A. Neuronal Antibodies in Patients with Suspected or Confirmed Sporadic Creutzfeldt-Jakob Disease: Table 1. J. Neurol. Neurosurg. Psychiatry 2015, 86, 692–694. [Google Scholar] [CrossRef] [PubMed]
- Zuhorn, F.; Hübenthal, A.; Rogalewski, A.; Dogan Onugoren, M.; Glatzel, M.; Bien, C.G.; Schäbitz, W.-R. Creutzfeldt-Jakob Disease Mimicking Autoimmune Encephalitis with CASPR2 Antibodies. BMC Neurol. 2014, 14, 227. [Google Scholar] [CrossRef] [PubMed]
- Grau-Rivera, O.; Sánchez-Valle, R.; Saiz, A.; Molinuevo, J.L.; Bernabé, R.; Munteis, E.; Pujadas, F.; Salvador, A.; Saura, J.; Ugarte, A.; et al. Determination of Neuronal Antibodies in Suspected and Definite Creutzfeldt-Jakob Disease. JAMA Neurol. 2014, 71, 74. [Google Scholar] [CrossRef]
- Çoban, A.; İsmail Küçükali, C.; Bilgiç, B.; Yalçınkaya, N.; Haytural, H.; Ulusoy, C.; Turan, S.; Çakır, S.; Üçok, A.; Ünübol, H.; et al. Evaluation of Incidence and Clinical Features of Antibody-Associated Autoimmune Encephalitis Mimicking Dementia. Behav. Neurol. 2014, 2014, 935379. [Google Scholar] [CrossRef]
- Gibson, L.L.; McKeever, A.; Cullen, A.E.; Nicholson, T.R.; Aarsland, D.; Zandi, M.S.; Pollak, T.A. Neuronal Surface Autoantibodies in Dementia: A Systematic Review and Meta-Analysis. J. Neurol. 2021, 268, 2769–2779. [Google Scholar] [CrossRef]
- Dahm, L.; Ott, C.; Steiner, J.; Stepniak, B.; Teegen, B.; Saschenbrecker, S.; Hammer, C.; Borowski, K.; Begemann, M.; Lemke, S.; et al. Seroprevalence of Autoantibodies against Brain Antigens in Health and Disease: Brain-Targeting Autoantibodies. Ann. Neurol. 2014, 76, 82–94. [Google Scholar] [CrossRef]
- Hopfner, F.; Müller, S.H.; Steppat, D.; Miller, J.; Schmidt, N.; Wandinger, K.-P.; Leypoldt, F.; Berg, D.; Franke, A.; Lieb, W.; et al. No Association between Parkinson Disease and Autoantibodies against NMDA-Type Glutamate Receptors. Transl. Neurodegener. 2019, 8, 11. [Google Scholar] [CrossRef]
- Lang, K.; Prüss, H. Frequencies of Neuronal Autoantibodies in Healthy Controls: Estimation of Disease Specificity. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e386. [Google Scholar] [CrossRef]
- Pollak, T.A.; Beck, K.; Irani, S.R.; Howes, O.D.; David, A.S.; McGuire, P.K. Autoantibodies to Central Nervous System Neuronal Surface Antigens: Psychiatric Symptoms and Psychopharmacological Implications. Psychopharmacology 2016, 233, 1605–1621. [Google Scholar] [CrossRef]
- Balint, B.; Vincent, A.; Meinck, H.-M.; Irani, S.R.; Bhatia, K.P. Movement Disorders with Neuronal Antibodies: Syndromic Approach, Genetic Parallels and Pathophysiology. Brain 2018, 141, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Ding, J.L. Natural Antibodies Bridge Innate and Adaptive Immunity. J. Immunol. 2015, 194, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Hammer, C.; Stepniak, B.; Schneider, A.; Papiol, S.; Tantra, M.; Begemann, M.; Sirén, A.-L.; Pardo, L.A.; Sperling, S.; Mohd Jofrry, S.; et al. Neuropsychiatric Disease Relevance of Circulating Anti-NMDA Receptor Autoantibodies Depends on Blood–Brain Barrier Integrity. Mol. Psychiatry 2014, 19, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Gómez, E.; Oliveira, B.; Tapken, D.; Bertrand, S.; Klein-Schmidt, C.; Pan, H.; Zafeiriou, P.; Steiner, J.; Jurek, B.; Trippe, R.; et al. All Naturally Occurring Autoantibodies against the NMDA Receptor Subunit NR1 Have Pathogenic Potential Irrespective of Epitope and Immunoglobulin Class. Mol. Psychiatry 2017, 22, 1776–1784. [Google Scholar] [CrossRef]
