
Enhanced Cytotoxic Effects in Human Oral Squamous Cell Carcinoma Cells Treated with Combined Methyltransferase Inhibitors and Histone Deacetylase Inhibitors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Measurement of Cell Viability
2.4. Measurement of Caspase Activity
2.5. Cell Cycle Analysis
2.6. Preparation of Cell Extracts
2.7. Western Blotting Analysis
2.8. Statistical Analysis
3. Results
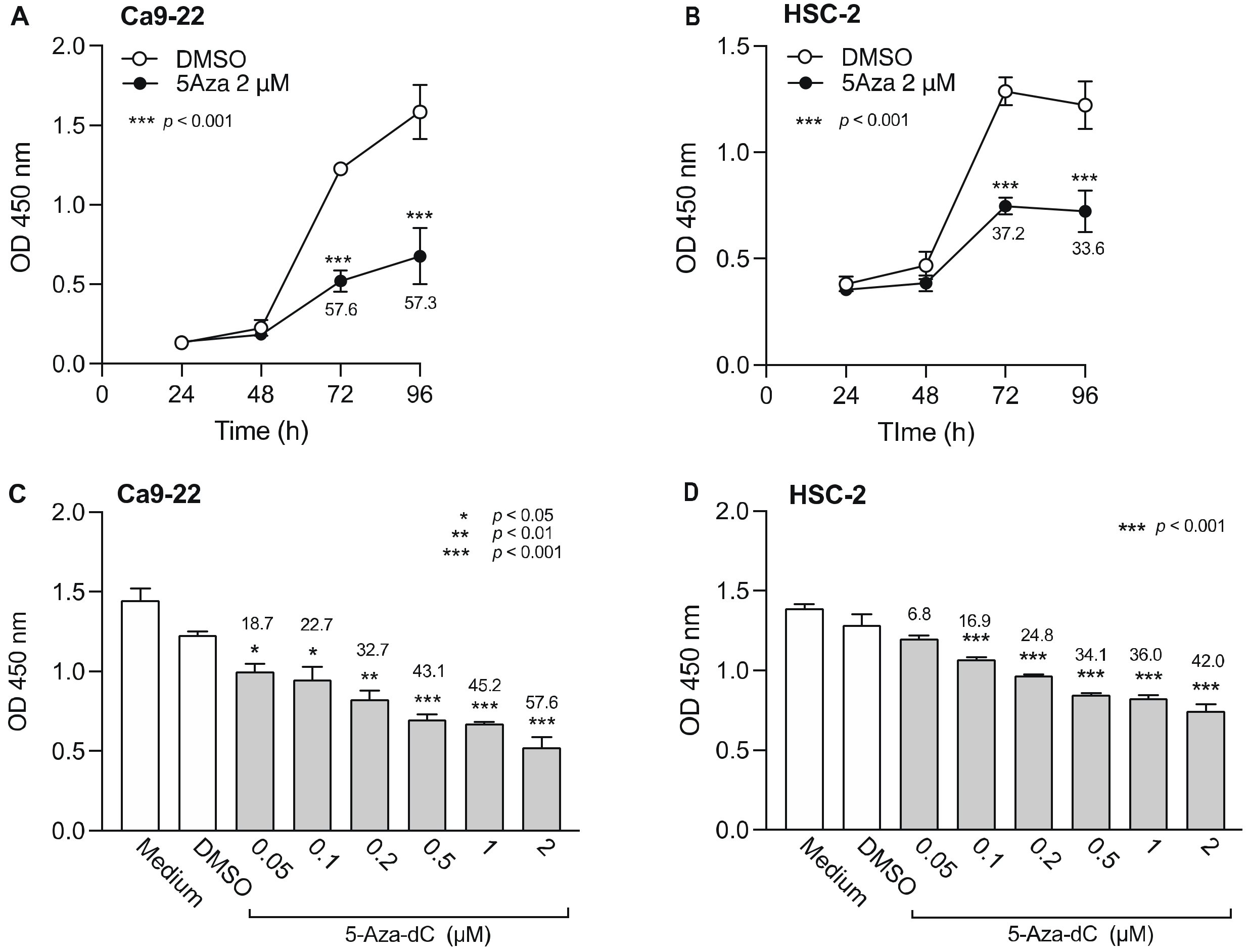
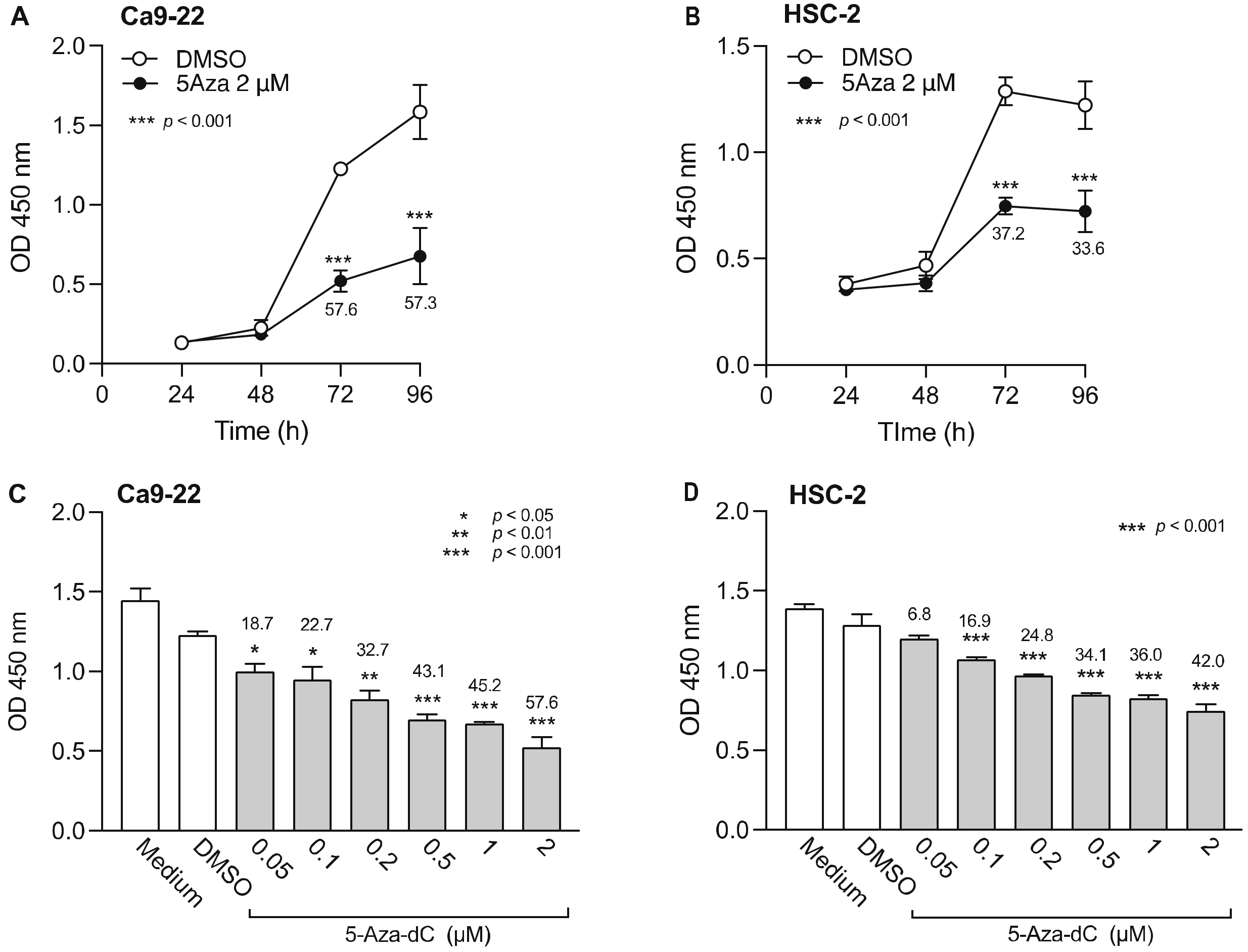
3.1. Effect of DNMTi on Cell Viability of OSCC Cells
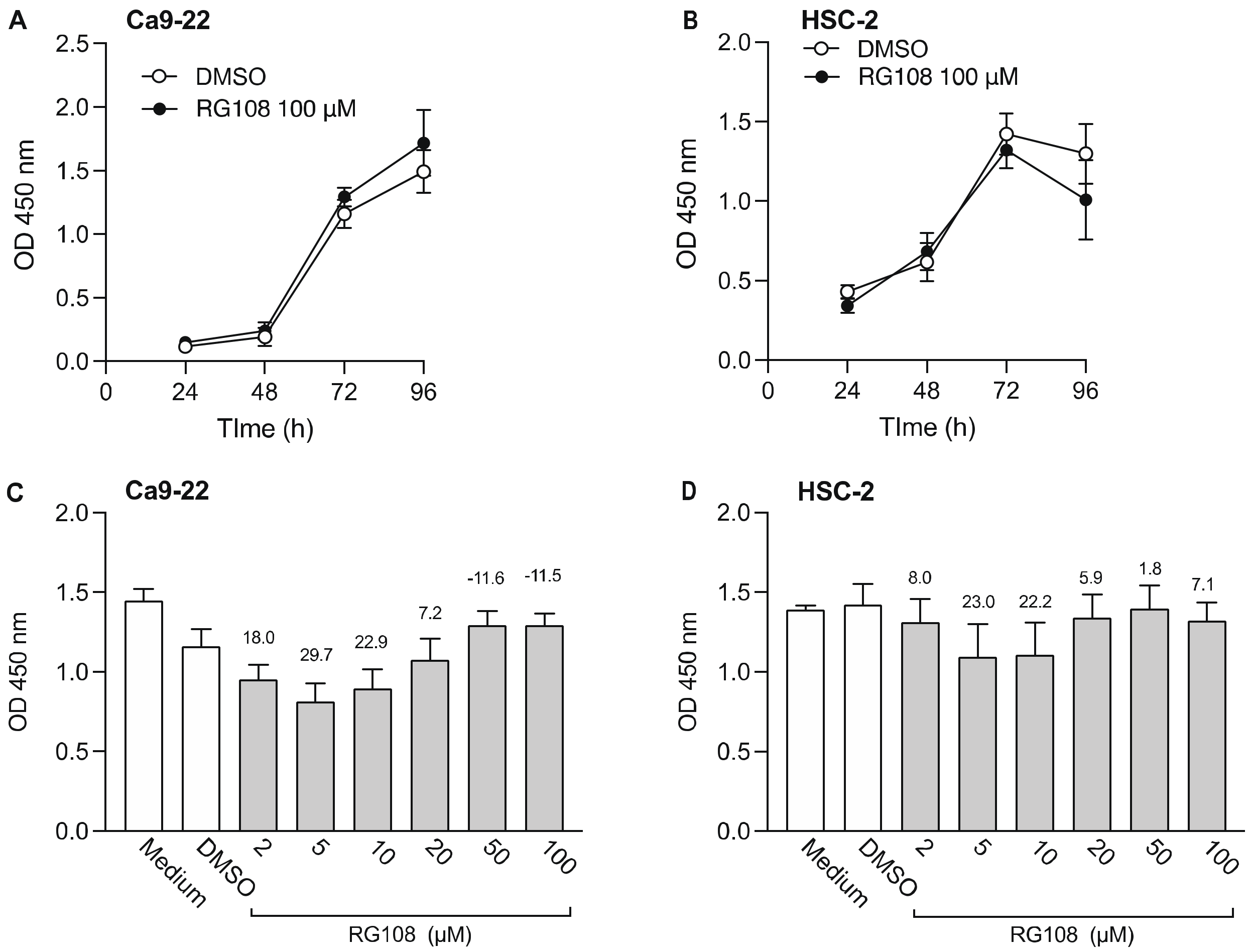
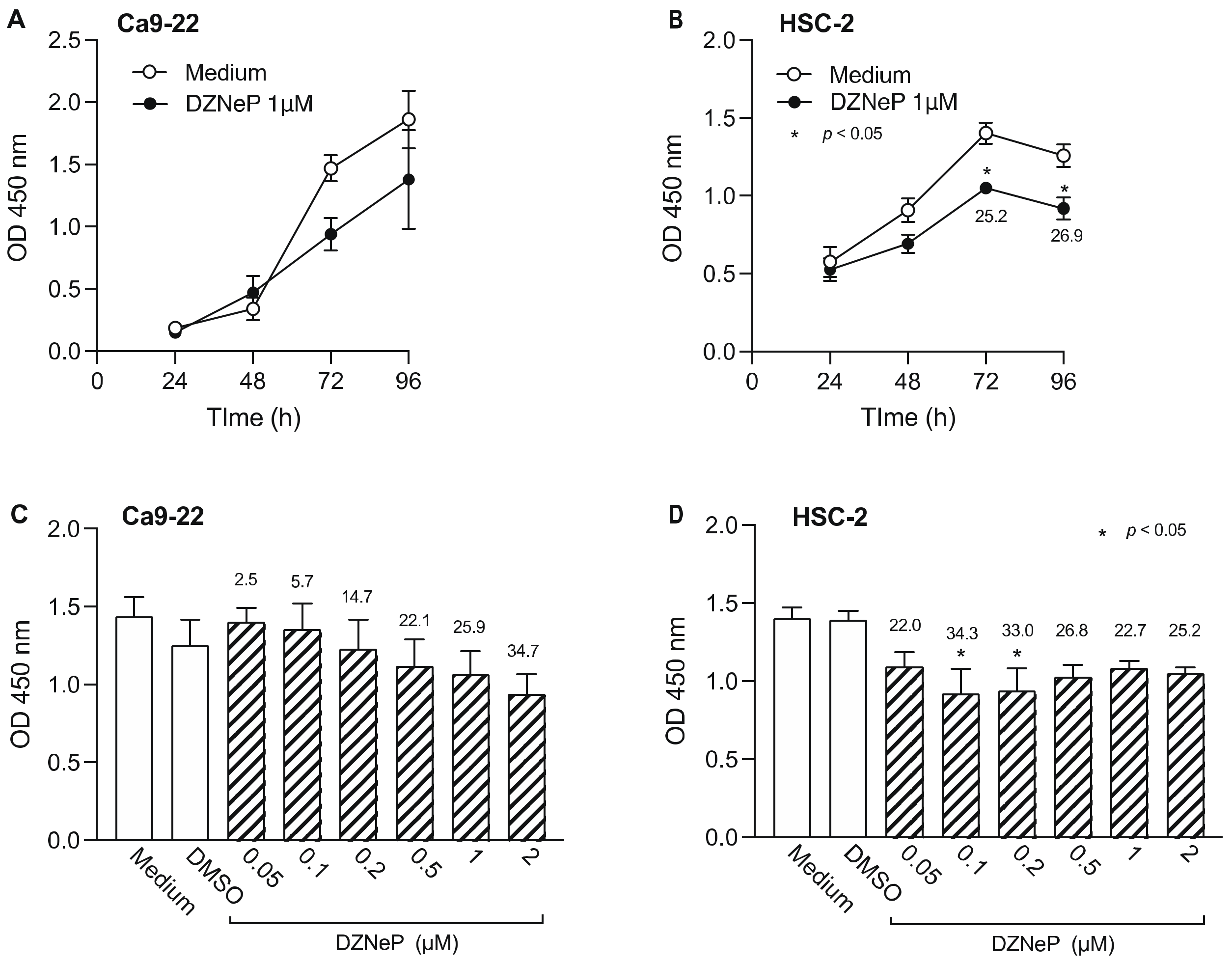
3.2. Effect of HMTi on Cell Viability of OSCC Cells
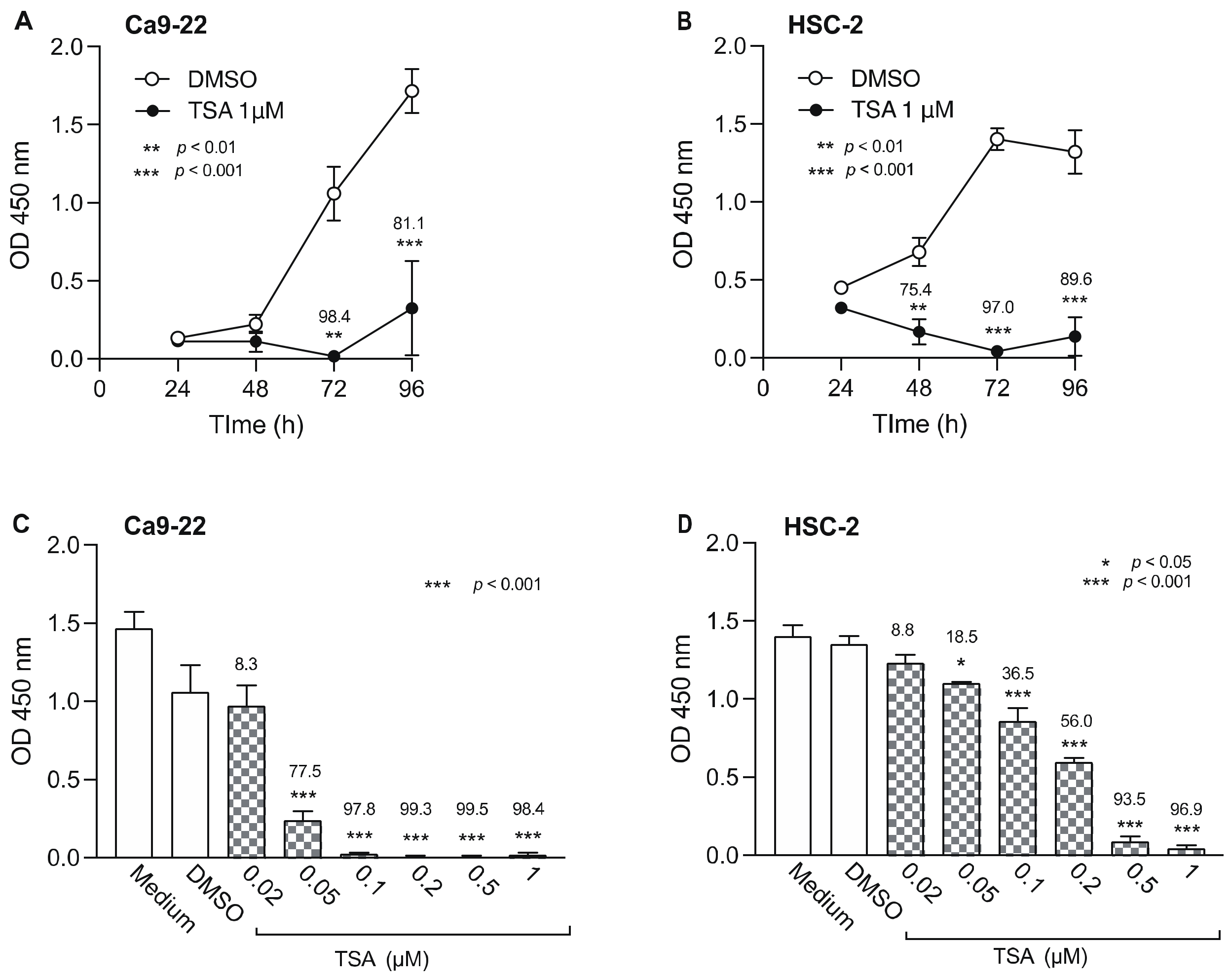
3.3. Effect of HDACis on the Viability of OSCC Cells
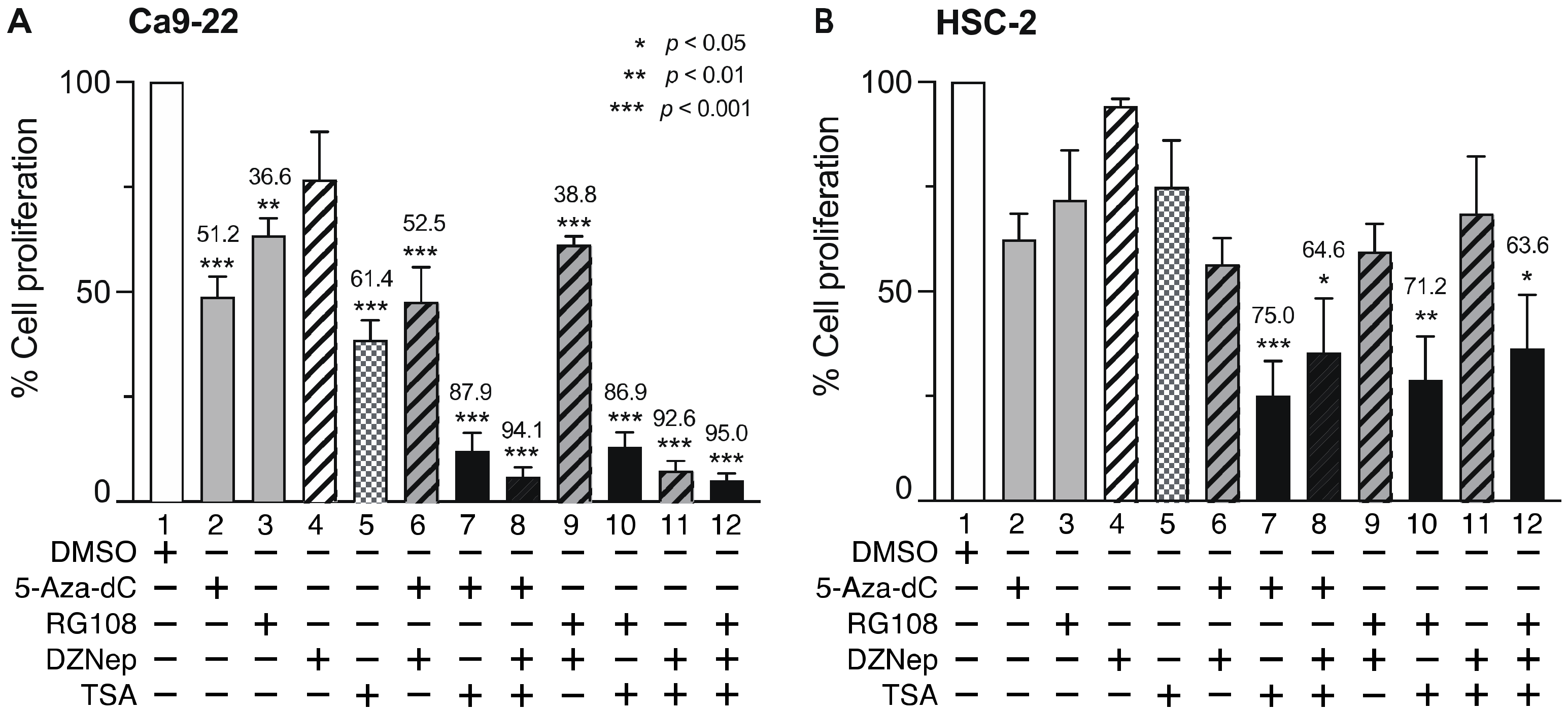
3.4. Combined DNMTi, HMTi, and HDACi Treatment Reduces OSCC Cell Viability
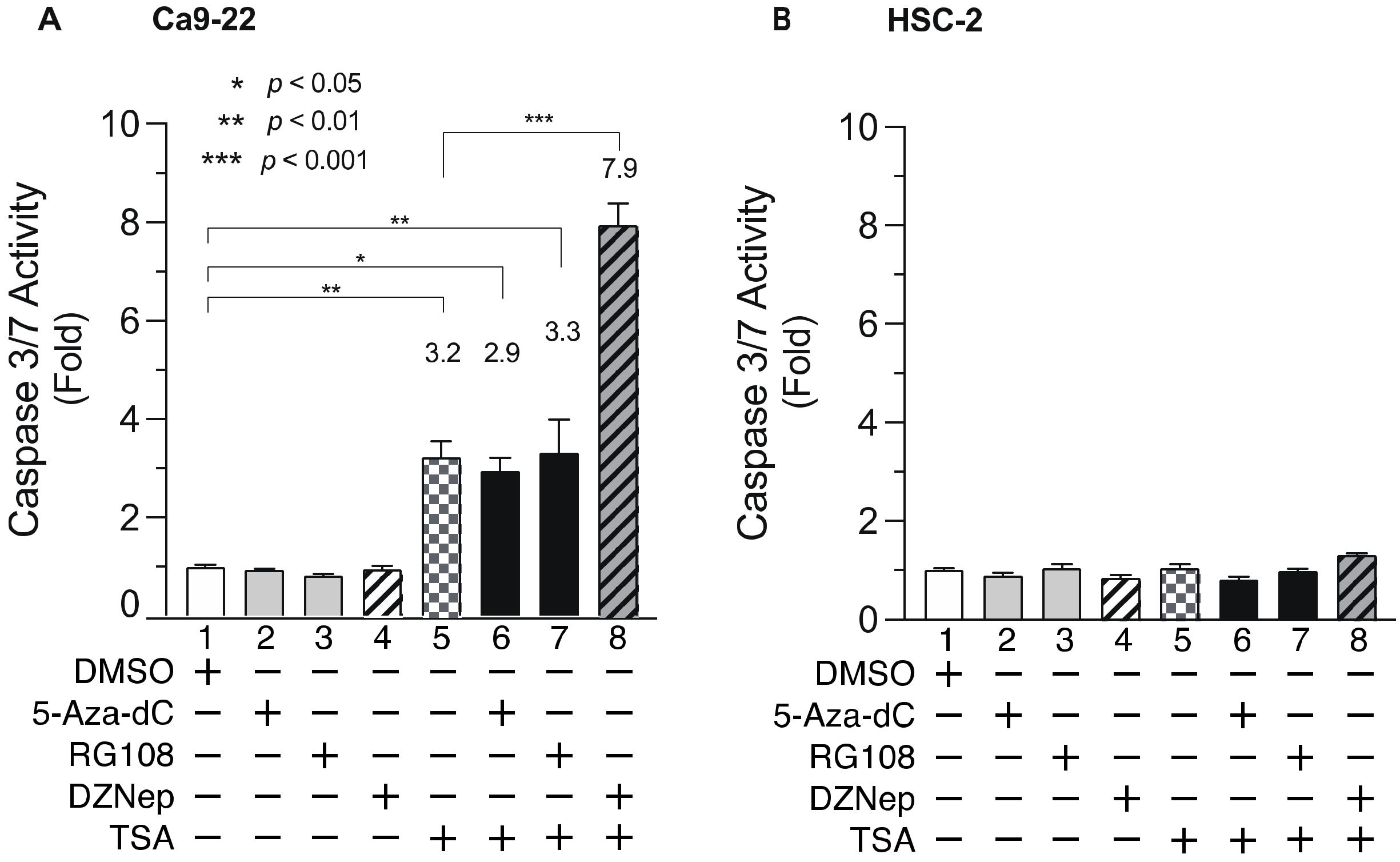
3.5. DNMTis, HMTis, and HDACis Alter Caspase 3/7 Activity in OSCC Cells
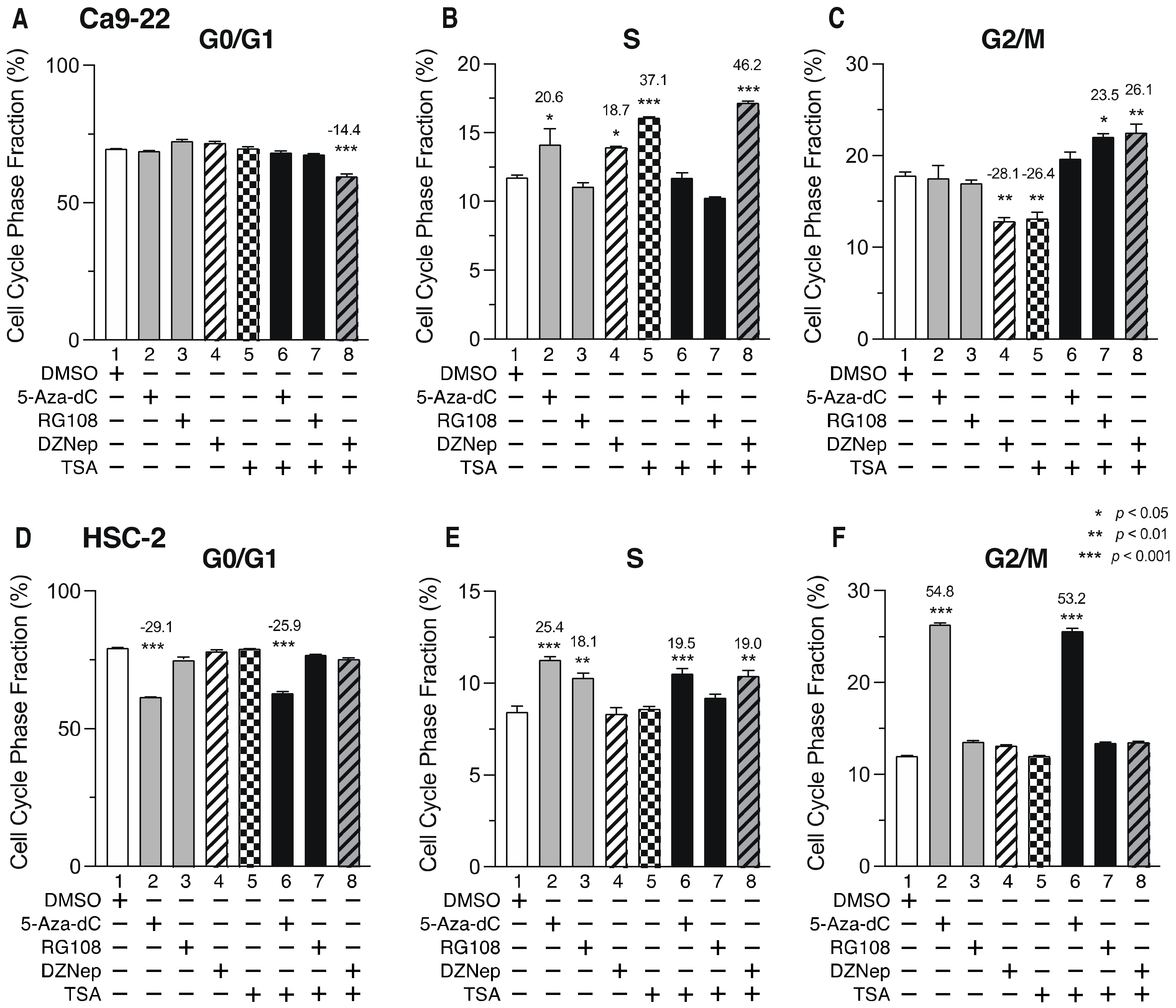
3.6. DNMTis, HMTis, and HDACis Induce Cell Cycle Arrest in OSCC Cells
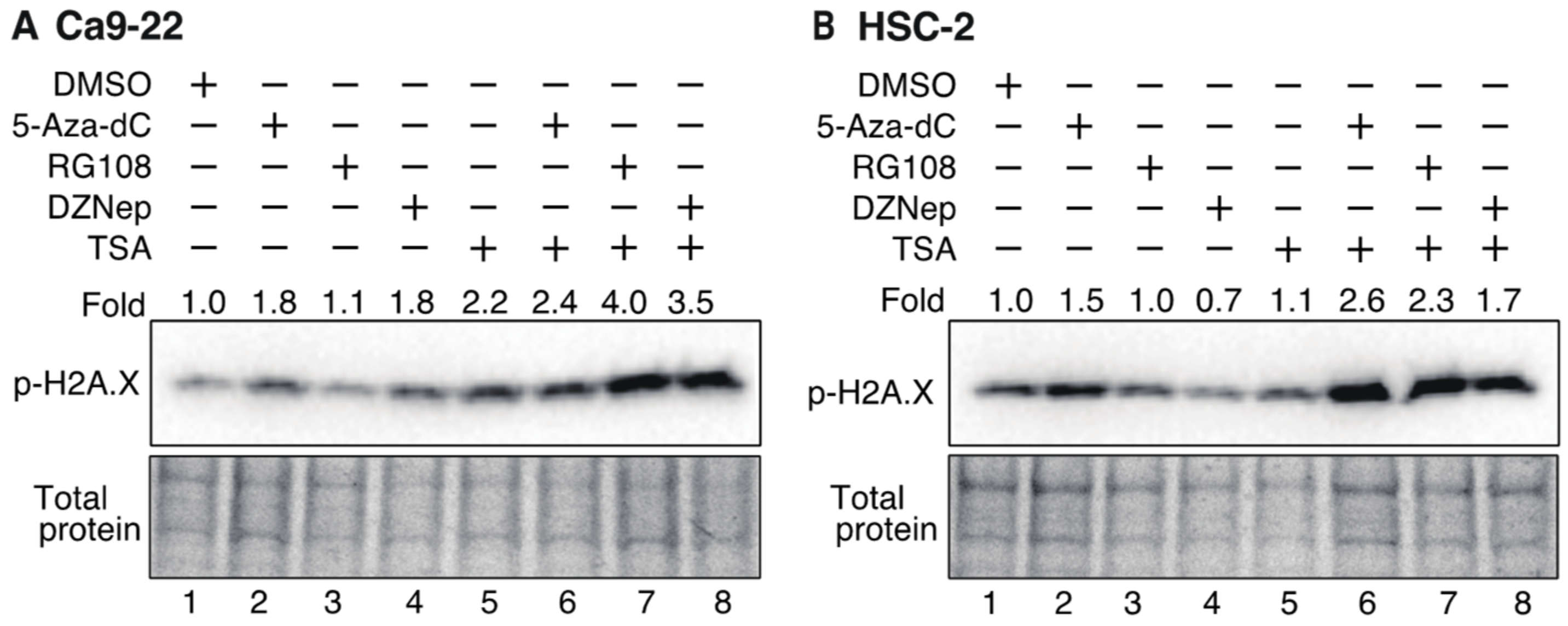
3.7. Histone H2AX Phosphorylation in OSCC Cells Increases after Treatment with DNMTis, HMTis, and HDACis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, R.; Gupta, S. Epigenetics and cancer. J. Appl. Physiol. 2010, 109, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Hillyar, C.; Rallis, K.S.; Varghese, J. Advances in epigenetic cancer therapeutics. Cureus 2020, 12, e11725. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P.; Wolffe, A.P. Methylation-induced repression-belts, braces, and chromatin. Cell 1999, 99, 451–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clouaire, T.; Stancheva, I. Methyl-CpG binding proteins: Specialized transcriptional repressors or structural components of chromatin? Cell Mol. Life Sci. 2008, 65, 1509–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [Green Version]
