
Cutting-Edge Therapies and Novel Strategies for Acute Intermittent Porphyria: Step-by-Step towards the Solution



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Link between Penetrance, Prevalence, and Genetic Traits in AIP
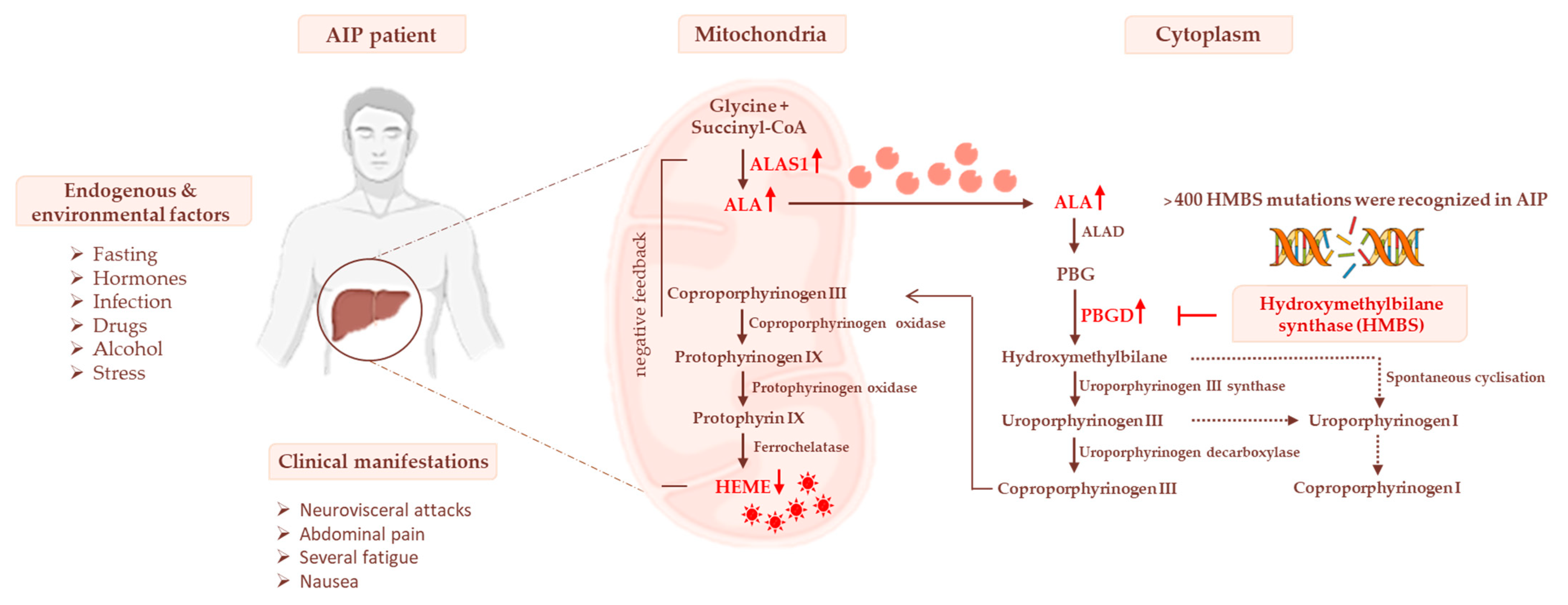
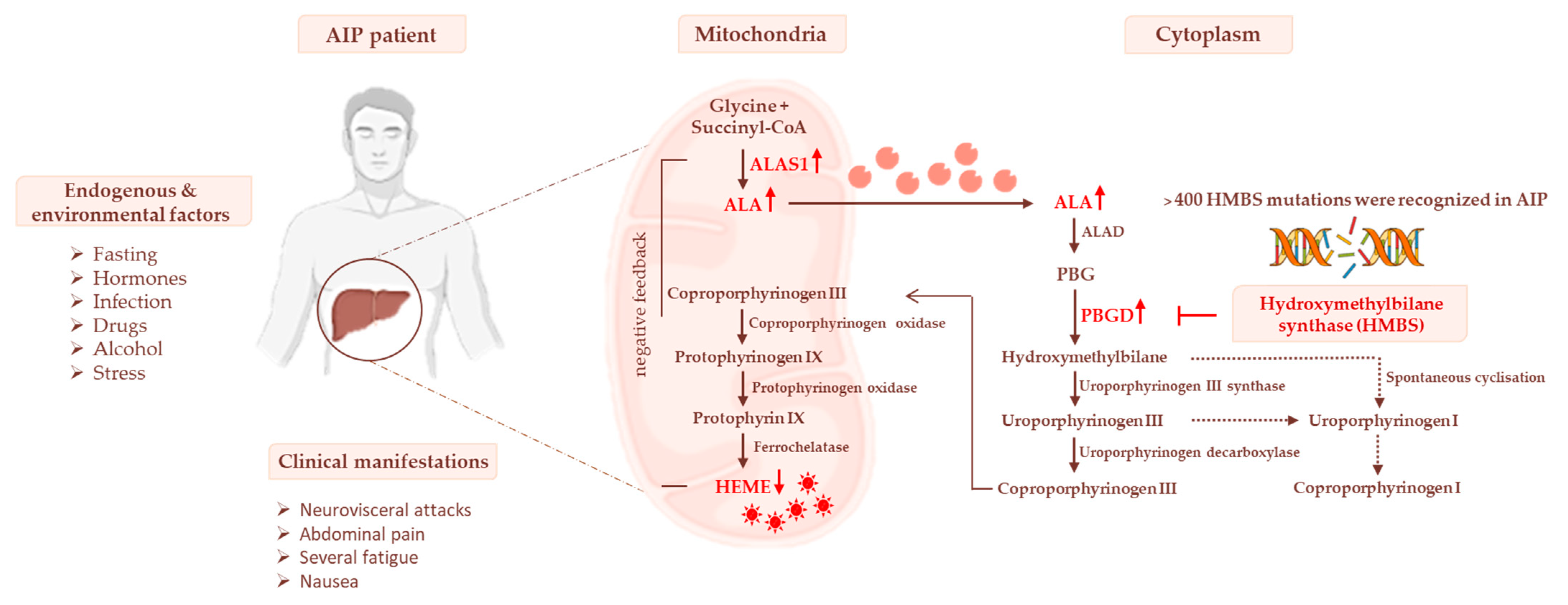
3. Heme Biosynthesis and AIP Pathogenesis
3.1. Impaired Glucose Homeostasis and IR Contribute to Acute Attacks of AIP
3.2. The Occurrence of Mitochondrial Failure in AIP Pathophysiology
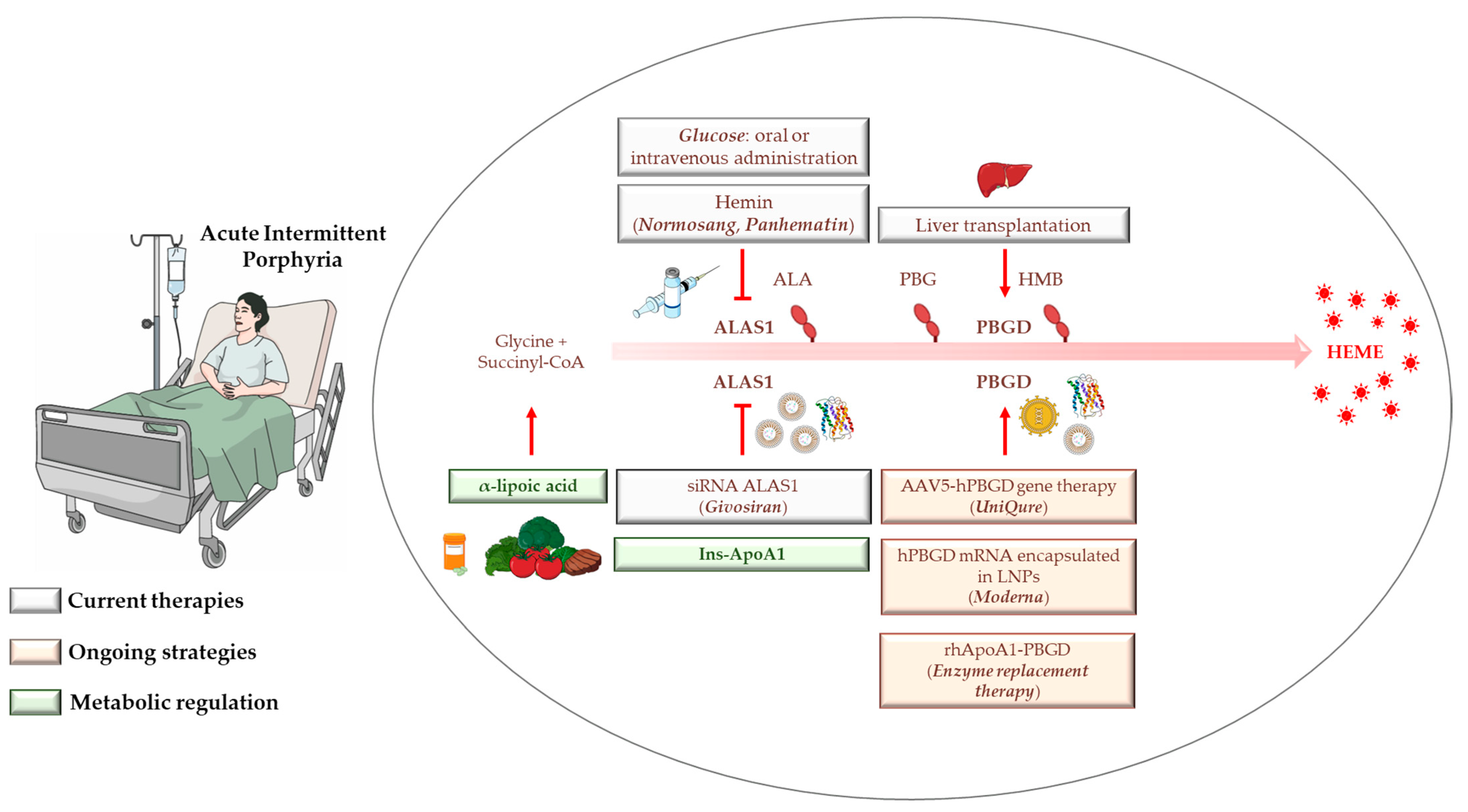
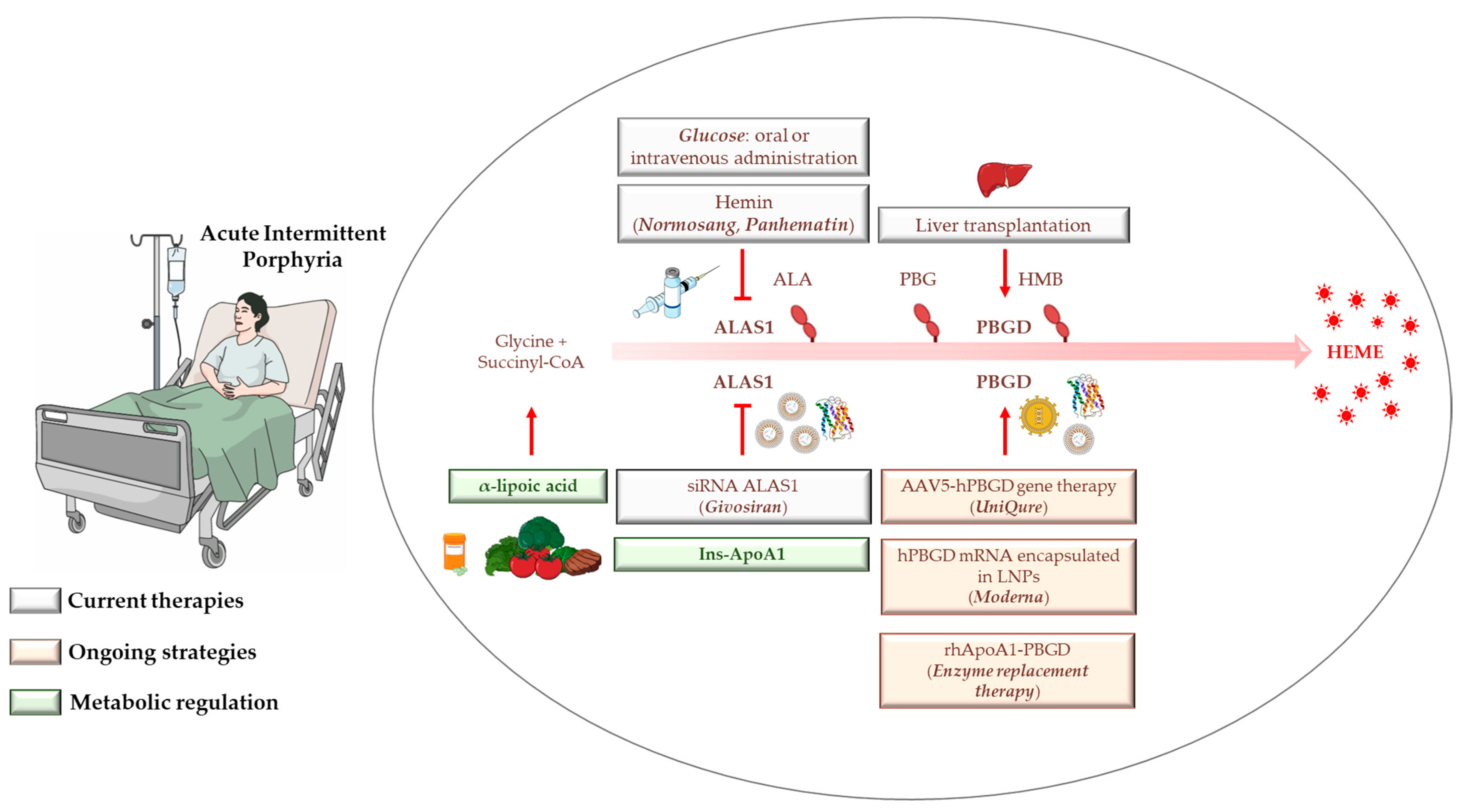
4. AIP Management, Diagnosis, and Standard Treatments
4.1. Hemin
4.2. Glucose
4.3. Liver Transplantation
4.4. Givosiran: From Preclinical Findings until Now
5. Gene Therapy, mRNA-Based Therapies, and Apolipoprotein for Protein/Enzyme deLIVERing: On the Verge of a Scientific Revolution
6. Insulin and Insulin-Mimetics: Possible Alternative Strategies for Hereditary Metabolic Disorders?
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Balwani, M.; Wang, B.; Anderson, K.E.; Bloomer, J.R.; Bissell, D.M.; Bonkovsky, H.L.; Phillips, J.D.; Desnick, R.J. Acute hepatic porphyrias: Recommendations for evaluation and long-term management. Hepatology 2017, 66, 1314–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, P.E.; Badminton, M.N.; Rees, D.C. Update review of the acute porphyrias. Br. J. Haematol. 2017, 176, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.D. Heme biosynthesis and the porphyrias. Mol. Genet. Metab. 2019, 128, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Bonkovsky, H.L.; Maddukuri, V.C.; Yazici, C.; Anderson, K.E.; Bissell, D.M.; Bloomer, J.R.; Phillips, J.D.; Naik, H.; Peter, I.; Baillargeon, G.; et al. Acute porphyrias in the USA: Features of 108 subjects from porphyrias consortium. Am. J. Med. 2014, 127, 1233–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauppinen, R. Porphyrias. Lancet 2005, 365, 241–252. [Google Scholar] [CrossRef]
- Puy, H.; Gouya, L.; Deybach, J.C. Porphyrias. Lancet 2010, 375, 924–937. [Google Scholar] [CrossRef]
- Frei, P.; Minder, E.I.; Corti, N.; Muellhaupt, B.; Geier, A.; Adams, H.; Dutertre, J.P.; Rudiger, A.; Dutkowski, P.; Maggiorini, M.; et al. Liver Transplantation because of Acute Liver Failure due to Heme Arginate Overdose in a Patient with Acute Intermittent Porphyria. Case Rep. Gastroenterol. 2012, 6, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Soonawalla, Z.F.; Orug, T.; Badminton, M.N.; Elder, G.H.; Rhodes, J.M.; Bramhall, S.R.; Elias, E. Liver transplantation as a cure for acute intermittent porphyria. Lancet 2004, 363, 705–706. [Google Scholar] [CrossRef]
- Stein, P.; Badminton, M.; Barth, J.; Rees, D.; Stewart, M.F. Best practice guidelines on clinical management of acute attacks of porphyria and their complications. Ann. Clin. Biochem. 2013, 50, 217–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singal, A.K.; Parker, C.; Bowden, C.; Thapar, M.; Liu, L.; McGuire, B.M. Liver transplantation in the management of porphyria. Hepatology 2014, 60, 1082–1089. [Google Scholar] [CrossRef] [Green Version]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Honor, A.; Rudnick, S.R.; Bonkovsky, H.L. Givosiran to treat acute porphyria. Drugs Today 2021, 57, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Fontanellas, A.; Ávila, M.A.; Anderson, K.E.; Deybach, J.C. Current and innovative emerging therapies for porphyrias with hepatic involvement. J. Hepatol. 2019, 71, 422–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontanellas, A.; Ávila, M.A.; Berraondo, P. Emerging therapies for acute intermittent porphyria. Expert Rev. Mol. Med. 2016, 18, e17. [Google Scholar] [CrossRef] [PubMed]