- Jézéquel, J.; Johansson, E.M.; Dupuis, J.P.; Rogemond, V.; Gréa, H.; Kellermayer, B.; Hamdani, N.; Le Guen, E.; Rabu, C.; Lepleux, M.; et al. Dynamic Disorganization of Synaptic NMDA Receptors Triggered by Autoantibodies from Psychotic Patients. Nat. Commun. 2017, 8, 1791. [Google Scholar] [CrossRef]
- Giannoccaro, M.P.; Wright, S.K.; Vincent, A. In Vivo Mechanisms of Antibody-Mediated Neurological Disorders: Animal Models and Potential Implications. Front. Neurol. 2020, 10, 1394. [Google Scholar] [CrossRef]
- Hirose, S.; Hara, M.; Kamei, S.; Dalmau, J.; Nakajima, H. Characteristics of Clinical Relapses and Patient-Oriented Long-Term Outcomes of Patients with Anti-N-Methyl-d-Aspartate Receptor Encephalitis. J. Neurol. 2022, 269, 2486–2492. [Google Scholar] [CrossRef]
- Finke, C.; Kopp, U.A.; Prüss, H.; Dalmau, J.; Wandinger, K.-P.; Ploner, C.J. Cognitive Deficits Following Anti-NMDA Receptor Encephalitis. J. Neurol. Neurosurg. Psychiatry 2012, 83, 195–198. [Google Scholar] [CrossRef]
- Finke, C.; Kopp, U.A.; Scheel, M.; Pech, L.-M.; Soemmer, C.; Schlichting, J.; Leypoldt, F.; Brandt, A.U.; Wuerfel, J.; Probst, C.; et al. Functional and Structural Brain Changes in Anti-N-Methyl-D-Aspartate Receptor Encephalitis: Finke et al.: MRI in Anti-NMDAR Encephalitis. Ann. Neurol. 2013, 74, 284–296. [Google Scholar] [CrossRef]
- McKeon, G.L.; Scott, J.G.; Spooner, D.M.; Ryan, A.E.; Blum, S.; Gillis, D.; Langguth, D.; Robinson, G.A. Cognitive and Social Functioning Deficits after Anti-N-Methyl-D-Aspartate Receptor Encephalitis: An Exploratory Case Series. J. Int. Neuropsychol. Soc. 2016, 22, 828–838. [Google Scholar] [CrossRef]
- Heine, J.; Kopp, U.A.; Klag, J.; Ploner, C.J.; Prüss, H.; Finke, C. Long-Term Cognitive Outcome in Anti–N-Methyl-D-Aspartate Receptor Encephalitis. Ann. Neurol. 2021, 90, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Finke, C.; Prüss, H.; Heine, J.; Reuter, S.; Kopp, U.A.; Wegner, F.; Then Bergh, F.; Koch, S.; Jansen, O.; Münte, T.; et al. Evaluation of Cognitive Deficits and Structural Hippocampal Damage in Encephalitis with Leucine-Rich, Glioma-Inactivated 1 Antibodies. JAMA Neurol. 2017, 74, 50. [Google Scholar] [CrossRef] [PubMed]
- Maureille, A.; Fenouil, T.; Joubert, B.; Picard, G.; Rogemond, V.; Pinto, A.-L.; Thomas, L.; Ducray, F.; Quadrio, I.; Psimaras, D.; et al. Isolated Seizures Are a Common Early Feature of Paraneoplastic Anti-GABAB Receptor Encephalitis. J. Neurol. 2019, 266, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, C.; Li, A.; Liu, X.; Chen, C.; Gong, X.; Zhou, D.; Hong, Z. Long-Term Cognitive and Neuropsychiatric Outcomes of Anti-GABABR Encephalitis Patients: A Prospective Study. J. Neuroimmunol. 2021, 351, 577471. [Google Scholar] [CrossRef] [PubMed]
- Nicolle, D.C.M.; Moses, J.L. A Systematic Review of the Neuropsychological Sequelae of People Diagnosed with Anti N-Methyl-D-Aspartate Receptor Encephalitis in the Acute and Chronic Phases. Arch. Clin. Neuropsychol. 2018, 33, 964–983. [Google Scholar] [CrossRef]
- McKeon, G.L.; Robinson, G.A.; Ryan, A.E.; Blum, S.; Gillis, D.; Finke, C.; Scott, J.G. Cognitive Outcomes Following Anti-N-Methyl-D-Aspartate Receptor Encephalitis: A Systematic Review. J. Clin. Exp. Neuropsychol. 2018, 40, 234–252. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, C.D.; Porter, J.N.; Waldron, E.J.; Klein, H.; Tranel, D.; Heffelfinger, A. Neuropsychological Characterization of Three Adolescent Females with Anti-NMDA Receptor Encephalitis in the Acute, Post-Acute, and Chronic Phases: An Inter-Institutional Case Series. Clin. Neuropsychol. 2017, 31, 268–288. [Google Scholar] [CrossRef]