- Brueckner, B.; Garcia Boy, R.; Siedlecki, P.; Musch, T.; Kliem, H.C.; Zielenkiewicz, P.; Suhai, S.; Wiessler, M.; Lyko, F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005, 65, 6305–6311. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef] [Green Version]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [Green Version]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 2009, 8, 1579–1588. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, Y.; Qiu, J.; Li, Q.; Yuan, C.; Zhang, W.; Wang, D.; Ye, J.; Jiang, H.; Yang, J.; et al. The polycomb group protein EZH2 is a novel therapeutic target in tongue cancer. Oncotarget 2013, 4, 2532–2549. [Google Scholar] [CrossRef] [Green Version]
- Hatta, M.; Naganuma, K.; Kato, K.; Yamazaki, J. 3-Deazaneplanocin A suppresses aggressive phenotype-related gene expression in an oral squamous cell carcinoma cell line. Biochem. Biophys. Res. Commun. 2015, 468, 269–273. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr. Opin. Genet. Dev. 2003, 13, 143–153. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- De Ruijter, A.J.; Van Gennip, A.H.; Caron, H.N.; Kemp, S.; Van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, W.G. Targeting histone deacetylases for cancer therapy: From molecular mechanisms to clinical implications. Int. J. Biol. Sci. 2014, 10, 757–770. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Yokozaki, H.; Kuniyasu, H.; Hayashi, K.; Naka, K.; Ono, S.; Ishikawa, T.; Tahara, E.; Yasui, W. Effect of trichostatin A on cell growth and expression of cell cycle- and apoptosis-related molecules in human gastric and oral carcinoma cell lines. Int. J. Cancer 2000, 88, 992–997. [Google Scholar] [CrossRef]