- Córdoba, K.M.; Serrano-Mendioroz, I. Recombinant porphobilinogen deaminase targeted to the liver corrects enzymopenia in a mouse model of acute intermittent porphyria. Sci. Transl. Med. 2022, 14, eabc0700. [Google Scholar] [CrossRef] [PubMed]
- Solares, I.; Izquierdo-Sánchez, L.; Morales-Conejo, M.; Jericó, D.; Castelbón, F.J.; Córdoba, K.M.; Sampedro, A.; Lumbreras, C.; Moreno-Aliaga, M.J.; Enríquez de Salamanca, R.; et al. High Prevalence of Insulin Resistance in Asymptomatic Patients with Acute Intermittent Porphyria and Liver-Targeted Insulin as a Novel Therapeutic Approach. Biomedicines 2021, 9, 255. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Paolini, E.; Meroni, M.; Duca, L.; Motta, I.; Fracanzani, A.L.; Di Pierro, E.; Dongiovanni, P. α-Lipoic Acid Improves Hepatic Metabolic Dysfunctions in Acute Intermittent Porphyria: A Proof-of-Concept Study. Diagnostics 2021, 11, 1628. [Google Scholar] [CrossRef]
- Vilas, G.L.; Aldonatti, C.; San Martín de Viale, L.C.; Ríos de Molina, M.C. Effect of alpha lipoic acid amide on hexachlorobenzene porphyria. Biochem. Mol. Biol. Int. 1999, 47, 815–823. [Google Scholar] [CrossRef]
- Yasuda, M.; Chen, B.; Desnick, R.J. Recent advances on porphyria genetics: Inheritance, penetrance & molecular heterogeneity, including new modifying/causative genes. Mol. Genet. Metab. 2019, 128, 320–331. [Google Scholar]
- Chen, C.H.; Astrin, K.H.; Lee, G.; Anderson, K.E.; Desnick, R.J. Acute intermittent porphyria: Identification and expression of exonic mutations in the hydroxymethylbilane synthase gene. An initiation codon missense mutation in the housekeeping transcript causes “variant acute intermittent porphyria” with normal expression of the erythroid-specific enzyme. J. Clin. Investig. 1994, 94, 1927–1937. [Google Scholar] [CrossRef]
- Brancaleoni, V.; Granata, F.; Colancecco, A.; Tavazzi, D.; Cappellini, M.D.; Di Pierro, E. Seven novel genetic mutations within the 5’UTR and the housekeeping promoter of HMBS gene responsible for the non-erythroid form of acute intermittent porphyria. Blood Cells Mol. Dis. 2012, 49, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N.; Youssoufian, H. The CpG dinucleotide and human genetic disease. Hum. Genet. 1988, 78, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Petersen, N.E.; Nissen, H.; Hørder, M.; Senz, J.; Jamani, A.; Schreiber, W.E. Mutation screening by denaturing gradient gel electrophoresis in North American patients with acute intermittent porphyria. Clin. Chem. 1998, 44 Pt 1, 1766–1768. [Google Scholar] [CrossRef] [PubMed]
- De Siervi, A.; Rossetti, M.V.; Parera, V.E.; Astrin, K.H.; Aizencang, G.I.; Glass, I.A.; Batlle, A.M.; Desnick, R.J. Identification and characterization of hydroxymethylbilane synthase mutations causing acute intermittent porphyria: Evidence for an ancestral founder of the common G111R mutation. Am. J. Med. Genet. 1999, 86, 366–375. [Google Scholar] [CrossRef]
