Secretin Receptor as a Target in Gastrointestinal Cancer: Expression Analysis and Ligand Development



Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Buffers, Reagents and Peptides
2.3. Software
2.4. Immunohistochemistry
2.5. Agonist-Induced β-arr2-GFP Translocation
2.6. Evaluation of Antagonistic Function/IDCC Internalization Assay
2.7. Measurement of Intracellular cAMP Concentrations in Transfected Cell Lines
2.8. Radiolabeling of Secretin
2.9. Isolation of SCTR-Bearing Membrane Fractions
2.10. Radioligand Binding Assay
3. Results
3.1. SCTR Expression in Human Tissues
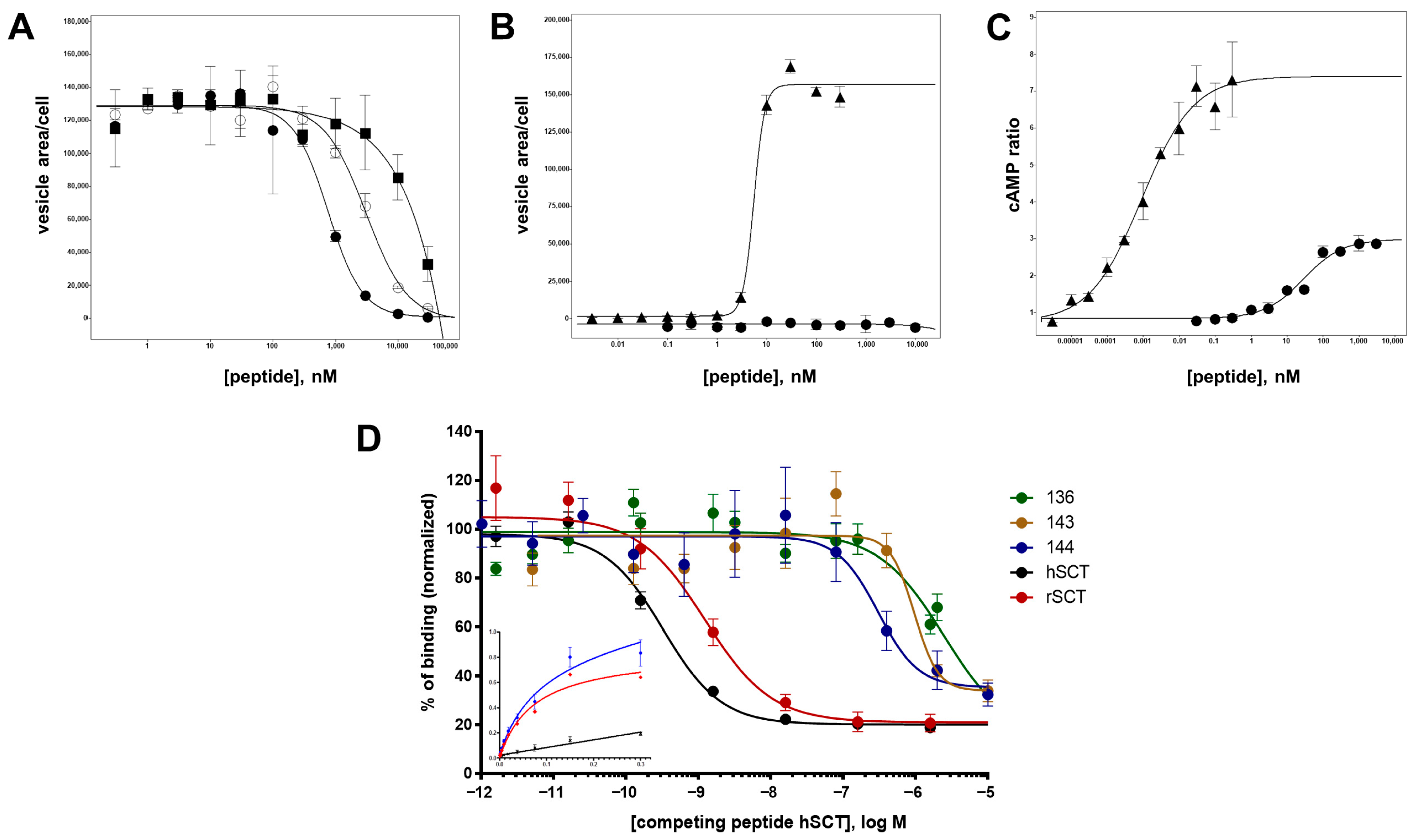
3.2. Assay Development
3.3. Alanine Walk
3.4. Additional Single Substitution Variants
3.5. Truncation Variants
3.6. Characterization of Antagonistic Properties of Secretin Analogs
3.7. Further Characterization of Selected Secretin Variants
3.8. Analysis of Binding Affinity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chey, W.Y.; Escoffery, R. Secretion cells in the gastrointestinal tract. Endocrinology 1976, 98, 1390–1395. [Google Scholar] [CrossRef]
- Bayliss, W.M.; Starling, E.H. The mechanism of pancreatic secretion. J. Physiol. 1902, 28, 325–353. [Google Scholar] [CrossRef]
- Kanno, N.; LeSage, G.; Glaser, S.; Alpini, G. Regulation of cholangiocyte bicarbonate secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G612–G625. [Google Scholar] [CrossRef]
- Jin, H.O.; Lee, K.Y.; Chang, T.M.; Chey, W.Y.; Dubois, A. Secretin: A physiological regulator of gastric emptying and acid output in dogs. Am. J. Physiol. 1994, 267, G702–G708. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Chu, J.Y.; Chow, B.K. Central and peripheral administration of secretin inhibits food intake in mice through the activation of the melanocortin system. Neuropsychopharmacology 2011, 36, 459–471. [Google Scholar] [CrossRef]
- Chu, J.Y.; Chung, S.C.; Lam, A.K.; Tam, S.; Chung, S.K.; Chow, B.K. Phenotypes developed in secretin receptor-null mice indicated a role for secretin in regulating renal water reabsorption. Mol. Cell. Biol. 2007, 27, 2499–2511. [Google Scholar] [CrossRef]
- Chu, J.Y.; Cheng, C.Y.; Lee, V.H.; Chan, Y.S.; Chow, B.K. Secretin and body fluid homeostasis. Kidney Int. 2011, 79, 280–287. [Google Scholar] [CrossRef]
- Lee, V.H.; Lee, L.T.; Chu, J.Y.; Lam, I.P.; Siu, F.K.; Vaudry, H.; Chow, B.K. An indispensable role of secretin in mediating the osmoregulatory functions of angiotensin II. FASEB J. 2010, 24, 5024–5032. [Google Scholar] [CrossRef]
- Zhang, L.; Chow, B.K. The central mechanisms of secretin in regulating multiple behaviors. Front. Endocrinol. 2014, 5, 77. [Google Scholar] [CrossRef]
- Sekar, R.; Chow, B.K. Metabolic effects of secretin. Gen. Comp. Endocrinol. 2013, 181, 18–24. [Google Scholar] [CrossRef]
