
Genetic Predisposition and Inflammatory Inhibitors in COVID-19: Where Do We Stand?

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
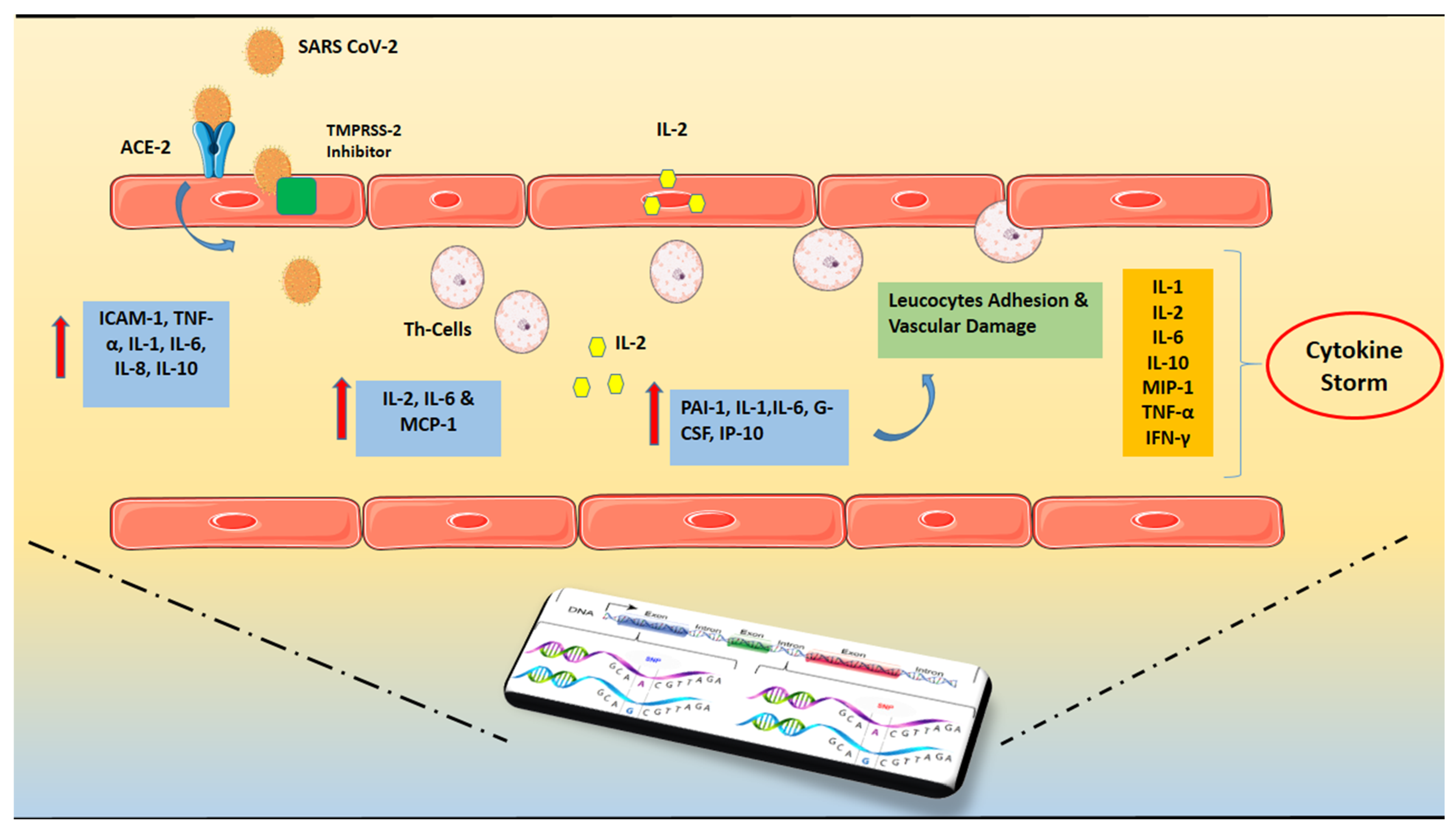
2. Inflammatory Mechanisms in COVID-19-Related Cardiovascular Disorders
3. Cytokine Storm and ARDS in COVID-19
4. Genetic Predisposition
5. Anti-Inflammatory Agents and Treatment Options
5.1. Inflammatory Inhibitors
5.1.1. Inhibition of IL-1 Signaling
5.1.2. Inhibition of IL-6 Signaling
5.1.3. Inhibition of TNF-α Signaling
5.1.4. Inhibition of IFN-γ Signaling
5.1.5. Inhibition of JAK Pathway
5.1.6. Inhibition of Granulocyte–Macrophage Colony-Stimulating Factor Signaling
5.2. Other Non-Specific Anti-Inflammatory Agents
5.2.1. Corticosteroids
5.2.2. Statins
5.2.3. Colchicine-Macrolides
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-up. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef] [PubMed]
- Harapan, H.; Itoh, N.; Yufika, A.; Winardi, W.; Keam, S.; Te, H.; Megawati, D.; Hayati, Z.; Wagner, A.L.; Mudatsir, M. Coronavirus disease 2019 (COVID-19): A literature review. J. Infect. Public Health 2020, 13, 667–673. [Google Scholar] [CrossRef]
- The WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group; Sterne, J.A.C.; Murthy, S.; Diaz, J.V.; Slutsky, A.S.; Villar, J.; Angus, D.C.; Annane, D.; Azevedo, L.C.P.; Berwanger, O.; et al. Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-analysis. JAMA 2020, 324, 1330–1341. [Google Scholar] [CrossRef]
- Abbasi-Oshaghi, E.; Mirzaei, F.; Farahani, F.; Khodadadi, I.; Tayebinia, H. Diagnosis and treatment of coronavirus disease 2019 (COVID-19): Laboratory, PCR, and chest CT imaging findings. Int. J. Surg. 2020, 79, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, H.K.; Mehra, M.R. COVID-19 illness in native and immunosuppressed states: A clinical-therapeutic staging proposal. J. Heart Lung Transpl. 2020, 39, 405–407. [Google Scholar] [CrossRef] [Green Version]
- Sagris, M.; Theofilis, P.; Antonopoulos, A.S.; Tsioufis, C.; Oikonomou, E.; Antoniades, C.; Crea, F.; Kaski, J.C.; Tousoulis, D. Inflammatory Mechanisms in COVID-19 and Atherosclerosis: Current Pharmaceutical Perspectives. Int. J. Mol. Sci. 2021, 22, 6607. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Bansal, M. Cardiovascular disease and COVID-19. Diabetes Metab. Syndr. 2020, 14, 247–250. [Google Scholar] [CrossRef]
- Dhakal, B.P.; Sweitzer, N.K.; Indik, J.H.; Acharya, D.; William, P. SARS-CoV-2 Infection and Cardiovascular Disease: COVID-19 Heart. Heart Lung Circ. 2020, 29, 973–987. [Google Scholar] [CrossRef]
- Hua, A.; O’Gallagher, K.; Sado, D.; Byrne, J. Life-threatening cardiac tamponade complicating myo-pericarditis in COVID-19. Eur. Heart J. 2020, 41, 2130. [Google Scholar] [CrossRef] [Green Version]
- Sagris, M.; Antonopoulos, A.S.; Theofilis, P.; Oikonomou, E.; Siasos, G.; Tsalamandris, S.; Antoniades, C.; Brilakis, E.S.; Kaski, J.C.; Tousoulis, D. Risk factors profile of young and older patients with Myocardial Infarction. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Manolis, A.S.; Manolis, A.A.; Manolis, T.A.; Apostolopoulos, E.J.; Papatheou, D.; Melita, H. COVID-19 infection and cardiac arrhythmias. Trends Cardiovasc. Med. 2020, 30, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Peppas, S.; Sagris, M.; Bikakis, I.; Giannopoulos, S.; Tzoumas, A.; Kokkinidis, D.G.; Ahmed, Z.; Korosoglou, G.; Malgor, E.A.; Malgor, R.D. A Systematic Review and Meta-analysis on the Efficacy and Safety of Direct Oral Anticoagulants in Patients with Peripheral Artery Disease. Ann. Vasc. Surg. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sagris, M.; Vardas, E.P.; Theofilis, P.; Antonopoulos, A.S.; Oikonomou, E.; Tousoulis, D. Atrial Fibrillation: Pathogenesis, Predisposing Factors, and Genetics. Int. J. Mol. Sci. 2021, 23, 6. [Google Scholar] [CrossRef] [PubMed]