- Ma, L.; Tian, Y.; Peng, C.; Zhang, Y.; Zhang, S. Recent advances in the epidemiology and genetics of acute intermittent porphyria. Intractable Rare Dis. Res. 2020, 9, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, H.; Schmitt, C.; Grange, T.; Manceau, H.; Karboul, N.; Bouchet-Crivat, F.; Robreau, A.M.; Nicolas, G.; Lamoril, J.; Simonin, S.; et al. From a dominant to an oligogenic model of inheritance with environmental modifiers in acute intermittent porphyria. Hum. Mol. Genet. 2018, 27, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.; Harper, P.; Badminton, M.; Sandberg, S.; Deybach, J.C. The incidence of inherited porphyrias in Europe. J. Inherit. Metab. Dis. 2013, 36, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Baumann, K.; Kauppinen, R. Penetrance and predictive value of genetic screening in acute porphyria. Mol. Genet. Metab. 2020, 130, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Andersson, C.; Floderus, Y.; Wikberg, A.; Lithner, F. The W198X and R173W mutations in the porphobilinogen deaminase gene in acute intermittent porphyria have higher clinical penetrance than R167W. A population-based study. Scand. J. Clin. Lab. Investig. 2000, 60, 643–648. [Google Scholar] [CrossRef]
- Fu, Y.; Jia, J.; Yue, L.; Yang, R.; Guo, Y.; Ni, X.; Shi, T. Systematically Analyzing the Pathogenic Variations for Acute Intermittent Porphyria. Front. Pharmacol. 2019, 10, 1018. [Google Scholar] [CrossRef] [Green Version]
- Szlendak, U.; Bykowska, K.; Lipniacka, A. Clinical, Biochemical and Molecular Characteristics of the Main Types of Porphyria. Adv. Clin. Exp. Med. 2016, 25, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, M.J.; Cantrill, R.C. Delta-Aminolaevulinic acid and amino acid neurotransmitters. Mol. Cell. Biochem. 1981, 38 Pt 1, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Laafi, J.; Homedan, C.; Jacques, C.; Gueguen, N.; Schmitt, C.; Puy, H.; Reynier, P.; Carmen Martinez, M.; Malthiery, Y. Pro-oxidant effect of ALA is implicated in mitochondrial dysfunction of HepG2 cells. Biochimie 2014, 106, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Lane, D.J.; Richardson, D.R. Mitochondrial mayhem: The mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid. Redox Signal. 2011, 15, 3003–3019. [Google Scholar] [CrossRef] [PubMed]
- Onuki, J.; Chen, Y.; Teixeira, P.C.; Schumacher, R.I.; Medeiros, M.H.; Van Houten, B.; Di Mascio, P. Mitochondrial and nuclear DNA damage induced by 5-aminolevulinic acid. Arch. Biochem. Biophys. 2004, 432, 178–187. [Google Scholar] [CrossRef]
- Dowman, J.K.; Gunson, B.K.; Mirza, D.F.; Bramhall, S.R.; Badminton, M.N.; Newsome, P.N.; on behalf of the UK Liver Selection and Allocation Working Party. Liver transplantation for acute intermittent porphyria is complicated by a high rate of hepatic artery thrombosis. Liver Transplant. 2012, 18, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Dowman, J.K.; Gunson, B.K.; Bramhall, S.; Badminton, M.N.; Newsome, P.N. Liver transplantation from donors with acute intermittent porphyria. Ann. Intern. Med. 2011, 154, 571–572. [Google Scholar] [CrossRef]
- Andersson, C.; Bylesjö, I.; Lithner, F. Effects of diabetes mellitus on patients with acute intermittent porphyria. J. Intern. Med. 1999, 245, 193–197. [Google Scholar] [CrossRef]
- Lin, C.-N.; Shiao, M.-S.; Cheng, M.-L.; Chen, C.-M.; Kuo, H.-C. Profiling of Serum Metabolites of Acute Intermittent Porphyria and Asymptomatic HMBS Mutation Carriers. Cells 2021, 10, 2579. [Google Scholar] [CrossRef] [PubMed]
- García-Diz, L.; Murcia, M.A.; Gris, J.L.; Pons, A.; Monteagudo, C.; Martínez-Tomé, M.; Jiménez-Monreal, A.M. Assessing nutritional status of acute intermittent porphyria patients. Eur. J. Clin. Investig. 2012, 42, 943–952. [Google Scholar] [CrossRef]