- Dong, M.; Wang, Y.; Hadac, E.M.; Pinon, D.I.; Holicky, E.; Miller, L.J. Identification of an interaction between residue 6 of the natural peptide ligand and a distinct residue within the amino-terminal tail of the secretin receptor. J. Biol. Chem. 1999, 274, 19161–19167. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.L.; Dong, M.; Miller, L.J. Differential determinants for coupling of distinct G proteins with the class B secretin receptor. Am. J. Physiol. Cell Physiol. 2012, 302, C1202–C1212. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.J.; Dong, M.; Harikumar, K.G.; Gao, F. Structural basis of natural ligand binding and activation of the Class II G-protein-coupled secretin receptor. Biochem. Soc. Trans. 2007, 35, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Bodansky, M.; Natarajan, S.; Gardner, J.D.; Makhlouf, G.M.; Said, S.I. Synthesis and some pharmacological properties of the 23-peptide 15-lysine-secretin-(5–27). Special role of the residue in position 15 in biological activity of the vasoactive intestinal polypeptide. J. Med. Chem. 1978, 21, 1171–1173. [Google Scholar] [CrossRef] [PubMed]
- Laburthe, M.; Couvineau, A.; Gaudin, P.; Maoret, J.J.; Rouyer-Fessard, C.; Nicole, P. Receptors for VIP, PACAP, secretin, GRF, glucagon, GLP-1, and other members of their new family of G protein-linked receptors: Structure-function relationship with special reference to the human VIP-1 receptor. Ann. N. Y. Acad. Sci. 1996, 805, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Te, J.A.; Dong, M.; Miller, L.J.; Bordner, A.J. Predicting the effects of amino acid replacements in peptide hormones on their binding affinities for class B GPCRs and application to the design of secretin receptor antagonists. J. Comput. Aided Mol. Des. 2012, 26, 835–845. [Google Scholar] [CrossRef][Green Version]
- Dong, M.; Pinon, D.I.; Bordner, A.J.; Miller, L.J. Elucidation of the active conformation of the amino terminus of receptor-bound secretin using intramolecular disulfide bond constraints. Bioorg. Med. Chem. Lett. 2010, 20, 6040–6044. [Google Scholar] [CrossRef]
- Dong, M.; Narang, P.; Pinon, D.I.; Bordner, A.J.; Miller, L.J. Refinement of the pharmacophore of an agonist ligand of the secretin receptor using conformationally constrained cyclic hexapeptides. Peptides 2010, 31, 1094–1098. [Google Scholar] [CrossRef][Green Version]
- Dong, M.; Te, J.A.; Xu, X.; Wang, J.; Pinon, D.I.; Storjohann, L.; Bordner, A.J.; Miller, L.J. Lactam constraints provide insights into the receptor-bound conformation of secretin and stabilize a receptor antagonist. Biochemistry 2011, 50, 8181–8192. [Google Scholar] [CrossRef]
- Dong, M.; Le, A.; Te, J.A.; Pinon, D.I.; Bordner, A.J.; Miller, L.J. Importance of each residue within secretin for receptor binding and biological activity. Biochemistry 2011, 50, 2983–2993. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Deganutti, G.; Piper, S.J.; Liang, Y.L.; Khoshouei, M.; Belousoff, M.J.; Harikumar, K.G.; Reynolds, C.A.; Glukhova, A.; Furness, S.G.B.; et al. Structure and dynamics of the active Gs-coupled human secretin receptor. Nat. Commun. 2020, 11, 4137. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, V.; Jornvall, H.; Mutt, V.; Sillard, R. Two alternative processing pathways for a preprohormone: A bioactive form of secretin. Proc. Natl. Acad. Sci. USA 1995, 92, 11985–11989. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.E.; Walsh, J.H.; Bussjaeger, L.; Zong, Y.; Hamilton, J.W.; Ho, F.J.; Lee, T.D.; Reeve, J.R., Jr. COOH-terminally extended secretins are potent stimulants of pancreatic secretion. Am. J. Physiol. 1999, 276, G808–G816. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, P.; De Neef, P.; Waelbroeck, M.; Camus, J.C.; Scemama, J.L.; Fourmy, D.; Pradayrol, L.; Vaysse, N.; Christophe, J. Secretin receptors in human pancreatic membranes. Pancreas 1988, 3, 529–535. [Google Scholar] [CrossRef]
- Haffar, B.M.; Hocart, S.J.; Coy, D.H.; Mantey, S.; Chiang, H.C.; Jensen, R.T. Reduced peptide bond pseudopeptide analogues of secretin. A new class of secretin receptor antagonists. J. Biol. Chem. 1991, 266, 316–322. [Google Scholar] [CrossRef]
- Dong, M.; Harikumar, K.G.; Raval, S.R.; Milburn, J.E.; Clark, C.; Alcala-Torano, R.; Mobarec, J.C.; Reynolds, C.A.; Ghirlanda, G.; Christopoulos, A.; et al. Rational development of a high-affinity secretin receptor antagonist. Biochem. Pharmacol. 2020, 177, 113929. [Google Scholar] [CrossRef]
- Cardoso, J.C.; Pinto, V.C.; Vieira, F.A.; Clark, M.S.; Power, D.M. Evolution of secretin family GPCR members in the metazoa. BMC Evol. Biol. 2006, 6, 108. [Google Scholar] [CrossRef]
- Ishihara, T.; Nakamura, S.; Kaziro, Y.; Takahashi, T.; Takahashi, K.; Nagata, S. Molecular cloning and expression of a cDNA encoding the secretin receptor. EMBO J. 1991, 10, 1635–1641. [Google Scholar] [CrossRef]
- Miller, L.J.; Dong, M.; Harikumar, K.G. Ligand binding and activation of the secretin receptor, a prototypic family B G protein-coupled receptor. Br. J. Pharmacol. 2012, 166, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H.A.; Au, M.; Hay, D.L. The structure of secretin family GPCR peptide ligands: Implications for receptor pharmacology and drug development. Drug Discov. Today 2012, 17, 1006–1014. [Google Scholar] [CrossRef]
- Patel, D.R.; Kong, Y.; Sreedharan, S.P. Molecular cloning and expression of a human secretin receptor. Mol. Pharmacol. 1995, 47, 467–473. [Google Scholar] [PubMed]
- Chow, B.K. Molecular cloning and functional characterization of a human secretin receptor. Biochem. Biophys. Res. Commun. 1995, 212, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Couvineau, A.; Laburthe, M. The family B1 GPCR: Structural aspects and interaction with accessory proteins. Curr. Drug Targets 2012, 13, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Harikumar, K.G.; Pinon, D.I.; Miller, L.J. Transmembrane segment IV contributes a functionally important interface for oligomerization of the Class II G protein-coupled secretin receptor. J. Biol. Chem. 2007, 282, 30363–30372. [Google Scholar] [CrossRef]
- Onori, P.; Wise, C.; Gaudio, E.; Franchitto, A.; Francis, H.; Carpino, G.; Lee, V.; Lam, I.; Miller, T.; Dostal, D.E.; et al. Secretin inhibits cholangiocarcinoma growth via dysregulation of the cAMP-dependent signaling mechanisms of secretin receptor. Int. J. Cancer 2010, 127, 43–54. [Google Scholar] [CrossRef]