- Duan, J.; Friedman, J.; Nottingham, L.; Chen, Z.; Ara, G.; Van Waes, C. Nuclear factor-kappaB p65 small interfering RNA or proteasome inhibitor bortezomib sensitizes head and neck squamous cell carcinomas to classic histone deacetylase inhibitors and novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 2007, 6, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Anh, T.D.; Ahn, M.Y.; Kim, S.A.; Yoon, J.H.; Ahn, S.G. The histone deacetylase inhibitor, Trichostatin A, induces G2/M phase arrest and apoptosis in YD-10B oral squamous carcinoma cells. Oncol. Rep. 2012, 27, 455–460. [Google Scholar] [CrossRef]
- Jang, B.; Kim, L.H.; Lee, S.Y.; Lee, K.E.; Shin, J.A.; Cho, S.D. Trichostatin A induces apoptosis in oral squamous cell carcinoma cell lines independent of hyperacetylation of histones. J. Cancer Res. Ther. 2018, 14, S576–S582. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Gao, L.; Shay, C.; Lang, L.; Lv, F.; Teng, Y. Histone deacetylase inhibitors suppress aggressiveness of head and neck squamous cell carcinoma via histone acetylation-independent blockade of the EGFR-Arf1 axis. J. Exp. Clin. Cancer Res. 2019, 38, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakiuchi, A.; Kakuki, T.; Ohwada, K.; Kurose, M.; Kondoh, A.; Obata, K.; Nomura, K.; Miyata, R.; Kaneko, Y.; Konno, T.; et al. HDAC inhibitors suppress the proliferation, migration and invasiveness of human head and neck squamous cell carcinoma cells via p63-mediated tight junction molecules and p21-mediated growth arrest. Oncol. Rep. 2021, 45, 46. [Google Scholar] [CrossRef] [PubMed]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; De Alava, E.; Hajji, N.; Garcia-Dominguez, D.J. Synergistic enhancement of cancer therapy using HDAC inhibitors: Opportunity for clinical trials. Front. Genet. 2020, 11, 578011. [Google Scholar] [CrossRef]
- Cameron, E.E.; Bachman, K.E.; Myohanen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef]
- Zhu, W.G.; Lakshmanan, R.R.; Beal, M.D.; Otterson, G.A. DNA methyltransferase inhibition enhances apoptosis induced by histone deacetylase inhibitors. Cancer Res. 2001, 61, 1327–1333. [Google Scholar]