- Collantes, M.; Serrano-Mendioroz, I.; Benito, M.; Molinet-Dronda, F.; Delgado, M.; Vinaixa, M.; Sampedro, A.; Enriquez de Salamanca, R.; Prieto, E.; Pozo, M.A.; et al. Glucose metabolism during fasting is altered in experimental porphobilinogen deaminase deficiency. Hum. Mol. Genet. 2016, 25, 1318–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrick, A.L.; Fisher, B.M.; Moore, M.R.; Cathcart, S.; McColl, K.E.L.; Goldberg, A. Elevation of blood lactate and pyruvate levels in acute intermittent porphyria—A reflection of haem deficiency? Clin. Chim. Acta 1990, 190, 157–162. [Google Scholar] [CrossRef]
- Matkovic, L.B.; D’Andrea, F.; Fornes, D.; San Martín de Viale, L.C.; Mazzetti, M.B. How porphyrinogenic drugs modeling acute porphyria impair the hormonal status that regulates glucose metabolism. Their relevance in the onset of this disease. Toxicology 2011, 290, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochemist. Rev. 2005, 26, 19–39. [Google Scholar]
- Lithner, F. Beneficial Effect of Diabetes on Acute Intermittent Porphyria. Diabetes Care 2002, 25, 797–798. [Google Scholar] [CrossRef] [Green Version]
- Waxman, A.D.; Berk, P.D.; Schalch, D.O.N.; Tschudy, D.P. Isolated Adrenocorticotrophic Hormone Deficiency in Acute Intermittent Porphyria. Ann. Intern. Med. 1969, 70, 317–323. [Google Scholar] [CrossRef]
- Oliveri, L.M.; Davio, C.; Batlle, A.M.; Gerez, E.N. ALAS1 gene expression is down-regulated by Akt-mediated phosphorylation and nuclear exclusion of FOXO1 by vanadate in diabetic mice. Biochem. J. 2012, 442, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Luck, M.; Schmitt, C.; Talbi, N.; Gouya, L.; Caradeuc, C.; Puy, H.; Bertho, G.; Pallet, N. Urinary metabolic profiling of asymptomatic acute intermittent porphyria using a rule-mining-based algorithm. Metabolomics 2018, 14, 10. [Google Scholar] [CrossRef] [Green Version]
- Thunell, S. Porphyrins, porphyrin metabolism and porphyrias. I. Update. Scand. J. Clin. Lab. Investig. 2000, 60, 509–540. [Google Scholar] [CrossRef]
- Bishop, D.F.; Tchaikovskii, V.; Hoffbrand, A.V.; Fraser, M.E.; Margolis, S. X-linked sideroblastic anemia due to carboxyl-terminal ALAS2 mutations that cause loss of binding to the β-subunit of succinyl-CoA synthetase (SUCLA2). J. Biol. Chem. 2012, 287, 28943–28955. [Google Scholar] [CrossRef] [Green Version]
- Balwani, M.; Desnick, R.J. The porphyrias: Advances in diagnosis and treatment. Blood 2012, 120, 4496–4504. [Google Scholar] [CrossRef] [PubMed]
- Homedan, C.; Laafi, J.; Schmitt, C.; Gueguen, N.; Lefebvre, T.; Karim, Z.; Desquiret-Dumas, V.; Wetterwald, C.; Deybach, J.C.; Gouya, L.; et al. Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model. Int. J. Biochem. Cell Biol. 2014, 51, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homedan, C.; Schmitt, C.; Laafi, J.; Gueguen, N.; Desquiret-Dumas, V.; Lenglet, H.; Karim, Z.; Gouya, L.; Deybach, J.-C.; Simard, G.; et al. Mitochondrial energetic defects in muscle and brain of a Hmbs−/− mouse model of acute intermittent porphyria. Hum. Mol. Genet. 2015, 24, 5015–5023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, S.; Stattmann, M.; Cicvaric, A.; Monje, F.J.; Coiro, P.; Hotka, M.; Ricken, G.; Hainfellner, J.; Greber-Platzer, S.; Yasuda, M.; et al. Severe hydroxymethylbilane synthase deficiency causes depression-like behavior and mitochondrial dysfunction in a mouse model of homozygous dominant acute intermittent porphyria. Acta Neuropathol. Commun. 2020, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.; Curi, R.; Kokubun, E.; Bechara, E.J. 5-aminolevulinic acid-induced alterations of oxidative metabolism in sedentary and exercise-trained rats. J. Appl. Physiol. 1992, 72, 226–230. [Google Scholar] [CrossRef]