- Dalmau, J.; Lancaster, E.; Martinez-Hernandez, E.; Rosenfeld, M.R.; Balice-Gordon, R. Clinical Experience and Laboratory Investigations in Patients with Anti-NMDAR Encephalitis. Lancet Neurol. 2011, 10, 63–74. [Google Scholar] [CrossRef]
- Hansen, N. Long-Term Memory Dysfunction in Limbic Encephalitis. Front. Neurol. 2019, 10, 330. [Google Scholar] [CrossRef]
- Hébert, J.; Day, G.S.; Steriade, C.; Wennberg, R.A.; Tang-Wai, D.F. Long-Term Cognitive Outcomes in Patients with Autoimmune Encephalitis. Can. J. Neurol. Sci. 2018, 45, 540–544. [Google Scholar] [CrossRef]
- Hanert, A.; Rave, J.; Granert, O.; Ziegler, M.; Pedersen, A.; Born, J.; Finke, C.; Bartsch, T. Hippocampal Dentate Gyrus Atrophy Predicts Pattern Separation Impairment in Patients with LGI1 Encephalitis. Neuroscience 2019, 400, 120–131. [Google Scholar] [CrossRef]
- Dogan Onugoren, M.; Golombeck, K.S.; Bien, C.; Abu-Tair, M.; Brand, M.; Bulla-Hellwig, M.; Lohmann, H.; Münstermann, D.; Pavenstädt, H.; Thölking, G.; et al. Immunoadsorption Therapy in Autoimmune Encephalitides. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e207. [Google Scholar] [CrossRef]
- Phillips, O.R.; Joshi, S.H.; Narr, K.L.; Shattuck, D.W.; Singh, M.; Di Paola, M.; Ploner, C.J.; Prüss, H.; Paul, F.; Finke, C. Superficial White Matter Damage in Anti-NMDA Receptor Encephalitis. J. Neurol. Neurosurg. Psychiatry 2018, 89, 518–525. [Google Scholar] [CrossRef]
- Finke, C.; Kopp, U.A.; Pajkert, A.; Behrens, J.R.; Leypoldt, F.; Wuerfel, J.T.; Ploner, C.J.; Prüss, H.; Paul, F. Structural Hippocampal Damage Following Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Biol. Psychiatry 2016, 79, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Cai, L.; Zhou, X.; Huang, H.; Zheng, J. Voxel-Based Analysis and Multivariate Pattern Analysis of Diffusion Tensor Imaging Study in Anti-NMDA Receptor Encephalitis. Neuroradiology 2020, 62, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xiang, Y.; Liu, J.; Feng, J.; Du, S.; Luo, T.; Li, Y.; Zeng, C. Diffusion Kurtosis Imaging and Diffusion Tensor Imaging Parameters Applied to White Matter and Gray Matter of Patients with Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Front. Neurosci. 2022, 16, 1030230. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Lv, X.; Zhang, J.; Li, C.; Wei, L.; Zhou, N.; Xu, J.; Tian, Y.; Wang, K. Gray Matter Atrophy and Corresponding Impairments in Connectivity in Patients with Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Brain Imaging Behav. 2022, 16, 2001–2010. [Google Scholar] [CrossRef]
- Xu, J.; Guo, Y.; Li, J.; Lv, X.; Zhang, J.; Zhang, J.; Hu, Q.; Wang, K.; Tian, Y. Progressive Cortical and Sub-Cortical Alterations in Patients with Anti-N-Methyl-d-Aspartate Receptor Encephalitis. J. Neurol. 2022, 269, 389–398. [Google Scholar] [CrossRef]
- Long, Q.; Lv, Z.; Zhao, J.; Shi, K.; Li, C.; Fan, B.; Zheng, J. Cerebral Gray Matter Volume Changes in Patients with Anti-N-Methyl-D-Aspartate Receptor Encephalitis: A Voxel-Based Morphometry Study. Front. Neurol. 2022, 13, 892242. [Google Scholar] [CrossRef]
- Peer, M.; Prüss, H.; Ben-Dayan, I.; Paul, F.; Arzy, S.; Finke, C. Functional Connectivity of Large-Scale Brain Networks in Patients with Anti-NMDA Receptor Encephalitis: An Observational Study. Lancet Psychiatry 2017, 4, 768–774. [Google Scholar] [CrossRef]
- Loane, C.; Argyropoulos, G.P.D.; Roca-Fernández, A.; Lage, C.; Sheerin, F.; Ahmed, S.; Zamboni, G.; Mackay, C.; Irani, S.R.; Butler, C.R. Hippocampal Network Abnormalities Explain Amnesia after VGKCC-Ab Related Autoimmune Limbic Encephalitis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 965–974. [Google Scholar] [CrossRef]