- Chai, G.; Li, L.; Zhou, W.; Wu, L.; Zhao, Y.; Wang, D.; Lu, S.; Yu, Y.; Wang, H.; McNutt, M.A.; et al. HDAC inhibitors act with 5-aza-2′-deoxycytidine to inhibit cell proliferation by suppressing removal of incorporated abases in lung cancer cells. PLoS ONE 2008, 3, e2445. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.Y.; Liao, W.S.; Lu, Z.; Bornmann, W.G.; Hennessey, V.; Washington, M.N.; Rosner, G.L.; Yu, Y.; Ahmed, A.A.; Bast, R.C., Jr. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011, 117, 4424–4438. [Google Scholar] [CrossRef] [Green Version]
- Ahrens, T.D.; Timme, S.; Hoeppner, J.; Ostendorp, J.; Hembach, S.; Follo, M.; Hopt, U.T.; Werner, M.; Busch, H.; Boerries, M.; et al. Selective inhibition of esophageal cancer cells by combination of HDAC inhibitors and Azacytidine. Epigenetics 2015, 10, 431–445. [Google Scholar] [CrossRef] [Green Version]
- Abou Najem, S.; Khawaja, G.; Hodroj, M.H.; Babikian, P.; Rizk, S. Adjuvant epigenetic therapy of decitabine and suberoylanilide hydroxamic acid exerts anti-neoplastic effects in acute myeloid leukemia cells. Cells 2019, 8, 1480. [Google Scholar] [CrossRef] [Green Version]
- De Schutter, H.; Kimpe, M.; Isebaert, S.; Nuyts, S. A systematic assessment of radiation dose enhancement by 5-Aza-2′-deoxycytidine and histone deacetylase inhibitors in head-and-neck squamous cell carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 904–912. [Google Scholar] [CrossRef]
- Cao, W.; Zhang, Z.Y.; Xu, Q.; Sun, Q.; Yan, M.; Zhang, J.; Zhang, P.; Han, Z.G.; Chen, W.T. Epigenetic silencing of MAL, a putative tumor suppressor gene, can contribute to human epithelium cell carcinoma. Mol. Cancer 2010, 9, 296. [Google Scholar] [CrossRef] [Green Version]
- Hodjat, M.; Jourshari, P.B.; Amirinia, F.; Asadi, N. 5-Azacitidine and Trichostatin A induce DNA damage and apoptotic responses in tongue squamous cell carcinoma: An in vitro study. Arch. Oral Biol. 2021, 133, 105296. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Linnekamp, J.F.; Butter, R.; Spijker, R.; Medema, J.P.; Van Laarhoven, H.W.M. Clinical and biological effects of demethylating agents on solid tumours—A systematic review. Cancer Treat. Rev. 2017, 54, 10–23. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, Z.; Zhu, W.G. Molecular mechanisms of epigenetic regulators as activatable targets in cancer theranostics. Curr. Med. Chem. 2019, 26, 1328–1350. [Google Scholar] [CrossRef]
- Kamata, N.; Chida, K.; Rikimaru, K.; Horikoshi, M.; Enomoto, S.; Kuroki, T. Growth-inhibitory effects of epidermal growth factor and overexpression of its receptors on human squamous cell carcinomas in culture. Cancer Res. 1986, 46, 1648–1653. [Google Scholar]
- Momose, F.; Araida, T.; Negishi, A.; Ichijo, H.; Shioda, S.; Sasaki, S. Variant sublines with different metastatic potentials selected in nude mice from human oral squamous cell carcinomas. J. Oral Pathol. Med. 1989, 18, 391–395. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar]
- Shi, Y. Caspase activation, inhibition, and reactivation: A mechanistic view. Protein Sci. 2004, 13, 1979–1987. [Google Scholar] [CrossRef] [Green Version]
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef]
- Georgoulis, A.; Vorgias, C.E.; Chrousos, G.P.; Rogakou, E.P. Genome instability and γH2AX. Int. J. Mol. Sci. 2017, 18, 1979. [Google Scholar] [CrossRef]
- Luszczek, W.; Cheriyath, V.; Mekhail, T.M.; Borden, E.C. Combinations of DNA methyltransferase and histone deacetylase inhibitors induce DNA damage in small cell lung cancer cells: Correlation of resistance with IFN-stimulated gene expression. Mol. Cancer Ther. 2010, 9, 2309–2321. [Google Scholar] [CrossRef] [Green Version]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol. Cancer Ther. 2013, 12, 2078–2087. [Google Scholar] [CrossRef] [Green Version]
- Sampath, D.; Rao, V.A.; Plunkett, W. Mechanisms of apoptosis induction by nucleoside analogs. Oncogene 2003, 22, 9063–9074. [Google Scholar] [CrossRef] [Green Version]