- Shetty, T.; Sishtla, K.; Park, B.; Repass, M.J.; Corson, T.W. Heme Synthesis Inhibition Blocks Angiogenesis via Mitochondrial Dysfunction. iScience 2020, 23, 101391. [Google Scholar] [CrossRef]
- Chen, B.; Wang, M.; Gan, L.; Zhang, B.; Desnick, R.J.; Yasuda, M. Characterization of the hepatic transcriptome following phenobarbital induction in mice with AIP. Mol. Genet. Metab. 2019, 128, 382–390. [Google Scholar] [CrossRef]
- Shapiro, S.H.; Wessely, Z.; Klavins, J.V. Hepatocyte mitochondrial alterations in griseofulvin fed mice. Ann. Clin. Lab. Sci. 1980, 10, 33–45. [Google Scholar]
- Le, T.H.; Caldwell, S.H.; Redick, J.A.; Sheppard, B.L.; Davis, C.A.; Arseneau, K.O.; Iezzoni, J.C.; Hespenheide, E.E.; Al-Osaimi, A.; Peterson, T.C. The zonal distribution of megamitochondria with crystalline inclusions in nonalcoholic steatohepatitis. Hepatology 2004, 39, 1423–1429. [Google Scholar] [CrossRef]
- Kashiwagi, R.; Okamoto, K.; Maeda, N.; Horie, Y.; Kawasaki, H.; Osatake, H.; Inoué, T. Crystalline inclusions in hepatocyte mitochondria of a patient with porphyria cutanea tarda. Yonago Acta Med. 1999, 42, 135–140. [Google Scholar]
- Ostrowski, J.; Kostrzewska, E.; Michalak, T.; Zawirska, B.; Medrzejewski, W.; Gregor, A. Abnormalities in liver function and morphology and impaired aminopyrine metabolism in hereditary hepatic porphyrias. Gastroenterology 1983, 85, 1131–1137. [Google Scholar] [CrossRef]
- Linenberger, M.; Fertrin, K.Y. Updates on the diagnosis and management of the most common hereditary porphyrias: AIP and EPP. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Mosquera, L.F.; Sonthalia, S. Acute Intermittent Porphyria. In StatPearls; internet; 2002. [Google Scholar]
- Kauppinen, R.; von und zu Fraunberg, M. Molecular and biochemical studies of acute intermittent porphyria in 196 patients and their families. Clin. Chem. 2002, 48, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Puy, H.; Deybach, J.C.; Lamoril, J.; Robreau, A.M.; Da Silva, V.; Gouya, L.; Grandchamp, B.; Nordmann, Y. Molecular epidemiology and diagnosis of PBG deaminase gene defects in acute intermittent porphyria. Am. J. Hum. Genet. 1997, 60, 1373–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pischik, E.; Kauppinen, R. An update of clinical management of acute intermittent porphyria. Appl. Clin. Genet. 2015, 8, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Berraondo, P.; Martini, P.G.V.; Avila, M.A. Messenger RNA therapy for rare genetic metabolic diseases. Gut 2019, 68, 1323–1330. [Google Scholar] [CrossRef] [Green Version]
- Herrero, C.; Badenas, C.; Aguilera, P.; To-Figueras, J. Acute intermittent porphyria: Long-term follow up of 35 patients. Med. Clin. 2015, 145, 332–337. [Google Scholar] [CrossRef]
- Schmitt, C.; Lenglet, H.; Yu, A.; Delaby, C.; Benecke, A.; Lefebvre, T.; Letteron, P.; Paradis, V.; Wahlin, S.; Sandberg, S.; et al. Recurrent attacks of acute hepatic porphyria: Major role of the chronic inflammatory response in the liver. J. Intern. Med. 2018, 284, 78–91. [Google Scholar] [CrossRef] [Green Version]
- Kuo, H.-C.; Lin, C.-N.; Tang, Y.-F. Prophylactic Heme Arginate Infusion for Acute Intermittent Porphyria. Front. Pharmacol. 2021, 12, 712305. [Google Scholar] [CrossRef]