- Miller, T.D.; Chong, T.T.-J.; Aimola Davies, A.M.; Ng, T.W.C.; Johnson, M.R.; Irani, S.R.; Vincent, A.; Husain, M.; Jacob, S.; Maddison, P.; et al. Focal CA3 Hippocampal Subfield Atrophy Following LGI1 VGKC-Complex Antibody Limbic Encephalitis. Brain 2017, 140, 1212–1219. [Google Scholar] [CrossRef]
- Szots, M.; Blaabjerg, M.; Orsi, G.; Iversen, P.; Kondziella, D.; Madsen, C.G.; Garde, E.; Magnusson, P.O.; Barsi, P.; Nagy, F.; et al. Global Brain Atrophy and Metabolic Dysfunction in LGI1 Encephalitis: A Prospective Multimodal MRI Study. J. Neurol. Sci. 2017, 376, 159–165. [Google Scholar] [CrossRef]
- Heine, J.; Prüss, H.; Kopp, U.A.; Wegner, F.; Then Bergh, F.; Münte, T.; Wandinger, K.-P.; Paul, F.; Bartsch, T.; Finke, C. Beyond the Limbic System: Disruption and Functional Compensation of Large-Scale Brain Networks in Patients with Anti-LGI1 Encephalitis. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Guery, D.; Cousyn, L.; Navarro, V.; Picard, G.; Rogemond, V.; Bani-Sadr, A.; Shor, N.; Joubert, B.; Muñiz-Castrillo, S.; Honnorat, J.; et al. Long-Term Evolution and Prognostic Factors of Epilepsy in Limbic Encephalitis with LGI1 Antibodies. J. Neurol. 2022, 269, 5061–5069. [Google Scholar] [CrossRef] [PubMed]
- Szots, M.; Marton, A.; Kover, F.; Kiss, T.; Berki, T.; Nagy, F.; Illes, Z. Natural Course of LGI1 Encephalitis: 3–5years of Follow-up without Immunotherapy. J. Neurol. Sci. 2014, 343, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.; David, B.; Ernst, L.; Becker, A.J.; Witt, J.; Helmstaedter, C.; Wagner, J.; Weber, B.; Elger, C.E.; Surges, R.; et al. Structural Network Topology in Limbic Encephalitis Is Associated with Amygdala Enlargement, Memory Performance and Serostatus. Epilepsia 2020, 61, e140–e145. [Google Scholar] [CrossRef]
- O’Connor, K.; Waters, P.; Komorowski, L.; Zekeridou, A.; Guo, C.-Y.; Mgbachi, V.C.; Probst, C.; Mindorf, S.; Teegen, B.; Gelfand, J.M.; et al. GABAA Receptor Autoimmunity: A Multicenter Experience. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e552. [Google Scholar] [CrossRef]
- Dogan Onugoren, M.; Deuretzbacher, D.; Haensch, C.A.; Hagedorn, H.J.; Halve, S.; Isenmann, S.; Kramme, C.; Lohner, H.; Melzer, N.; Monotti, R.; et al. Limbic Encephalitis Due to GABA B and AMPA Receptor Antibodies: A Case Series. J. Neurol. Neurosurg. Psychiatry 2015, 86, 965–972. [Google Scholar] [CrossRef]
- Gaetani, L.; Blennow, K.; Calabresi, P.; Di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament Light Chain as a Biomarker in Neurological Disorders. J. Neurol. Neurosurg. Psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. Biomarkers for Alzheimer’s Disease: Current Status and Prospects for the Future. J. Intern. Med. 2018, 284, 643–663. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.K.; Day, G.S.; Graff-Radford, J.; McKeon, A.; Flanagan, E.P.; Algeciras-Schimnich, A.; Mielke, M.M.; Nguyen, A.; Jones, D.T.; Toledano, M.; et al. Alzheimer’s Disease Cerebrospinal Fluid Biomarkers Differentiate Patients with Creutzfeldt–Jakob Disease and Autoimmune Encephalitis. Eur. J. Neurol. 2022, 29, 2905–2912. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, R.; Krýsl, D.; Bergquist, F.; Andrén, K.; Malmeström, C.; Asztély, F.; Axelsson, M.; Menachem, E.B.; Blennow, K.; Rosengren, L.; et al. Cerebrospinal Fluid Markers of Neuronal and Glial Cell Damage to Monitor Disease Activity and Predict Long-Term Outcome in Patients with Autoimmune Encephalitis. Eur. J. Neurol. 2016, 23, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, R.; Krýsl, D.; Andrén, K.; Asztély, F.; Bergquist, F.; Zetterberg, H.; Andreasson, U.; Axelsson, M.; Menachem, E.B.; Jons, D.; et al. Cerebrospinal Fluid Markers of Neuronal and Glial Cell Damage in Patients with Autoimmune Neurologic Syndromes with and without Underlying Malignancies. J. Neuroimmunol. 2017, 306, 25–30. [Google Scholar] [CrossRef]