- Ewald, B.; Sampath, D.; Plunkett, W. Nucleoside analogs: Molecular mechanisms signaling cell death. Oncogene 2008, 27, 6522–6537. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Tan, J.; Li, J.; Kivimae, S.; Yang, X.; Zhuang, L.; Lee, P.L.; Chan, M.T.; Stanton, L.W.; Liu, E.T.; et al. DACT3 is an epigenetic regulator of Wnt/beta-catenin signaling in colorectal cancer and is a therapeutic target of histone modifications. Cancer Cell 2008, 13, 529–541. [Google Scholar] [CrossRef] [Green Version]
- Hayden, A.; Johnson, P.W.; Packham, G.; Crabb, S.J. S-adenosylhomocysteine hydrolase inhibition by 3-deazaneplanocin A analogues induces anti-cancer effects in breast cancer cell lines and synergy with both histone deacetylase and HER2 inhibition. Breast Cancer Res. Treat. 2011, 127, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Valiuliene, G.; Stirblyte, I.; Jasnauskaite, M.; Borutinskaite, V.; Navakauskiene, R. Anti-leukemic effects of HDACi Belinostat and HMTi 3-Deazaneplanocin A on human acute promyelocytic leukemia cells. Eur. J. Pharmacol. 2017, 799, 143–153. [Google Scholar] [CrossRef]
- Savickiene, J.; Treigyte, G.; Borutinskaite, V.V.; Navakauskiene, R. Antileukemic activity of combined epigenetic agents, DNMT inhibitors zebularine and RG108 with HDAC inhibitors, against promyelocytic leukemia HL-60 cells. Cell Mol. Biol. Lett. 2012, 17, 501–525. [Google Scholar] [CrossRef]
- Yang, L.; Hou, J.; Cui, X.H.; Suo, L.N.; Lv, Y.W. RG108 induces the apoptosis of endometrial cancer Ishikawa cell lines by inhibiting the expression of DNMT3B and demethylation of HMLH1. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5056–5064. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Zhang, Q.; Tang, Y.; Lu, Z.; Lu, X.; Zhou, X.; Liu, C. DNA methylation enzyme inhibitor RG108 suppresses the radioresistance of esophageal cancer. Oncol. Rep. 2018, 39, 993–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.S.; Kim, M.J. Expression of multidrug resistance-related genes in oral squamous cell carcinomas. Oral Oncol. 