- Clerkin, K.J.; Fried, J.A.; Raikhelkar, J.; Sayer, G.; Griffin, J.M.; Masoumi, A.; Jain, S.S.; Burkhoff, D.; Kumaraiah, D.; Rabbani, L.; et al. COVID-19 and Cardiovascular Disease. Circulation 2020, 141, 1648–1655. [Google Scholar] [CrossRef] [Green Version]
- Siripanthong, B.; Nazarian, S.; Muser, D.; Deo, R.; Santangeli, P.; Khanji, M.Y.; Cooper, L.T., Jr.; Chahal, C.A.A. Recognizing COVID-19-related myocarditis: The possible pathophysiology and proposed guideline for diagnosis and management. Heart Rhythm 2020, 17, 1463–1471. [Google Scholar] [CrossRef]
- Sagris, M.; Theofilis, P.; Antonopoulos, A.S.; Oikonomou, E.; Paschaliori, C.; Galiatsatos, N.; Tsioufis, K.; Tousoulis, D. Inflammation in Coronary Microvascular Dysfunction. Int. J. Mol. Sci. 2021, 22, 13471. [Google Scholar] [CrossRef]
- Sagris, M.; Giannopoulos, S.; Giannopoulos, S.; Tzoumas, A.; Texakalidis, P.; Charisis, N.; Kokkinidis, D.G.; Malgor, R.D.; Mouawad, N.J.; Bakoyiannis, C. Transcervical carotid artery revascularization: A systematic review and meta-analysis of outcomes. J. Vasc. Surg. 2021, 74, 657–665.e612. [Google Scholar] [CrossRef]
- Esenwa, C.; Cheng, N.T.; Lipsitz, E.; Hsu, K.; Zampolin, R.; Gersten, A.; Antoniello, D.; Soetanto, A.; Kirchoff, K.; Liberman, A.; et al. COVID-19-Associated Carotid Atherothrombosis and Stroke. AJNR Am. J. Neuroradiol. 2020, 41, 1993–1995. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machowicz, R.; Janka, G.; Wiktor-Jedrzejczak, W. Similar but not the same: Differential diagnosis of HLH and sepsis. Crit. Rev. Oncol. Hematol. 2017, 114, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.C.; Alves, J.D. Pathogenesis of multi-organic failure in autoimmune diseases. Autoimmun. Rev. 2009, 8, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Theofilis, P.; Sagris, M.; Antonopoulos, A.S.; Oikonomou, E.; Tsioufis, C.; Tousoulis, D. Inflammatory Mediators of Platelet Activation: Focus on Atherosclerosis and COVID-19. Int. J. Mol. Sci. 2021, 22, 11170. [Google Scholar] [CrossRef]
- Alunno, A.; Carubbi, F.; Rodríguez-Carrio, J. Storm, typhoon, cyclone or hurricane in patients with COVID-19? Beware of the same storm that has a different origin. RMD Open 2020, 6, e001295. [Google Scholar] [CrossRef]
- Mazodier, K.; Marin, V.; Novick, D.; Farnarier, C.; Robitail, S.; Schleinitz, N.; Veit, V.; Paul, P.; Rubinstein, M.; Dinarello, C.A.; et al. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood 2005, 106, 3483–3489. [Google Scholar] [CrossRef]
- Cheung, C.Y.; Poon, L.L.; Ng, I.H.; Luk, W.; Sia, S.F.; Wu, M.H.; Chan, K.H.; Yuen, K.Y.; Gordon, S.; Guan, Y.; et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: Possible relevance to pathogenesis. J. Virol. 2005, 79, 7819–7826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Frydas, I.; Kritas, S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34, 1. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Lau, C.C.Y.; Chan, K.H.; Li, C.P.Y.; Chen, H.; Jin, D.Y.; Chan, J.F.W.; Woo, P.C.Y.; Yuen, K.Y. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: Implications for pathogenesis and treatment. J. Gen. Virol. 2013, 94, 2679–2690. [Google Scholar] [CrossRef] [PubMed]
- Law, H.K.; Cheung, C.Y.; Ng, H.Y.; Sia, S.F.; Chan, Y.O.; Luk, W.; Nicholls, J.M.; Peiris, J.S.; Lau, Y.L. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood 2005, 106, 2366–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Hu, R.; Gu, X. A close-up on COVID-19 and cardiovascular diseases. Nutr. Metab. Cardiovasc. Dis. NMCD 2020, 30, 1057–1060. [Google Scholar] [CrossRef] [PubMed]
- Hogner, K.; Wolff, T.; Pleschka, S.; Plog, S.; Gruber, A.D.; Kalinke, U.; Walmrath, H.D.; Bodner, J.; Gattenlohner, S.; Lewe-Schlosser, P.; et al. Correction: Macrophage-expressed IFN-beta Contributes to Apoptotic Alveolar Epithelial Cell Injury in Severe Influenza Virus Pneumonia. PLoS Pathog. 2016, 12, e1005716. [Google Scholar] [CrossRef] [Green Version]