- Shetzline, M.A.; Premont, R.T.; Walker, J.K.; Vigna, S.R.; Caron, M.G. A role for receptor kinases in the regulation of class II G protein-coupled receptors. Phosphorylation and desensitization of the secretin receptor. J. Biol. Chem. 1998, 273, 6756–6762. [Google Scholar] [CrossRef]
- Walker, J.K.; Premont, R.T.; Barak, L.S.; Caron, M.G.; Shetzline, M.A. Properties of secretin receptor internalization differ from those of the beta(2)-adrenergic receptor. J. Biol. Chem. 1999, 274, 31515–31523. [Google Scholar] [CrossRef]
- Holtmann, M.H.; Hadac, E.M.; Miller, L.J. Critical contributions of amino-terminal extracellular domains in agonist binding and activation of secretin and vasoactive intestinal polypeptide receptors. Studies of chimeric receptors. J. Biol. Chem. 1995, 270, 14394–14398. [Google Scholar] [CrossRef]
- Izzo, R.S.; Chen, A.I.; Pellecchia, C.; Praissman, M. Secretin internalization and adenosine 3’,5’-monophosphate levels in pancreatic acinar cells. Endocrinology 1989, 124, 2252–2260. [Google Scholar] [CrossRef]
- Ding, W.Q.; Cheng, Z.J.; McElhiney, J.; Kuntz, S.M.; Miller, L.J. Silencing of secretin receptor function by dimerization with a misspliced variant secretin receptor in ductal pancreatic adenocarcinoma. Cancer Res. 2002, 62, 5223–5229. [Google Scholar]
- Harikumar, K.G.; Ball, A.M.; Sexton, P.M.; Miller, L.J. Importance of lipid-exposed residues in transmembrane segment four for family B calcitonin receptor homo-dimerization. Regul. Pept. 2010, 164, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Harikumar, K.G.; Happs, R.M.; Miller, L.J. Dimerization in the absence of higher-order oligomerization of the G protein-coupled secretin receptor. Biochim. Biophys. Acta 2008, 1778, 2555–2563. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; Ulrich, C.D., 2nd; Phillips, J.O.; Pham, L.D.; Miller, L.J.; LaRusso, N.F. Upregulation of secretin receptor gene expression in rat cholangiocytes after bile duct ligation. Am. J. Physiol. 1994, 266, G922–G928. [Google Scholar] [CrossRef] [PubMed]
- Guerrier, M.; Attili, F.; Alpini, G.; Glaser, S. Prolonged administration of secretin to normal rats increases biliary proliferation and secretin-induced ductal secretory activity. Hepatobiliary Surg. Nutr. 2014, 3, 118–125. [Google Scholar] [CrossRef]
- Glaser, S.; Lam, I.P.; Franchitto, A.; Gaudio, E.; Onori, P.; Chow, B.K.; Wise, C.; Kopriva, S.; Venter, J.; White, M.; et al. Knockout of secretin receptor reduces large cholangiocyte hyperplasia in mice with extrahepatic cholestasis induced by bile duct ligation. Hepatology 2010, 52, 204–214. [Google Scholar] [CrossRef]
- Kennedy, L.; Francis, H.; Invernizzi, P.; Venter, J.; Wu, N.; Carbone, M.; Gershwin, M.E.; Bernuzzi, F.; Franchitto, A.; Alvaro, D.; et al. Secretin/secretin receptor signaling mediates biliary damage and liver fibrosis in early-stage primary biliary cholangitis. FASEB J. 2019, 33, 10269–10279. [Google Scholar] [CrossRef]
- Chen, L.; Wu, N.; Kennedy, L.; Francis, H.; Ceci, L.; Zhou, T.; Samala, N.; Kyritsi, K.; Wu, C.; Sybenga, A.; et al. Inhibition of Secretin/Secretin Receptor Axis Ameliorates NAFLD Phenotypes. Hepatology 2021, 74, 1845–1863. [Google Scholar] [CrossRef]