- Day, G.S.; Yarbrough, M.Y.; Körtvelyessy, P.; Prüss, H.; Bucelli, R.C.; Fritzler, M.J.; Mason, W.; Tang-Wai, D.F.; Steriade, C.; Hébert, J.; et al. Prospective Quantification of CSF Biomarkers in Antibody-Mediated Encephalitis. Neurology 2021, 96, e2546–e2557. [Google Scholar] [CrossRef]
- Guasp, M.; Martín-Aguilar, L.; Sabater, L.; Bioque, M.; Armangué, T.; Martínez-Hernández, E.; Landa, J.; Maudes, E.; Borràs, R.; Muñoz-Lopetegi, A.; et al. Neurofilament Light Chain Levels in Anti-NMDAR Encephalitis and Primary Psychiatric Psychosis. Neurology 2022, 98, e1489–e1498. [Google Scholar] [CrossRef]
- Körtvelyessy, P.; Prüss, H.; Thurner, L.; Maetzler, W.; Vittore-Welliong, D.; Schultze-Amberger, J.; Heinze, H.-J.; Reinhold, D.; Leypoldt, F.; Schreiber, S.; et al. Biomarkers of Neurodegeneration in Autoimmune-Mediated Encephalitis. Front. Neurol. 2018, 9, 668. [Google Scholar] [CrossRef]
- Lardeux, P.; Fourier, A.; Peter, E.; Dorey, A.; Muñiz-Castrillo, S.; Vogrig, A.; Picard, G.; Rogemond, V.; Verdurand, M.; Formaglio, M.; et al. Core Cerebrospinal Fluid Biomarker Profile in Anti-LGI1 Encephalitis. J. Neurol. 2022, 269, 377–388. [Google Scholar] [CrossRef]
- Ma, X.; Lu, Y.; Peng, F.; Wang, Y.; Sun, X.; Luo, W.; Shen, S.; Liu, Z.; Kermode, A.G.; Qiu, W.; et al. Serum NfL Associated with Anti-NMDA Receptor Encephalitis. Neurol. Sci. 2022, 43, 3893–3899. [Google Scholar] [CrossRef]
- Nissen, M.S.; Ryding, M.; Nilsson, A.C.; Madsen, J.S.; Olsen, D.A.; Halekoh, U.; Lydolph, M.; Illes, Z.; Blaabjerg, M. CSF-Neurofilament Light Chain Levels in NMDAR and LGI1 Encephalitis: A National Cohort Study. Front. Immunol. 2021, 12, 719432. [Google Scholar] [CrossRef]
- Vakrakou, A.G.; Tzartos, J.S.; Strataki, E.; Boufidou, F.; Dimou, E.; Pyrgelis, E.-S.; Constantinides, V.C.; Paraskevas, G.P.; Kapaki, E. Neuronal and Neuroaxonal Damage Cerebrospinal Fluid Biomarkers in Autoimmune Encephalitis Associated or Not with the Presence of Tumor. Biomedicines 2022, 10, 1262. [Google Scholar] [CrossRef] [PubMed]
- Juhl, A.L.; Grenzer, I.M.; Teegen, B.; Wiltfang, J.; Fitzner, D.; Hansen, N. Biomarkers of Neurodegeneration in Neural Autoantibody-Associated Psychiatric Syndromes: A Retrospective Cohort Study. J. Transl. Autoimmun. 2022, 5, 100169. [Google Scholar] [CrossRef]
- Mariotto, S.; Gajofatto, A.; Zuliani, L.; Zoccarato, M.; Gastaldi, M.; Franciotta, D.; Cantalupo, G.; Piardi, F.; Polo, A.; Alberti, D.; et al. Serum and CSF Neurofilament Light Chain Levels in Antibody-Mediated Encephalitis. J. Neurol. 2019, 266, 1643–1648. [Google Scholar] [CrossRef]
- Kammeyer, R.; Mizenko, C.; Sillau, S.; Richie, A.; Owens, G.; Nair, K.V.; Alvarez, E.; Vollmer, T.L.; Bennett, J.L.; Piquet, A.L. Evaluation of Plasma Neurofilament Light Chain Levels as a Biomarker of Neuronal Injury in the Active and Chronic Phases of Autoimmune Neurologic Disorders. Front. Neurol. 2022, 13, 689975. [Google Scholar] [CrossRef] [PubMed]
- Macher, S.; Zrzavy, T.; Höftberger, R.; Altmann, P.; Pataraia, E.; Zimprich, F.; Berger, T.; Rommer, P. Longitudinal Measurement of Cerebrospinal Fluid Neurofilament Light in Anti-N-methyl-D-aspartate Receptor Encephalitis. Eur. J. Neurol. 2021, 28, 1401–1405. [Google Scholar] [CrossRef]