- Rodrigue-Gervais, I.G.; Labbe, K.; Dagenais, M.; Dupaul-Chicoine, J.; Champagne, C.; Morizot, A.; Skeldon, A.; Brincks, E.L.; Vidal, S.M.; Griffith, T.S.; et al. Cellular inhibitor of apoptosis protein cIAP2 protects against pulmonary tissue necrosis during influenza virus infection to promote host survival. Cell Host. Microbe 2014, 15, 23–35. [Google Scholar] [CrossRef] [Green Version]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Severe Covid, G.G.; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernandez, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe COVID-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef]
- Wu, Y.; Feng, Z.; Li, P.; Yu, Q. Relationship between ABO blood group distribution and clinical characteristics in patients with COVID-19. Clin. Chim. Acta 2020, 509, 220–223. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, Y.; Huang, H.; Li, D.; Gu, D.; Lu, X.; Zhang, Z.; Liu, L.; Liu, T.; Liu, Y.; et al. Relationship between the ABO Blood Group and the Coronavirus Disease 2019 (COVID-19) Susceptibility. Clin. Infect. Dis. 2021, 73, 328–331. [Google Scholar] [CrossRef]
- Diavati, S.; Sagris, M.; Terentes-Printzios, D.; Vlachopoulos, C. Anticoagulation Treatment in Venous Thromboembolism: Options and Optimal Duration. Curr. Pharm. Des. 2021. [Google Scholar] [CrossRef]
- Wu, B.B.; Gu, D.Z.; Yu, J.N.; Yang, J.; Shen, W.Q. Association between ABO blood groups and COVID-19 infection, severity and demise: A systematic review and meta-analysis. Infect. Genet. Evol. 2020, 84, 104485. [Google Scholar] [CrossRef]
- Hou, Y.; Zhao, J.; Martin, W.; Kallianpur, A.; Chung, M.K.; Jehi, L.; Sharifi, N.; Erzurum, S.; Eng, C.; Cheng, F. New insights into genetic susceptibility of COVID-19: An ACE2 and TMPRSS2 polymorphism analysis. BMC Med. 2020, 18, 216. [Google Scholar] [CrossRef]
- Suryamohan, K.; Diwanji, D.; Stawiski, E.W.; Gupta, R.; Miersch, S.; Liu, J.; Chen, C.; Jiang, Y.P.; Fellouse, F.A.; Sathirapongsasuti, J.F.; et al. Human ACE2 receptor polymorphisms and altered susceptibility to SARS-CoV-2. Commun. Biol. 2021, 4, 475. [Google Scholar] [CrossRef] [PubMed]
- Pouladi, N.; Abdolahi, S. Investigating the ACE2 polymorphisms in COVID-19 susceptibility: An in silico analysis. Mol. Genet. Genom. Med. 2021, 9, e1672. [Google Scholar] [CrossRef] [PubMed]
- Novelli, A.; Biancolella, M.; Borgiani, P.; Cocciadiferro, D.; Colona, V.L.; D’Apice, M.R.; Rogliani, P.; Zaffina, S.; Leonardis, F.; Campana, A.; et al. Analysis of ACE2 genetic variants in 131 Italian SARS-CoV-2-positive patients. Hum. Genom. 2020, 14, 29. [Google Scholar] [CrossRef]
- Vargas-Alarcon, G.; Posadas-Sanchez, R.; Ramirez-Bello, J. Variability in genes related to SARS-CoV-2 entry into host cells (ACE2, TMPRSS2, TMPRSS11A, ELANE, and CTSL) and its potential use in association studies. Life Sci. 2020, 260, 118313. [Google Scholar] [CrossRef]
- Siasos, G.; Skotsimara, G.; Oikonomou, E.; Sagris, M.; Vasiliki-Chara, M.; Bletsa, E.; Stampouloglou, P.; Theofilis, P.; Charalampous, G.; Tousoulis, D. Antithrombotic Treatment in Diabetes Mellitus: A Review of the Literature about Antiplatelet and Anticoagulation Strategies Used for Diabetic Patients in Primary and Secondary Prevention. Curr. Pharm. Des. 2020, 26, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Kasparian, K.; Graykowski, D.; Cudaback, E. Commentary: APOE e4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. Front. Immunol. 2020, 11, 1939. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Kuchel, G.A.; Melzer, D. APOE e4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2231–2232. [Google Scholar] [CrossRef]