- Ding, W.Q.; Kuntz, S.; Bohmig, M.; Wiedenmann, B.; Miller, L.J. Dominant negative action of an abnormal secretin receptor arising from mRNA missplicing in a gastrinoma. Gastroenterology 2002, 122, 500–511. [Google Scholar] [CrossRef]
- Hayes, G.M.; Carrigan, P.E.; Dong, M.; Reubi, J.C.; Miller, L.J. A novel secretin receptor splice variant potentially useful for early diagnosis of pancreatic carcinoma. Gastroenterology 2007, 133, 853–861. [Google Scholar] [CrossRef]
- Long, S.H.; Berna, M.J.; Thill, M.; Pace, A.; Pradhan, T.K.; Hoffmann, K.M.; Serrano, J.; Jensen, R.T. Secretin-receptor and secretin-receptor-variant expression in gastrinomas: Correlation with clinical and tumoral features and secretin and calcium provocative test results. J. Clin. Endocrinol. Metab. 2007, 92, 4394–4402. [Google Scholar] [CrossRef]
- Korner, M.; Hayes, G.M.; Rehmann, R.; Zimmermann, A.; Friess, H.; Miller, L.J.; Reubi, J.C. Secretin receptors in normal and diseased human pancreas: Marked reduction of receptor binding in ductal neoplasia. Am. J. Pathol. 2005, 167, 959–968. [Google Scholar] [CrossRef]
- Korner, M.; Hayes, G.M.; Rehmann, R.; Zimmermann, A.; Scholz, A.; Wiedenmann, B.; Miller, L.J.; Reubi, J.C. Secretin receptors in the human liver: Expression in biliary tract and cholangiocarcinoma, but not in hepatocytes or hepatocellular carcinoma. J. Hepatol. 2006, 45, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Korner, M.U.; Hayes, G.M.; Carrigan, P.E.; Rehmann, R.; Miller, L.J.; Reubi, J.C. Wild-type and splice-variant secretin receptors in lung cancer: Overexpression in carcinoid tumors and peritumoral lung tissue. Mod. Pathol. 2008, 21, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Buchsbaum, D.J. Experimental tumor targeting with radiolabeled ligands. Cancer 1997, 80, 2371–2377. [Google Scholar] [CrossRef]
- Schlom, J.; Hand, P.H.; Greiner, J.W.; Colcher, D.; Shrivastav, S.; Carrasquillo, J.A.; Reynolds, J.C.; Larson, S.M.; Raubitschek, A. Innovations that influence the pharmacology of monoclonal antibody guided tumor targeting. Cancer Res. 1990, 50, 820s–827s. [Google Scholar] [PubMed]
- Heppeler, A.; Froidevaux, S.; Eberle, A.N.; Maecke, H.R. Receptor targeting for tumor localisation and therapy with radiopeptides. Curr. Med. Chem. 2000, 7, 971–994. [Google Scholar] [CrossRef]
- Lee, S.T.; Kulkarni, H.R.; Singh, A.; Baum, R.P. Theranostics of Neuroendocrine Tumors. Visc. Med. 2017, 33, 358–366. [Google Scholar] [CrossRef]
- Hunter, W.M.; Greenwood, F.C. Preparation of iodine-131 labelled human growth hormone of high specific activity. Nature 1962, 194, 495–496. [Google Scholar] [CrossRef]
- Barak, L.S.; Ferguson, S.S.; Zhang, J.; Caron, M.G. A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J. Biol. Chem. 1997, 272, 27497–27500. [Google Scholar] [CrossRef]
- Oakley, R.H.; Hudson, C.C.; Cruickshank, R.D.; Meyers, D.M.; Payne, R.E., Jr.; Rhem, S.M.; Loomis, C.R. The cellular distribution of fluorescently labeled arrestins provides a robust, sensitive, and universal assay for screening G protein-coupled receptors. Assay Drug Dev. Technol. 2002, 1, 21–30. [Google Scholar] [CrossRef]