- Willandt, B.; Langendonk, J.G.; Biermann, K.; Meersseman, W.; D’Heygere, F.; George, C.; Verslype, C.; Monbaliu, D.; Cassiman, D. Liver Fibrosis Associated with Iron Accumulation Due to Long-Term Heme-Arginate Treatment in Acute Intermittent Porphyria: A Case Series. JIMD Rep. 2016, 25, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Storjord, E.; Dahl, J.A.; Landsem, A.; Ludviksen, J.K.; Karlsen, M.B.; Karlsen, B.O.; Brekke, O.-L. Lifestyle factors including diet and biochemical biomarkers in acute intermittent porphyria: Results from a case-control study in northern Norway. Mol. Genet. Metab. 2019, 128, 254–270. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.H.; Hift, R.J. Treatment of acute porphyria. Hosp. Med. 2001, 62, 422–425. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, R.; Rao, G.; Stein, P.; Bandmann, O. Porphyria: Often discussed but too often missed. Pract. Neurol. 2018, 18, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, E.; Granata, F. Nutrients and Porphyria: An Intriguing Crosstalk. Int. J. Mol. Sci. 2020, 21, 3462. [Google Scholar] [CrossRef] [PubMed]
- Solares, I.; Tejedor, M.; Jericó, D.; Morales-Conejo, M.; Enríquez de Salamanca, R.; Fontanellas, A.; Tejedor-Jorge, A. Management of hyponatremia associated with acute porphyria-proposal for the use of tolvaptan. Ann. Transl. Med. 2020, 8, 1098. [Google Scholar] [CrossRef] [PubMed]
- Gilles, A.; Vermeersch, S.; Vermeersch, P.; Wolff, F.; Cotton, F.; Tilleux, S.; Cassiman, D. Expert consensus statement on acute hepatic porphyria in Belgium. Acta Clin. Belg. 2021, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lissing, M.; Nowak, G.; Adam, R.; Karam, V.; Boyd, A.; Gouya, L.; Meersseman, W.; Melum, E.; Ołdakowska-Jedynak, U.; Reiter, F.P.; et al. Liver Transplantation for Acute Intermittent Porphyria. Liver Transplant. Acute Intermittent Porphyria 2021, 27, 491–501. [Google Scholar] [CrossRef]
- D’Avola, D.; Gonzalez Aseguinolaza, G. Prospect and progress of gene therapy in acute intermittent porphyria. Expert Opin. Orphan Drugs 2016, 4, 711–717. [Google Scholar] [CrossRef]
- Yasuda, M.; Gan, L.; Chen, B.; Kadirvel, S.; Yu, C.; Phillips, J.D.; New, M.I.; Liebow, A.; Fitzgerald, K.; Querbes, W.; et al. RNAi-mediated silencing of hepatic Alas1 effectively prevents and treats the induced acute attacks in acute intermittent porphyria mice. Proc. Natl. Acad. Sci. USA 2014, 111, 7777–7782. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.; Liebow, A.; Yasuda, M.; Gan, L.; Racie, T.; Maier, M.; Kuchimanchi, S.; Foster, D.; Milstein, S.; Charisse, K.; et al. Preclinical Development of a Subcutaneous ALAS1 RNAi Therapeutic for Treatment of Hepatic Porphyrias Using Circulating RNA Quantification. Mol. Ther. Nucleic Acids 2015, 4, e263. [Google Scholar] [CrossRef]
- Sardh, E.; Harper, P.; Balwani, M.; Stein, P.; Rees, D.; Bissell, D.M.; Desnick, R.; Parker, C.; Phillips, J.; Bonkovsky, H.L.; et al. Phase 1 Trial of an RNA Interference Therapy for Acute Intermittent Porphyria. N. Engl. J. Med. 2019, 380, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.; Rees, D.; Anderson, K.E.; Bonkovsky, D.H.L.; Sardh, E.; Harper, P.; Balwani, M.; Parker, C.; Phillips, J.; Vassilliou, D.; et al. A Phase 1/2 Open Label Extension Study of Givosiran, An Investigational Rnai Therapeutic, in Patients with Acute Intermittent Porphyria. J. Hepatol. 2020, 73, S553. [Google Scholar]