- Nguyen, A.; David, J.K.; Maden, S.K.; Wood, M.A.; Weeder, B.R.; Nellore, A.; Thompson, R.F. Human Leukocyte Antigen Susceptibility Map for Severe Acute Respiratory Syndrome Coronavirus 2. J. Virol. 2020, 94, e00510-20. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Huang, S.; Gao, R.; Zhou, Y.; Lai, C.; Li, Z.; Xian, W.; Qian, X.; Li, Z.; Huang, Y.; et al. Initial whole-genome sequencing and analysis of the host genetic contribution to COVID-19 severity and susceptibility. Cell Discov. 2020, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Delanghe, J.R.; De Buyzere, M.L.; Speeckaert, M.M. Genetic Polymorphisms in the Host and COVID-19 Infection. Adv. Exp. Med. Biol. 2021, 1318, 109–118. [Google Scholar] [CrossRef]
- Thevarajan, I.; Nguyen, T.H.O.; Koutsakos, M.; Druce, J.; Caly, L.; van de Sandt, C.E.; Jia, X.; Nicholson, S.; Catton, M.; Cowie, B.; et al. Breadth of concomitant immune responses prior to patient recovery: A case report of non-severe COVID-19. Nat. Med. 2020, 26, 453–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; van den Heuvel, G.; Mantere, T.; Kersten, S.; van Deuren, R.C.; Steehouwer, M.; van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of Genetic Variants Among Young Men with Severe COVID-19. JAMA 2020, 324, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Borcherding, N.; Kolb, R. IL-1 Signaling in Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1240, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakoory, B.; Carcillo, J.A.; Chatham, W.W.; Amdur, R.L.; Zhao, H.; Dinarello, C.A.; Cron, R.Q.; Opal, S.M. Interleukin-1 Receptor Blockade Is Associated with Reduced Mortality in Sepsis Patients with Features of Macrophage Activation Syndrome: Reanalysis of a Prior Phase III Trial. Crit. Care Med. 2016, 44, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, G.; De Luca, G.; Campochiaro, C.; Della-Torre, E.; Ripa, M.; Canetti, D.; Oltolini, C.; Castiglioni, B.; Tassan Din, C.; Boffini, N.; et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: A retrospective cohort study. Lancet Rheumatol. 2020, 2, e325–e331. [Google Scholar] [CrossRef]
- Bozzi, G.; Mangioni, D.; Minoia, F.; Aliberti, S.; Grasselli, G.; Barbetta, L.; Castelli, V.; Palomba, E.; Alagna, L.; Lombardi, A.; et al. Anakinra combined with methylprednisolone in patients with severe COVID-19 pneumonia and hyperinflammation: An observational cohort study. J. Allergy Clin. Immunol. 2020, 147, 561–566. [Google Scholar] [CrossRef]
- Landi, L.; Ravaglia, C.; Russo, E.; Cataleta, P.; Fusari, M.; Boschi, A.; Giannarelli, D.; Facondini, F.; Valentini, I.; Panzini, I.; et al. Blockage of interleukin-1beta with canakinumab in patients with COVID-19. Sci. Rep. 2020, 10, 21775. [Google Scholar] [CrossRef]
- Cacciapaglia, F.; Anelli, M.G.; Rinaldi, A.; Fornaro, M.; Lopalco, G.; Scioscia, C.; Lapadula, G.; Iannone, F. Lipids and Atherogenic Indices Fluctuation in Rheumatoid Arthritis Patients on Long-Term Tocilizumab Treatment. Mediat. Inflamm. 2018, 2018, 2453265. [Google Scholar] [CrossRef] [PubMed]
- Rossotti, R.; Travi, G.; Ughi, N.; Corradin, M.; Baiguera, C.; Fumagalli, R.; Bottiroli, M.; Mondino, M.; Merli, M.; Bellone, A.; et al. Safety and efficacy of anti-il6-receptor tocilizumab use in severe and critical patients affected by coronavirus disease 2019: A comparative analysis. J. Infect. 2020, 81, e11–e17. [Google Scholar] [CrossRef] [PubMed]
- Toniati, P.; Piva, S.; Cattalini, M.; Garrafa, E.; Regola, F.; Castelli, F.; Franceschini, F.; Airo, P.; Bazzani, C.; Beindorf, E.A.; et al. Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia, Italy. Autoimmun. Rev. 2020, 102568. [Google Scholar] [CrossRef] [PubMed]
- van Kraaij, T.D.; Mostard, R.L.; Ramiro, S.; Magro Checa, C.; van Dongen, C.M.; van Haren, E.H.; Buijs, J.; Landewe, R.B. Tocilizumab in Severe COVID-19 Pneumonia and Concomitant Cytokine Release Syndrome. Eur. J. Case Rep. Intern. Med. 2020, 7, 001675. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, C.; Della-Torre, E.; Cavalli, G.; De Luca, G.; Ripa, M.; Boffini, N.; Tomelleri, A.; Baldissera, E.; Rovere-Querini, P.; Ruggeri, A.; et al. Efficacy and safety of tocilizumab in severe COVID-19 patients: A single-centre retrospective cohort study. Eur. J. Intern. Med. 2020, 76, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Hossen, M.S.; Barek, M.A.; Jahan, N.; Safiqul Islam, M. A Review on Current Repurposing Drugs for the Treatment of COVID-19: Reality and Challenges. SN Compr. Clin. Med. 2020, 2, 1777–1789. [Google Scholar] [CrossRef]