- Sikora, K. Personalized medicine for cancer: From molecular signature to therapeutic choice. Adv. Cancer Res. 2007, 96, 345–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, H.Y.; Zheng, Y.Q. Personalized medicine of esophageal cancer. J. Cancer Res. Ther. 2012, 8, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Di Federico, A.; Tateo, V.; Parisi, C.; Formica, F.; Carloni, R.; Frega, G.; Rizzo, A.; Ricci, D.; Di Marco, M.; Palloni, A.; et al. Hacking Pancreatic Cancer: Present and Future of Personalized Medicine. Pharmaceuticals 2021, 14, 677. [Google Scholar] [CrossRef] [PubMed]
- Vaseghi Maghvan, P.; Jeibouei, S.; Akbari, M.E.; Niazi, V.; Karami, F.; Rezvani, A.; Ansarinejad, N.; Abbasinia, M.; Sarvari, G.; Zali, H.; et al. Personalized medicine in colorectal cancer. Gastroenterol. Hepatol. Bed Bench 2020, 13, S18–S28. [Google Scholar] [PubMed]
- Rijpkema, M.; Boerman, O.C.; Oyen, W.J. Tumor Targeting Using Radiolabeled Antibodies for Image-Guided Drug Delivery. Curr. Drug Targets 2015, 16, 625–633. [Google Scholar] [CrossRef]
- Eberle, A.N.; Mild, G.; Froidevaux, S. Receptor-mediated tumor targeting with radiopeptides. Part 1. General concepts and methods: Applications to somatostatin receptor-expressing tumors. J. Recept. Signal Transduct. Res. 2004, 24, 319–455. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review (CSR), 1975–2018; National Cancer Institute: Bethesda, MD, USA, 2021.
- Beermann, S.; Seifert, R.; Neumann, D. Commercially available antibodies against human and murine histamine H(4)-receptor lack specificity. Naunyn Schmiedebergs Arch. Pharmacol. 2012, 385, 125–135. [Google Scholar] [CrossRef]
- Michel, M.C.; Wieland, T.; Tsujimoto, G. How reliable are G-protein-coupled receptor antibodies? Naunyn Schmiedebergs Arch. Pharmacol. 2009, 379, 385–388. [Google Scholar] [CrossRef]
- Jensen, B.C.; Swigart, P.M.; Simpson, P.C. Ten commercial antibodies for alpha-1-adrenergic receptor subtypes are nonspecific. Naunyn Schmiedebergs Arch. Pharmacol. 2009, 379, 409–412. [Google Scholar] [CrossRef]
- Hamdani, N.; van der Velden, J. Lack of specificity of antibodies directed against human beta-adrenergic receptors. Naunyn Schmiedebergs Arch. Pharmacol. 2009, 379, 403–407. [Google Scholar] [CrossRef]
- Tang, C.; Biemond, I.; Offerhaus, G.J.; Verspaget, W.; Lamers, C.B. Expression of receptors for gut peptides in human pancreatic adenocarcinoma and tumour-free pancreas. Br. J. Cancer 1997, 75, 1467–1473. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Korner, M.; Waser, B.; Reubi, J.C. Does somatostatin or gastric inhibitory peptide receptor expression correlate with tumor grade and stage in gut neuroendocrine tumors? Neuroendocrinology 2015, 101, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Waser, B.; Rehmann, R.; Sanchez, C.; Fourmy, D.; Reubi, J.C. Glucose-dependent insulinotropic polypeptide receptors in most gastroenteropancreatic and bronchial neuroendocrine tumors. J. Clin. Endocrinol. Metab. 2012, 97, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, L.; Fu, J.; Huang, H.; Sun, S.; Zhang, D.; Zhao, L.; Ucheojor Onwuka, J.; Zhao, Y.; Cui, B. SCTR hypermethylation is a diagnostic biomarker in colorectal cancer. Cancer Sci. 2020, 111, 4558–4566. [Google Scholar] [CrossRef] [PubMed]