- Li, J.; Gu, Y.; An, H.; Zhou, Z.; Zheng, D.; Wang, Z.; Wen, Z.; Shen, H.; Wang, Q.; Wang, H. Cerebrospinal Fluid Light and Heavy Neurofilament Level Increased in Anti-N-methyl-d-aspartate Receptor Encephalitis. Brain Behav. 2019, 9, e01354. [Google Scholar] [CrossRef]
- Freund, B.; Probasco, J.C.; Cervenka, M.C.; Sutter, R.; Kaplan, P.W. EEG Differences in Two Clinically Similar Rapid Dementias: Voltage-Gated Potassium Channel Complex–Associated Autoimmune Encephalitis and Creutzfeldt-Jakob Disease. Clin. EEG Neurosci. 2019, 50, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Fominykh, V.; Brylev, L.; Gaskin, V.; Luzin, R.; Yakovlev, A.; Komoltsev, I.; Belousova, I.; Rosliakova, A.; Guekht, A.; Gulyaeva, N. Neuronal Damage and Neuroinflammation Markers in Patients with Autoimmune Encephalitis and Multiple Sclerosis. Metab. Brain Dis. 2019, 34, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ding, Y.; Zheng, D.; Wang, Z.; Pan, S.; Ji, T.; Shen, H.-Y.; Wang, H. Elevation of YKL-40 in the CSF of Anti-NMDAR Encephalitis Patients Is Associated with Poor Prognosis. Front. Neurol. 2018, 9, 727. [Google Scholar] [CrossRef]
- Mikasova, L.; De Rossi, P.; Bouchet, D.; Georges, F.; Rogemond, V.; Didelot, A.; Meissirel, C.; Honnorat, J.; Groc, L. Disrupted Surface Cross-Talk between NMDA and Ephrin-B2 Receptors in Anti-NMDA Encephalitis. Brain 2012, 135, 1606–1621. [Google Scholar] [CrossRef]
- Newcomer, J.W.; Farber, N.B.; Olney, J.W. NMDA Receptor Function, Memory, and Brain Aging. Dialogues Clin. Neurosci. 2000, 2, 219–232. [Google Scholar] [CrossRef]
- Tanaka, M.; Bohár, Z.; Vécsei, L. Are Kynurenines Accomplices or Principal Villains in Dementia? Maintenance of Kynurenine Metabolism. Molecules 2020, 25, 564. [Google Scholar] [CrossRef]
- Jorratt, P.; Hoschl, C.; Ovsepian, S.V. Endogenous Antagonists of N-Methyl-d-Aspartate Receptor in Schizophrenia. Alzheimer’s Dement. 2021, 17, 888–905. [Google Scholar] [CrossRef]
- Planagumà, J.; Leypoldt, F.; Mannara, F.; Gutiérrez-Cuesta, J.; Martín-García, E.; Aguilar, E.; Titulaer, M.J.; Petit-Pedrol, M.; Jain, A.; Balice-Gordon, R.; et al. Human N-Methyl D-Aspartate Receptor Antibodies Alter Memory and Behaviour in Mice. Brain 2015, 138, 94–109. [Google Scholar] [CrossRef]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef]
- Matute, C.; Palma, A.; Serrano-Regal, M.P.; Maudes, E.; Barman, S.; Sánchez-Gómez, M.V.; Domercq, M.; Goebels, N.; Dalmau, J. N-Methyl-D-Aspartate Receptor Antibodies in Autoimmune Encephalopathy Alter Oligodendrocyte Function. Ann. Neurol. 2020, 87, 670–676. [Google Scholar] [CrossRef]
- Jones, B.E.; Tovar, K.R.; Goehring, A.; Jalali-Yazdi, F.; Okada, N.J.; Gouaux, E.; Westbrook, G.L. Autoimmune Receptor Encephalitis in Mice Induced by Active Immunization with Conformationally Stabilized Holoreceptors. Sci. Transl. Med. 2019, 11, eaaw0044. [Google Scholar] [CrossRef]
- Zrzavy, T.; Endmayr, V.; Bauer, J.; Macher, S.; Mossaheb, N.; Schwaiger, C.; Ricken, G.; Winklehner, M.; Glatter, S.; Breu, M.; et al. Neuropathological Variability within a Spectrum of NMDAR-Encephalitis. Ann. Neurol. 2021, 90, 725–737. [Google Scholar] [CrossRef]
- Ohkawa, T.; Fukata, Y.; Yamasaki, M.; Miyazaki, T.; Yokoi, N.; Takashima, H.; Watanabe, M.; Watanabe, O.; Fukata, M. Autoantibodies to Epilepsy-Related LGI1 in Limbic Encephalitis Neutralize LGI1-ADAM22 Interaction and Reduce Synaptic AMPA Receptors. J. Neurosci. 2013, 33, 18161–18174. [Google Scholar] [CrossRef]
- Klang, A.; Schmidt, P.; Kneissl, S.; Bagó, Z.; Vincent, A.; Lang, B.; Moloney, T.; Bien, C.G.; Halász, P.; Bauer, J.; et al. IgG and Complement Deposition and Neuronal Loss in Cats and Humans with Epilepsy and Voltage-Gated Potassium Channel Complex Antibodies. J. Neuropathol. Exp. Neurol. 2014, 73, 403–413. [Google Scholar] [CrossRef]