- Idriss, H.T.; Naismith, J.H. TNF alpha and the TNF receptor superfamily: Structure-function relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Bergstrom, U.; Jovinge, S.; Persson, J.; Jacobsson, L.T.H.; Turesson, C. Effects of Treatment with Adalimumab on Blood Lipid Levels and Atherosclerosis in Patients with Rheumatoid Arthritis. Curr. Ther. Res. Clin. Exp. 2018, 89, 1–6. [Google Scholar] [CrossRef]
- Chen, X.Y.; Yan, B.X.; Man, X.Y. TNFalpha inhibitor may be effective for severe COVID-19: Learning from toxic epidermal necrolysis. Ther. Adv. Respir. Dis. 2020, 14, 1753466620926800. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Boisson-Dupuis, S.; Chapgier, A.; Yang, K.; Bustamante, J.; Puel, A.; Picard, C.; Abel, L.; Jouanguy, E.; Casanova, J.L. Inborn errors of interferon (IFN)-mediated immunity in humans: Insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol. Rev. 2008, 226, 29–40. [Google Scholar] [CrossRef]
- Cure, E.; Kucuk, A.; Cure, M.C. Can emapalumab be life saving for refractory, recurrent, and progressive cytokine storm caused by COVID-19, which is resistant to anakinra, tocilizumab, and Janus kinase inhibitors. Indian J. Pharmacol. 2021, 53, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elli, E.M.; Barate, C.; Mendicino, F.; Palandri, F.; Palumbo, G.A. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front. Oncol. 2019, 9, 1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Rawling, M.; Savory, E.; Stebbing, J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, e30–e31. [Google Scholar] [CrossRef] [Green Version]
- Becher, B.; Tugues, S.; Greter, M. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 2016, 45, 963–973. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef]
- Zhou, Y.; Fu, B.; Zheng, X.; Wang, D.; Zhao, C.; Qi, Y.; Sun, R.; Tian, Z.; Xu, X.; Wei, H. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020, 7, 998–1002. [Google Scholar] [CrossRef] [Green Version]
- Rice, J.B.; White, A.G.; Scarpati, L.M.; Wan, G.; Nelson, W.W. Long-term Systemic Corticosteroid Exposure: A Systematic Literature Review. Clin. Ther. 2017, 39, 2216–2229. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.M.; Vinetz, J.M. Dexamethasone in the management of COVID-19. BMJ 2020, 370, m2648. [Google Scholar] [CrossRef]
- Saghazadeh, A.; Rezaei, N. Towards treatment planning of COVID-19: Rationale and hypothesis for the use of multiple immunosuppressive agents: Anti-antibodies, immunoglobulins, and corticosteroids. Int. Immunopharmacol. 2020, 84, 106560. [Google Scholar] [CrossRef]
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F.; et al. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020, 116, 1666–1687. [Google Scholar] [CrossRef] [PubMed]
- Sagris, M.; Kokkinidis, D.G.; Lempesis, I.G.; Giannopoulos, S.; Rallidis, L.; Mena-Hurtado, C.; Bakoyiannis, C. Nutrition, dietary habits, and weight management to prevent and treat patients with peripheral artery disease. Rev. Cardiovasc. Med. 2020, 21, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.O.; Budoff, M. Effect of statins on atherosclerotic plaque. Trends Cardiovasc. Med. 2019, 29, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.; Morrison, T.E.; Funkhouser, W.; Uematsu, S.; Akira, S.; Baric, R.S.; Heise, M.T. MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathog. 2008, 4, e1000240. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Deng, Y.; Guo, X.; Shang, J.; Zhu, D.; Liu, H. Atorvastatin attenuates myocardial remodeling induced by chronic intermittent hypoxia in rats: Partly involvement of TLR-4/MYD88 pathway. Biochem. Biophys. Res. Commun. 2014, 446, 292–297. [Google Scholar] [CrossRef]