- Pang, R.T.; Lee, L.T.; Ng, S.S.; Yung, W.H.; Chow, B.K. CpG methylation and transcription factors Sp1 and Sp3 regulate the expression of the human secretin receptor gene. Mol. Endocrinol. 2004, 18, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Werth, B.; Hildebrand, P.; Gyr, K.; Stalder, G.A.; Beglinger, C. Human secretin. Biologic effects and plasma kinetics in humans. Gastroenterology 1988, 94, 311–316. [Google Scholar] [CrossRef]
- Beglinger, C.; Gyr, K.; Werth, B.; Keller, U.; Girard, J. Comparative effects of synthetic and natural secretin on pancreatic secretion and on secretin, insulin, and glucagon levels in man. Dig. Dis. Sci. 1982, 27, 231–233. [Google Scholar] [CrossRef]
- Schaffalitzky de Muckadell, O.B.; Fahrenkrug, J.; Watt-Boolsen, S.; Worning, H. Pancreatic response and plasma secretin concentration during infusion of low dose secretin in man. Scand. J. Gastroenterol. 1978, 13, 305–311. [Google Scholar] [CrossRef]
- Kolts, B.E.; McGuigan, J.E. Radioimmunoassay measurement of secretin half-life in man. Gastroenterology 1977, 72, 55–60. [Google Scholar] [CrossRef]
- Dong, M.; Pinon, D.I.; Miller, L.J. Insights into the impact of phenolic residue incorporation at each position along secretin for receptor binding and biological activity. Regul. Pept. 2013, 180, 5–11. [Google Scholar] [CrossRef][Green Version]
- Gardner, J.D.; Rottman, A.J.; Natarajan, S.; Bodanszky, M. Interaction of secretin5-27 and its analogues with hormone receptors on pancreatic acini. Biochim. Biophys. Acta 1979, 583, 491–503. [Google Scholar] [CrossRef]
- Kim, C.D.; Li, P.; Lee, K.Y.; Coy, D.H.; Chey, W.Y. Effect of [(CH2NH)4,5]secretin on pancreatic exocrine secretion in guinea pigs and rats. Am. J. Physiol. 1993, 265, G805–G810. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. Biased GPCR signaling: Possible mechanisms and inherent limitations. Pharmacol. Ther. 2020, 211, 107540. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Grade | Strong SCTR Staining | Weak SCTR Staining | No SCTR Staining |
|---|---|---|---|---|
| PDAC | All grades | 65/70 (93%) | 1/70 (1%) | 4/70 (6%) |
| PDAC | G1 | 10/10 (100%) | 0/10 (0%) | 0/10 (0%) |
| PDAC | G2 | 36/37 (97%) | 1/37 (3%) | 0/37 (0%) |
| PDAC | G3 | 19/23 (83%) | 0/23 (0%) | 4/23 (17%) |
| Adenosquamous carcinoma | 2/3 (67%) | 1/3 (33%) | 0/3 (0%) | |
| Islet cell carcinoma (malignant) | 0/1 (0%) | 0/1 (0%) | 1/1 (100% | |
| Islet cell tumor (benign) | 0/10 (0%) | 1/10 (10%) | 9/10 (90%) | |
| Ductal hyperplasia | 2/2 (100%) | 0/2 (0%) | 0/2 (0%) | |
| Chronic pancreatitis | 0/10 (0%) | 0/10 (0%) | 10/10 (100%) |
| Peptide # | Sequence | IC50 [nM] |
|---|---|---|
| 115 | SDGTFTSELSRLREGARLQRLLQGLV | 1283 ± 703 |
| 116 | DGTFTSELSRLREGARLQRLLQGLV | 1735 ± 590 |