- Patterson, K.R.; Dalmau, J.; Lancaster, E. Mechanisms of Caspr2 Antibodies in Autoimmune Encephalitis and Neuromyotonia: Caspr2 Antibody Mechanisms. Ann. Neurol. 2018, 83, 40–51. [Google Scholar] [CrossRef]
- Saint-Martin, M.; Pieters, A.; Déchelotte, B.; Malleval, C.; Pinatel, D.; Pascual, O.; Karagogeos, D.; Honnorat, J.; Pellier-Monnin, V.; Noraz, N. Impact of Anti-CASPR2 Autoantibodies from Patients with Autoimmune Encephalitis on CASPR2/TAG-1 Interaction and Kv1 Expression. J. Autoimmun. 2019, 103, 102284. [Google Scholar] [CrossRef]
- Sundal, C.; Vedeler, C.; Miletic, H.; Andersen, O. Morvan Syndrome with Caspr2 Antibodies. Clinical and Autopsy Report. J. Neurol. Sci. 2017, 372, 453–455. [Google Scholar] [CrossRef]
- Liguori, R. Morvan’s Syndrome: Peripheral and Central Nervous System and Cardiac Involvement with Antibodies to Voltage-Gated Potassium Channels. Brain 2001, 124, 2417–2426. [Google Scholar] [CrossRef]
- Körtvelyessy, P.; Bauer, J.; Stoppel, C.M.; Brück, W.; Gerth, I.; Vielhaber, S.; Wiedemann, F.R.; Heinze, H.J.; Bartels, C.; Bien, C.G. Complement-Associated Neuronal Loss in a Patient with CASPR2 Antibody–Associated Encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e75. [Google Scholar] [CrossRef]
- Golombeck, K.S.; Bönte, K.; Mönig, C.; van Loo, K.M.; Hartwig, M.; Schwindt, W.; Widman, G.; Lindenau, M.; Becker, A.J.; Glatzel, M.; et al. Evidence of a Pathogenic Role for CD8 + T Cells in Anti-GABA B Receptor Limbic Encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e232. [Google Scholar] [CrossRef]
- Wei, Y.-C.; Liu, C.-H.; Lin, J.-J.; Lin, K.-J.; Huang, K.-L.; Lee, T.-H.; Chang, Y.-J.; Peng, T.-I.; Lin, K.-L.; Chang, T.-Y.; et al. Rapid Progression and Brain Atrophy in Anti-AMPA Receptor Encephalitis. J. Neuroimmunol. 2013, 261, 129–133. [Google Scholar] [CrossRef]
- Simabukuro, M.M.; Sabater, L.; Adoni, T.; Cury, R.G.; Haddad, M.S.; Moreira, C.H.; Oliveira, L.; Boaventura, M.; Alves, R.C.; Azevedo Soster, L.; et al. Sleep Disorder, Chorea, and Dementia Associated with IgLON5 Antibodies. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e136. [Google Scholar] [CrossRef]
- Brüggemann, N.; Wandinger, K.-P.; Gaig, C.; Sprenger, A.; Junghanns, K.; Helmchen, C.; Münchau, A. Dystonia, Lower Limb Stiffness, and Upward Gaze Palsy in a Patient with IgLON5 Antibodies. Mov. Disord. 2016, 31, 762–764. [Google Scholar] [CrossRef]
- Haitao, R.; Yingmai, Y.; Yan, H.; Fei, H.; Xia, L.; Honglin, H.; Chaiyan, L.; Stöcker, W.; Liying, C.; Hongzhi, G. Chorea and Parkinsonism Associated with Autoantibodies to IgLON5 and Responsive to Immunotherapy. J. Neuroimmunol. 2016, 300, 9–10. [Google Scholar] [CrossRef]
- Montagna, M.; Amir, R.; De Volder, I.; Lammens, M.; Huyskens, J.; Willekens, B. IgLON5-Associated Encephalitis with Atypical Brain Magnetic Resonance Imaging and Cerebrospinal Fluid Changes. Front. Neurol. 2018, 9, 329. [Google Scholar] [CrossRef]
- Bonello, M.; Jacob, A.; Ellul, M.A.; Barker, E.; Parker, R.; Jefferson, S.; Alusi, S. IgLON5 Disease Responsive to Immunotherapy. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e383. [Google Scholar] [CrossRef]
- Honorat, J.A.; Komorowski, L.; Josephs, K.A.; Fechner, K.; St Louis, E.K.; Hinson, S.R.; Lederer, S.; Kumar, N.; Gadoth, A.; Lennon, V.A.; et al. IgLON5 Antibody: Neurological Accompaniments and Outcomes in 20 Patients. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e385. [Google Scholar] [CrossRef]