- Yuan, S. Statins May Decrease the Fatality Rate of Middle East Respiratory Syndrome Infection. mBio 2015, 6, e01120. [Google Scholar] [CrossRef] [Green Version]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, M.; Thompson, P.L. Effect of colchicine (0.5 mg twice daily) on high-sensitivity C-reactive protein independent of aspirin and atorvastatin in patients with stable coronary artery disease. Am. J. Cardiol. 2007, 99, 805–807. [Google Scholar] [CrossRef]
- Bouabdallaoui, N.; Tardif, J.C.; Waters, D.D.; Pinto, F.J.; Maggioni, A.P.; Diaz, R.; Berry, C.; Koenig, W.; Lopez-Sendon, J.; Gamra, H.; et al. Time-to-treatment initiation of colchicine and cardiovascular outcomes after myocardial infarction in the Colchicine Cardiovascular Outcomes Trial (COLCOT). Eur. Heart J. 2020, 41, 4092–4099. [Google Scholar] [CrossRef]
- Bonam, S.R.; Kaveri, S.V.; Sakuntabhai, A.; Gilardin, L.; Bayry, J. Adjunct Immunotherapies for the Management of Severely Ill COVID-19 Patients. Cell Rep. Med. 2020, 1, 100016. [Google Scholar] [CrossRef]
- Cumhur Cure, M.; Kucuk, A.; Cure, E. Colchicine may not be effective in COVID-19 infection; it may even be harmful? Clin. Rheumatol. 2020, 39, 2101–2102. [Google Scholar] [CrossRef] [PubMed]
- Arabi, Y.M.; Deeb, A.M.; Al-Hameed, F.; Mandourah, Y.; Almekhlafi, G.A.; Sindi, A.A.; Al-Omari, A.; Shalhoub, S.; Mady, A.; Alraddadi, B.; et al. Macrolides in critically ill patients with Middle East Respiratory Syndrome. Int. J. Infect. Dis. 2019, 81, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishaqui, A.A.; Khan, A.H.; Sulaiman, S.A.S.; Alsultan, M.T.; Khan, I.; Naqvi, A.A. Assessment of efficacy of Oseltamivir-Azithromycin combination therapy in prevention of Influenza-A (H1N1)pdm09 infection complications and rapidity of symptoms relief. Expert Rev. Respir. Med. 2020, 14, 533–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, T.; Imaoka, H. Azithromycin and the risk of cardiovascular death. N. Engl. J. Med. 2012, 367, 774. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Gene | Polymorphism | Result |
|---|---|---|
| ABO | rs657152 | Higher risk of infection for blood group A vs. non-A and lower risk of infection for blood group O vs. non-O [37]. |
| HLA | HLA-B*46:01, HLA-A*11:01, -B*51:01, -C*14:02, HLA-DRB1*15:01, -DQB1*06:02, and -B*27:07 | Vulnerable to disease for HLA-B*46:01 and cross-protective T cell-based immunity for HLA-B*15:03 [50]. |
| TMPRSS2 | p.Val160Met (rs12329760) | Increased susceptibility to SARS-CoV-2 [46]. |
| ACE2 | K31R, N33I, H34R, E35K, E37K, D38V, Y50F, N51S, M62V, K68E, F72V, Y83H, G326E, G352V, D355N, Q388L, and D509Y rs233574 rs2074192 rs4646188 (p.(Asn720Asp) p.(Lys26Arg) p.(Gly211Arg) p.(Leu351Val) p.(Pro389His) | Better cardiovascular and pulmonary course of the disease, less susceptibility to SARS-CoV-2 [42,43,44,45]. |
| ApoE | rs429358-C-C (e4e4) | Severe course of the disease [48,49]. |
| SLC6A20, LZTFL1, CCR9, FYCO1, CXCR6, XCR1 | rs11385942-GA | Severe course of the disease and potentially higher odds for ARDS [53,54]. |
| Of Complement proteins | C3 FF, C3 FS, C3 SS | Severe course of the disease, Increased susceptibility to SARS-CoV-2 [52]. |
| TMEM189- UBE2V1 | rs6020298-A | Severe course of the disease [51]. |
| TLR7 | g.12905756_12905759del and g.12906010G > T | Severe course of the disease [50]. |
| Agent | Dose-Route of Administration | Action | Specific Populations | Adverse Events | Contraindications |
|---|---|---|---|---|---|