- To-Figueras, J.; Wijngaard, R.; García-Villoria, J.; Aarsand, A.K.; Aguilera, P.; Deulofeu, R.; Brunet, M.; Gómez-Gómez, À.; Pozo, O.J.; Sandberg, S. Dysregulation of homocysteine homeostasis in acute intermittent porphyria patients receiving heme arginate or givosiran. J. Inherit. Metab. Dis. 2021, 44, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Ventura, P.; Corradini, E.; Di Pierro, E.; Marchini, S.; Marcacci, M.; Cuoghi, C.; Buzzetti, E.; Pietrangelo, A. Hyperhomocysteinemia in patients with acute porphyrias: A potentially dangerous metabolic crossroad? Eur. J. Intern. Med. 2020, 79, 101–107. [Google Scholar] [CrossRef]
- Fontanellas, A.; Ávila, M.A.; Arranz, E.; Enríquez de Salamanca, R.; Morales-Conejo, M. Acute intermittent porphyria, givosiran, and homocysteine. J. Inherit. Metab. Dis. 2021, 44, 790–791. [Google Scholar] [CrossRef]
- Unzu, C.; Sampedro, A.; Mauleón, I.; Vanrell, L.; Dubrot, J.; de Salamanca, R.E.; González-Aseguinolaza, G.; Melero, I.; Prieto, J.; Fontanellas, A. Porphobilinogen deaminase over-expression in hepatocytes, but not in erythrocytes, prevents accumulation of toxic porphyrin precursors in a mouse model of acute intermittent porphyria. J. Hepatol. 2010, 52, 417–424. [Google Scholar] [CrossRef]
- Meroni, M.; Dongiovanni, P.; Longo, M.; Carli, F.; Baselli, G.; Rametta, R.; Pelusi, S.; Badiali, S.; Maggioni, M.; Gaggini, M.; et al. Mboat7 down-regulation by hyper-insulinemia induces fat accumulation in hepatocytes. EBioMedicine 2020, 52, 102658. [Google Scholar] [CrossRef] [Green Version]
- Longo, M.; Meroni, M.; Paolini, E.; Erconi, V.; Carli, F.; Fortunato, F.; Ronchi, D.; Piciotti, R.; Sabatini, S.; Macchi, C.; et al. TM6SF2/PNPLA3/MBOAT7 Loss-of-Function Genetic Variants Impact on NAFLD Development and Progression Both in Patients and in In Vitro Models. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 759–788. [Google Scholar] [CrossRef]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef]
- Unzu, C.; Hervás-Stubbs, S.; Sampedro, A.; Mauleón, I.; Mancheño, U.; Alfaro, C.; de Salamanca, R.E.; Benito, A.; Beattie, S.G.; Petry, H.; et al. Transient and intensive pharmacological immunosuppression fails to improve AAV-based liver gene transfer in non-human primates. J. Transl. Med. 2012, 10, 122. [Google Scholar] [CrossRef] [Green Version]
- Unzu, C.; Melero, I.; Hervás-Stubbs, S.; Sampedro, A.; Mancheño, U.; Morales-Kastresana, A.; Serrano-Mendioroz, I.; de Salamanca, R.E.; Benito, A.; Fontanellas, A. Helper-dependent adenovirus achieve more efficient and persistent liver transgene expression in non-human primates under immunosuppression. Gene Ther. 2015, 22, 856–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unzu, C.; Sampedro, A.; Mauleon, I.; Alegre, M.; Beattie, S.G.; de Salamanca, R.E.; Snapper, J.; Twisk, J.; Petry, H.; Gonzalez-Aseguinolaza, G.; et al. Sustained enzymatic correction by rAAV-mediated liver gene therapy protects against induced motor neuropathy in acute porphyria mice. Mol. Ther. 2011, 19, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Pañeda, A.; Lopez-Franco, E.; Kaeppel, C.; Unzu, C.; Gil-Royo, A.G.; D’Avola, D.; Beattie, S.G.; Olagüe, C.; Ferrero, R.; Sampedro, A.; et al. Safety and liver transduction efficacy of rAAV5-cohPBGD in nonhuman primates: A potential therapy for acute intermittent porphyria. Hum. Gene Ther. 2013, 24, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.L.; Shirley, J.L.; Zolotukhin, I.; Kumar, S.R.P.; Sherman, A.; Perrin, G.Q.; Hoffman, B.E.; Srivastava, A.; Basner-Tschakarjan, E.; Wallet, M.A.; et al. Plasmacytoid and conventional dendritic cells cooperate in crosspriming AAV capsid-specific CD8(+) T cells. Blood 2017, 129, 3184–3195. [Google Scholar] [CrossRef] [Green Version]