| 117 | GTFTSELSRLREGARLQRLLQGLV | 1917 ± 355 |
| 118 | TFTSELSRLREGARLQRLLQGLV | 2588 ± 317 |
| 119 | FTSELSRLREGARLQRLLQGLV | 2338 ± 366 |
| 120 | TSELSRLREGARLQRLLQGLV | - |
| 121 | SELSRLREGARLQRLLQGLV | 2727 ± 60 |
| 122 | ELSRLREGARLQRLLQGLV | - |
| 123 | LSRLREGARLQRLLQGLV | - |
| Peptide # | Sequence | IC50 [nM] |
|---|---|---|
| 124 | RFTSELSRLREGARLQRLLQGLV | 924 ± 237 |
| 125 | rFTSELSRLREGARLQRLLQGLV | 1361 ± 322 |
| 126 | rATSELSRLREGARLQRLLQGLV | 1757 ± 322 |
| 127 | rFASELSRLREGARLQRLLQGLV | 1612 ± 185 |
| 128 | rFTAELSRLREGARLQRLLQGLV | 897 ± 108 |
| 129 | rFTSALSRLREGARLQRLLQGLV | 1141 ± 281 |
| 130 | rFTSEASRLREGARLQRLLQGLV | - |
| 131 | rFTSELARLREGARLQRLLQGLV | 998 ± 142 |
| 132 | rFTSELSALREGARLQRLLQGLV | 2479 ± 579 |
| 133 | rFTSELSRAREGARLQRLLQGLV | 884 ± 169 |
| 134 | rFTSELSRLAEGARLQRLLQGLV | 1580 ± 77 |
| 135 | rFTSELSRLRAGARLQRLLQGLV | - |
| 136 | rFTSELSRLREAARLQRLLQGLV | 309 ± 74 |
| 137 | rFTSELSRLREGAALQRLLQGLV | 1858 ± 159 |
| 138 | rFTSELSRLREGARAQRLLQGLV | - |
| 139 | rFTSELSRLREGARLARLLQGLV | 1017 ± 107 |
| 140 | rFTSELSRLREGARLQALLQGLV | 1492 ± 218 |
| 141 | rFTSELSRLREGARLQRALQGLV | - |
| 142 | rFTSELSRLREGARLQRLAQGLV | - |
| 143 | rFTSELSRLREGARLQRLLAGLV | 697 ± 55 |
| 144 | rFTSELSRLREGARLQRLLQALV | 570 ± 30 |
| 145 | rFTSELSRLREGARLQRLLQGAV | - |
| 146 | rFTSELSRLREGARLQRLLQGLA | 2057 ± 760 |
| Peptide # | Sequence | Inhibition of β-arr2-GFP Translocation; IC50 [nM] | Inhibition of Radioligand Binding; IC50 [nM] | cAMP Production; EC50 [nM] | cAMP Production, Intrinsic Activity [% of Secretin] |
|---|---|---|---|---|---|
| 136 | rFTSELSRLREAARLQRLLQGLV | 309 ± 74 | 2472 ± 1621 | 38.4 ± 18.6 | 33.6 ± 0.9 |
| 143 | rFTSELSRLREGARLQRLLAGLV | 697 ± 55 | 963 ± 476 | 23.4 ± 16.2 | 27.5 ± 8.8 |
| 144 | rFTSELSRLREGARLQRLLQALV | 570 ± 30 | 304 ± 142 | 29.4 ± 17.4 | 31.1 ± 2.2 |
| 120 | TSELSRLREGARLQRLLQGLV | >3000 | ND | 3366 ± 1570 | 90.6 ± 11.5 |
| 1 | HSDGTFTSELSRLREGARLQRLLQGLV | ND | 0.3 ± 0.1 | 0.0012 ± 0.0003 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klussmeier, A.; Aurich, S.; Niederstadt, L.; Wiedenmann, B.; Grötzinger, C. Secretin Receptor as a Target in Gastrointestinal Cancer: Expression Analysis and Ligand Development. Biomedicines 2022, 10, 536. https://doi.org/10.3390/biomedicines10030536
Klussmeier A, Aurich S, Niederstadt L, Wiedenmann B, Grötzinger C. Secretin Receptor as a Target in Gastrointestinal Cancer: Expression Analysis and Ligand Development. Biomedicines. 2022; 10(3):536. https://doi.org/10.3390/biomedicines10030536
Chicago/Turabian StyleKlussmeier, Anja, Stefan Aurich, Lars Niederstadt, Bertram Wiedenmann, and Carsten Grötzinger. 2022. "Secretin Receptor as a Target in Gastrointestinal Cancer: Expression Analysis and Ligand Development" Biomedicines 10, no. 3: 536. https://doi.org/10.3390/biomedicines10030536
APA StyleKlussmeier, A., Aurich, S., Niederstadt, L., Wiedenmann, B., & Grötzinger, C. (2022). Secretin Receptor as a Target in Gastrointestinal Cancer: Expression Analysis and Ligand Development. Biomedicines, 10(3), 536. https://doi.org/10.3390/biomedicines10030536