| Anakinra | IV: 100 mg every 6 h (total daily dose: 400 mg) for 15 days; 200 mg every 8 h for 7 days; 300 mg od for 4 days, followed by 100 mg od SC: 100 mg od for 10 or 28 days. Alternative regimen: 100 mg every 12 h on days 1–3, then 100 mg od from days 4–10 | IL-1 receptor antagonist | Higher rates of infections in the elderly population In patients with CrCl < 30 and ESRD, use extended dosing intervals (every other day) | Injection site reactions, upper respiratory tract infections, headache, nausea, diarrhea, sinusitis, flu-like symptoms, abdominal pain | Hypersensitivity to Escherichia coli-derived proteins |
| Canakinumab | Undefined/IV/It is administered every eight weeks as a single dose via subcutaneous injection | IL-1 receptor antagonist | Canakinumab has not been studied in patients with hepatic impairment | Respiratory tract infections (including pneumonia, bronchitis, influenza, viral infection, sinusitis, rhinitis, pharyngitis, tonsillitis, nasopharyngitis, upper respiratory tract infection) Ear infection Cellulitis Gastroenteritis Urinary tract infection | Hypersensitivity |
| Tocilizumab | IV: 4–8 mg/kg (maximum single dose: 800 mg), may repeat after 12 h | IL-6 receptor antagonist | Safety during pregnancy and lactation is unknown | Injection site reactions, upper respiratory tract infections (including tuberculosis), nasopharyngitis, headache, hypertension, increased ALT, hematological effects | Hypersensitivity |
| Sarilumab | Undefined/IV | IL-6 receptor antagonist | Safety during pregnancy and lactation is unknown | Neutropenia, increased ALT, injection site erythema, upper respiratory infections, urinary tract infections | Hypersensitivity |
| Etanercept | Undefined/IV | TNF-a inhibitor | High awareness in pediatric population | Pain, swelling, itching, reddening, and bleeding at the puncture site, infections (such as upper respiratory infections, bronchitis, bladder infections, and skin infections), headache, allergic reactions, development of autoantibodies, itching, and fever | Hypersensitivity, Sepsis, Not be initiated in patients with active infections, including chronic or localized infections |
| Adalimumab | Undefined/IV | TNF-a inhibitor | Use with caution in patients with heart failure or decreased left ventricular function; may cause myocardial toxicity or exacerbate underlying myocardial dysfunction Use caution in elderly infection risk patients; may increase | Upper respiratory tract infections, sinusitis, increased macrophage- dependent infection, tuberculosis, opportunistic infections, injection site reactions, increased creatine phosphokinase, headache, rash | None |
| Emapalumab | Undefined/IV | TNF-a inhibitor | None | Infections, hypertension, infusion-related reactions, and pyrexia. | None |
| Ruxolitinib | Various regimens under investigation: PO: 5 mg bid for 14 days; 10 mg bid; 2 × 10 mg bid dose at day 1 and can be increased up to 2 × 15 mg bid from day 2 to day 28; 5 mg bid from day 1 to day 3, then 10 mg bid from day 4 to day 10; 10 mg bid, for 14 days followed by 5 mg bid for 2 days and 5 mg od for 1 day | JAK1/ JAK2 inhibitor | Use in pregnant and lactating women is not recommended May require starting dose reduction in hepatic and renal impairment | Thrombocytopenia, neutropenia, anemia, infections, edema, headache, dizziness | None |
| Baricitinib | PO: 2 or 4 mg od for 14 days | JAK1/ JAK2 inhibitor | Avoid use in patients with severe hepatic impairment, and in patients with moderate or severe renal impairment | Upper respiratory tract infections, nausea, herpes simplex, herpes zoster | None |
| Gimsilumab | IV: High dose on day 1 and low dose on day 8, specifics not described | Anti-GM–CSF | None | None | None |
| Dexamethasone | IV or PO: RECOVERY trial: 6 mg daily for 10 days; DEXACOVID19 trial: 20 mg od from day 1 to day 5, followed by 10 mg od from day 6 to day 10 | Anti-inflammatory and anti-fibrotic effects | Use with caution in the elderly with the smallest possible effective dose for the shortest duration | Sodium and water retention (less than methylprednisolone), hypertension, hyperglycemia, osteoporosis, cardiac hypertrophy, edema, hypokalemia, bruising, diaphoresis, urticaria, allergic rash, euphoria, psychosis, infections, myasthenia gravis | Hypersensitivity to corticosteroids or any component of the formulation, systemic fungal infection |