- Meliani, A.; Boisgerault, F.; Hardet, R.; Marmier, S.; Collaud, F.; Ronzitti, G.; Leborgne, C.; Costa Verdera, H.; Simon Sola, M.; Charles, S.; et al. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat. Commun. 2018, 9, 4098. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Mendioroz, I.; Sampedro, A.; Alegre, M.; Enríquez de Salamanca, R.; Berraondo, P.; Fontanellas, A. An Inducible Promoter Responsive to Different Porphyrinogenic Stimuli Improves Gene Therapy Vectors for Acute Intermittent Porphyria. Hum. Gene Ther. 2018, 29, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Mendioroz, I.; Sampedro, A.; Serna, N.; de Salamanca, R.E.; Sanz-Parra, A.; Corrales, F.; Berraondo, P.; Millet, O.; Fontanellas, A. Bioengineered PBGD variant improves the therapeutic index of gene therapy vectors for acute intermittent porphyria. Hum. Mol. Genet. 2018, 27, 3688–3696. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Berraondo, P. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat. Med. 2018, 24, 1899–1909. [Google Scholar] [CrossRef]
- Jericó, D.; Córdoba, K.M.; Jiang, L.; Schmitt, C.; Morán, M.; Sampedro, A.; Alegre, M.; Collantes, M.; Santamaría, E.; Alegre, E.; et al. mRNA-based therapy in a rabbit model of variegate porphyria offers new insights into the pathogenesis of acute attacks. Mol. Ther.-Nucleic Acids 2021, 25, 207–219. [Google Scholar] [CrossRef]
- Ardaiz, N.; Gomar, C.; Vasquez, M.; Tenesaca, S.; Fernandez-Sendin, M.; Di Trani, C.A.; Belsué, V.; Escalada, J.; Werner, U.; Tennagels, N.; et al. Insulin Fused to Apolipoprotein A-I Reduces Body Weight and Steatosis in DB/DB Mice. Front. Pharmacol. 2021, 11, 591293. [Google Scholar] [CrossRef]
- Székely, E.; Szentmihályi, K.; Tasnádi, G.; Várnai, K.; Blázovics, A. Effect of alpha-lipoic acid on the porphyria cutanea tarda patients with type 2 diabetes mellitus and heavy drinkers. Z. Für Gastroenterol. 2005, 43, 136. [Google Scholar] [CrossRef]
- Székely, E.; Szentmihályi, K.; Bor, M.; Pusztai, A.; Kurucz, T.; Pallai, Z.; Blázovics, A. Secunder prevention with alpha-lipoic acid and vitamin E in porphyria cutanea tarda patients. Z. Gastroenterol. 2008, 46, A103. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Longo, M.; Paolini, E.; Meroni, M.; Dongiovanni, P. Cutting-Edge Therapies and Novel Strategies for Acute Intermittent Porphyria: Step-by-Step towards the Solution. Biomedicines 2022, 10, 648. https://doi.org/10.3390/biomedicines10030648
Longo M, Paolini E, Meroni M, Dongiovanni P. Cutting-Edge Therapies and Novel Strategies for Acute Intermittent Porphyria: Step-by-Step towards the Solution. Biomedicines. 2022; 10(3):648. https://doi.org/10.3390/biomedicines10030648
Chicago/Turabian StyleLongo, Miriam, Erika Paolini, Marica Meroni, and Paola Dongiovanni. 2022. "Cutting-Edge Therapies and Novel Strategies for Acute Intermittent Porphyria: Step-by-Step towards the Solution" Biomedicines 10, no. 3: 648. https://doi.org/10.3390/biomedicines10030648
APA StyleLongo, M., Paolini, E., Meroni, M., & Dongiovanni, P. (2022). Cutting-Edge Therapies and Novel Strategies for Acute Intermittent Porphyria: Step-by-Step towards the Solution. Biomedicines, 10(3), 648. https://doi.org/10.3390/biomedicines10030648