- Gaig, C.; Compta, Y.; Heidbreder, A.; Marti, M.J.; Titulaer, M.J.; Crijnen, Y.; Högl, B.; Lewerenz, J.; Erro, M.E.; García-Moncó, J.C.; et al. Frequency and Characterization of Movement Disorders in Anti-IgLON5 Disease. Neurology 2021, 97, e1367–e1381. [Google Scholar] [CrossRef]
- Gelpi, E.; Höftberger, R.; Graus, F.; Ling, H.; Holton, J.L.; Dawson, T.; Popovic, M.; Pretnar-Oblak, J.; Högl, B.; Schmutzhard, E.; et al. Neuropathological Criteria of Anti-IgLON5-Related Tauopathy. Acta Neuropathol. 2016, 132, 531–543. [Google Scholar] [CrossRef]
- Morales-Briceño, H.; Cruse, B.; Fois, A.F.; Lin, M.-W.; Jiang, J.; Banerjee, D.; Grunstein, R.; Varikatt, W.; Rodriguez, M.; Shepherd, C.; et al. IgLON5-Mediated Neurodegeneration Is a Differential Diagnosis of CNS Whipple Disease. Neurology 2018, 90, 1113–1115. [Google Scholar] [CrossRef]
- Erro, M.E.; Sabater, L.; Martínez, L.; Herrera, M.; Ostolaza, A.; García de Gurtubay, I.; Tuñón, T.; Graus, F.; Gelpi, E. Anti-IGLON5 Disease: A New Case without Neuropathologic Evidence of Brainstem Tauopathy. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e651. [Google Scholar] [CrossRef] [PubMed]
- Schöberl, F.; Levin, J.; Remi, J.; Goldschagg, N.; Eren, O.; Okamura, N.; Unterrainer, M.; Rominger, A.; Albert, N.; Brendel, M. IgLON5: A Case with Predominant Cerebellar Tau Deposits and Leptomeningeal Inflammation. Neurology 2018, 91, 180–182. [Google Scholar] [CrossRef]
- Strippel, C.; Heidbreder, A.; Schulte-Mecklenbeck, A.; Korn, L.; Warnecke, T.; Melzer, N.; Wiendl, H.; Pawlowski, M.; Gross, C.C.; Kovac, S. Increased Intrathecal B and Plasma Cells in Patients with Anti-IgLON5 Disease: A Case Series. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1137. [Google Scholar] [CrossRef]
- Sabater, L.; Planagumà, J.; Dalmau, J.; Graus, F. Cellular Investigations with Human Antibodies Associated with the Anti-IgLON5 Syndrome. J. Neuroinflamm. 2016, 13, 226. [Google Scholar] [CrossRef]
- Landa, J.; Gaig, C.; Plagumà, J.; Saiz, A.; Antonell, A.; Sanchez-Valle, R.; Dalmau, J.; Graus, F.; Sabater, L. Effects of IgLON5 Antibodies on Neuronal Cytoskeleton: A Link between Autoimmunity and Neurodegeneration. Ann. Neurol. 2020, 88, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Ryding, M.; Gamre, M.; Nissen, M.S.; Nilsson, A.C.; Okarmus, J.; Poulsen, A.A.E.; Meyer, M.; Blaabjerg, M. Neurodegeneration Induced by Anti-IgLON5 Antibodies Studied in Induced Pluripotent Stem Cell-Derived Human Neurons. Cells 2021, 10, 837. [Google Scholar] [CrossRef] [PubMed]
- Alvente, S.; Matteoli, G.; Molina-Porcel, L.; Landa, J.; Alba, M.; Bastianini, S.; Berteotti, C.; Graus, F.; Lo Martire, V.; Sabater, L.; et al. Pilot Study of the Effects of Chronic Intracerebroventricular Infusion of Human Anti-IgLON5 Disease Antibodies in Mice. Cells 2022, 11, 1024. [Google Scholar] [CrossRef] [PubMed]
- Giil, L.M.; Aarsland, D.; Hellton, K.; Lund, A.; Heidecke, H.; Schulze-Forster, K.; Riemekasten, G.; Vik-Mo, A.O.; Kristoffersen, E.K.; Vedeler, C.A.; et al. Antibodies to Multiple Receptors Are Associated with Neuropsychiatric Symptoms and Mortality in Alzheimer’s Disease: A Longitudinal Study. J. Alzheimer’s Dis. 2018, 64, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.-M.; Koh, Y.H.; Kim, J.-H.; Jeon, J.-P. A Combination of Multiple Autoantibodies Is Associated with the Risk of Alzheimer’s Disease and Cognitive Impairment. Sci. Rep. 2022, 12, 1312. [Google Scholar] [CrossRef]
- Prüss, H. Autoantibodies in Neurological Disease. Nat. Rev. Immunol. 2021, 21, 798–813. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).