2001, 37, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Hiroi, M.; Ohmori, Y. Constitutive nuclear factor kappaB activity is required to elicit interferon-gamma-induced expression of chemokine CXC ligand 9 (CXCL9) and CXCL10 in human tumour cell lines. Biochem. J. 2003, 376, 393–402. [Google Scholar]
- Almeida, L.O.; Abrahao, A.C.; Rosselli-Murai, L.K.; Giudice, F.S.; Zagni, C.; Leopoldino, A.M.; Squarize, C.H.; Castilho, R.M. NFkappaB mediates cisplatin resistance through histone modifications in head and neck squamous cell carcinoma (HNSCC). FEBS Open Bio 2014, 4, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Sethi, G. Targeting transcription factor NF-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim. Biophys. Acta 2010, 1805, 167–180. [Google Scholar] [CrossRef]
- Lindemann, A.; Takahashi, H.; Patel, A.A.; Osman, A.A.; Myers, J.N. Targeting the DNA damage response in OSCC with TP53 mutations. J. Dent. Res. 2018, 97, 635–644. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Sakai, E.; Tsuchida, N. Most human squamous cell carcinomas in the oral cavity contain mutated p53 tumor-suppressor genes. Oncogene 1992, 7, 927–933. [Google Scholar]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in cancer: Accumulation, gain-of-function, and therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef]
- Yamamoto, S.; Iwakuma, T. Regulators of oncogenic mutant TP53 gain of function. Cancers 2018, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Udaka, K.; Yokoyama, A. Simultaneous inhibition of DNA methyltransferase and histone deacetylase induces p53-independent apoptosis via down-regulation of Mcl-1 in acute myelogenous leukemia cells. Leuk. Res. 2011, 35, 932–939. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [Green Version]