| Methylprednisolone | IV: 0.5–1 mg/kg daily or 1–2 mg/kg daily (of methylprednisolone or equivalent) have been proposed Higher doses (cytokine storm): 60–125 mg (methylprednisolone) every 6 h for up to 3 days | Anti-inflammatory and anti-fibrotic effects | Use with caution in the elderly with the smallest possible effective dose for the shortest duration | Sodium and water retention, hypertension, hyperglycemia, osteoporosis, cardiac hypertrophy, edema, hypokalemia, bruising, diaphoresis, urticaria, allergic rash, euphoria, psychosis, infections, myasthenia gravis | Hypersensitivity to corticosteroids or any component of the formulation, systemic fungal infection |
| Statins | PO: Simvastatin 40 mg od for 14 days, simvastatin 80 mg od, atorvastatin 40 mg od | Anti-inflammatory and pleiotropic effects | Use with caution in elderly patients; may be at higher risk for myopathy | Hepatotoxicity, myopathies, GI effects, rhabdomyolysis, increased risk of diabetes | Hypersensitivity to Statin, active liver disease; unexplained persistent elevations of serum transaminases; pregnancy, breastfeeding |
| Colchicine | PO: 0.5 mg bid for 3 days, then 0.5 mg od for 27 days | Anti-inflammatory and immunomodulatory effects | Dose adjustment is required in patients with renal or hepatic function | GI symptoms (diarrhea, nausea, vomiting, abdominal pain), neuromuscular toxicity, hematological effects, elevated AST and ALT | Renal or hepatic impairment in conjunction with drugs that inhibit both CYP3A4 and P-gp (e.g., clarithromycin) |
| Azithromycin | PO: 500 mg on day 1, then 250 mg od on days 2–5 | Anti-inflammatory and bacteriostatic effects | Torsades de pointes Arrhythmias in elderly patients | QTc prolongation and ventricular arrhythmias, diarrhea, nausea, abdominal pain, vomiting | Hypersensitivity to azithromycin or other macrolides, history of cholestatic jaundice/ hepatic dysfunction associated with prior azithromycin use |
| Clarithromycin | PO: 250 mg twice daily or 500 mg twice daily in severe cases | Anti-inflammatory and bacteriostatic effects | Parallel administrarion with astemizole, cisapride, pimozide, and terfenadine may result in QT prolongation and cardiac arrhythmias, including ventricular tachycardia, ventricular fibrillation, and Torsades de pointes | Abdominal pain, diarrhoea, nausea, vomiting and taste perversion | Hypersensitivity to Clarithromycin or other macrolides, Clarithromycin should not be used in patients who suffer from severe hepatic failure in combination with renal impairment |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sagris, M.; Theofilis, P.; Antonopoulos, A.S.; Oikonomou, E.; Tsioufis, K.; Tousoulis, D. Genetic Predisposition and Inflammatory Inhibitors in COVID-19: Where Do We Stand? Biomedicines 2022, 10, 242. https://doi.org/10.3390/biomedicines10020242
Sagris M, Theofilis P, Antonopoulos AS, Oikonomou E, Tsioufis K, Tousoulis D. Genetic Predisposition and Inflammatory Inhibitors in COVID-19: Where Do We Stand? Biomedicines. 2022; 10(2):242. https://doi.org/10.3390/biomedicines10020242
Chicago/Turabian StyleSagris, Marios, Panagiotis Theofilis, Alexios S. Antonopoulos, Evangelos Oikonomou, Kostas Tsioufis, and Dimitris Tousoulis. 2022. "Genetic Predisposition and Inflammatory Inhibitors in COVID-19: Where Do We Stand?" Biomedicines 10, no. 2: 242. https://doi.org/10.3390/biomedicines10020242
APA StyleSagris, M., Theofilis, P., Antonopoulos, A. S., Oikonomou, E., Tsioufis, K., & Tousoulis, D. (2022). Genetic Predisposition and Inflammatory Inhibitors in COVID-19: Where Do We Stand? Biomedicines, 10(2), 242. https://doi.org/10.3390/biomedicines10020242