- Jensen, C.; Teng, Y. Is it time to start transitioning from 2D to 3D cell culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Chitturi Suryaprakash, R.T.; Kujan, O.; Shearston, K.; Farah, C.S. Three-dimensional cell culture models to investigate oral carcinogenesis: A scoping review. Int. J. Mol. Sci. 2020, 21, 9520. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, X. Study and analysis of antitumor resistance mechanism of PD1/PD-L1 immune checkpoint blocker. Cancer Med. 2020, 9, 8086–8121. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef]
- Mazzone, R.; Zwergel, C.; Mai, A.; Valente, S. Epi-drugs in combination with immunotherapy: A new avenue to improve anticancer efficacy. Clin. Epigenetics 2017, 9, 59. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ushio, R.; Hiroi, M.; Matsumoto, A.; Mori, K.; Yamamoto, N.; Ohmori, Y. Enhanced Cytotoxic Effects in Human Oral Squamous Cell Carcinoma Cells Treated with Combined Methyltransferase Inhibitors and Histone Deacetylase Inhibitors. Biomedicines 2022, 10, 763. https://doi.org/10.3390/biomedicines10040763
Ushio R, Hiroi M, Matsumoto A, Mori K, Yamamoto N, Ohmori Y. Enhanced Cytotoxic Effects in Human Oral Squamous Cell Carcinoma Cells Treated with Combined Methyltransferase Inhibitors and Histone Deacetylase Inhibitors. Biomedicines. 2022; 10(4):763. https://doi.org/10.3390/biomedicines10040763
Chicago/Turabian StyleUshio, Ryosuke, Miki Hiroi, Ari Matsumoto, Kazumasa Mori, Nobuharu Yamamoto, and Yoshihiro Ohmori. 2022. "Enhanced Cytotoxic Effects in Human Oral Squamous Cell Carcinoma Cells Treated with Combined Methyltransferase Inhibitors and Histone Deacetylase Inhibitors" Biomedicines 10, no. 4: 763. https://doi.org/10.3390/biomedicines10040763
APA StyleUshio, R., Hiroi, M., Matsumoto, A., Mori, K., Yamamoto, N., & Ohmori, Y. (2022). Enhanced Cytotoxic Effects in Human Oral Squamous Cell Carcinoma Cells Treated with Combined Methyltransferase Inhibitors and Histone Deacetylase Inhibitors. Biomedicines, 10(4), 763. https://doi.org/10.3390/biomedicines10040763