
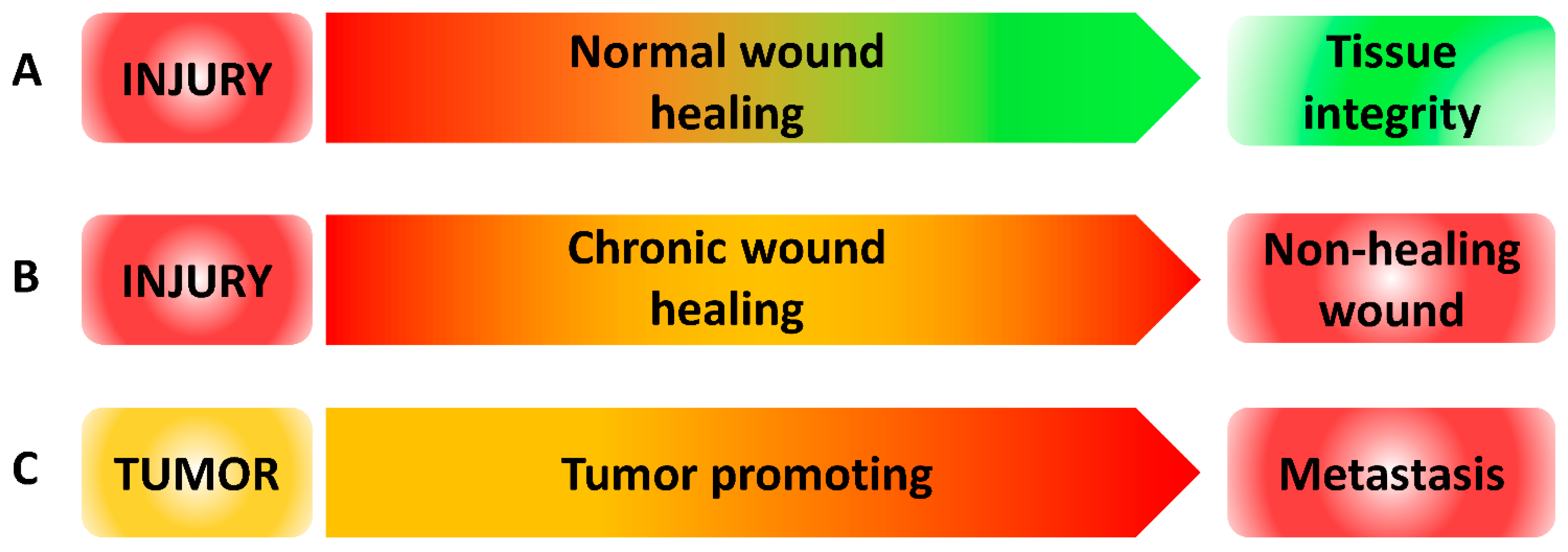
Wound Healing versus Metastasis: Role of Oxidative Stress

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
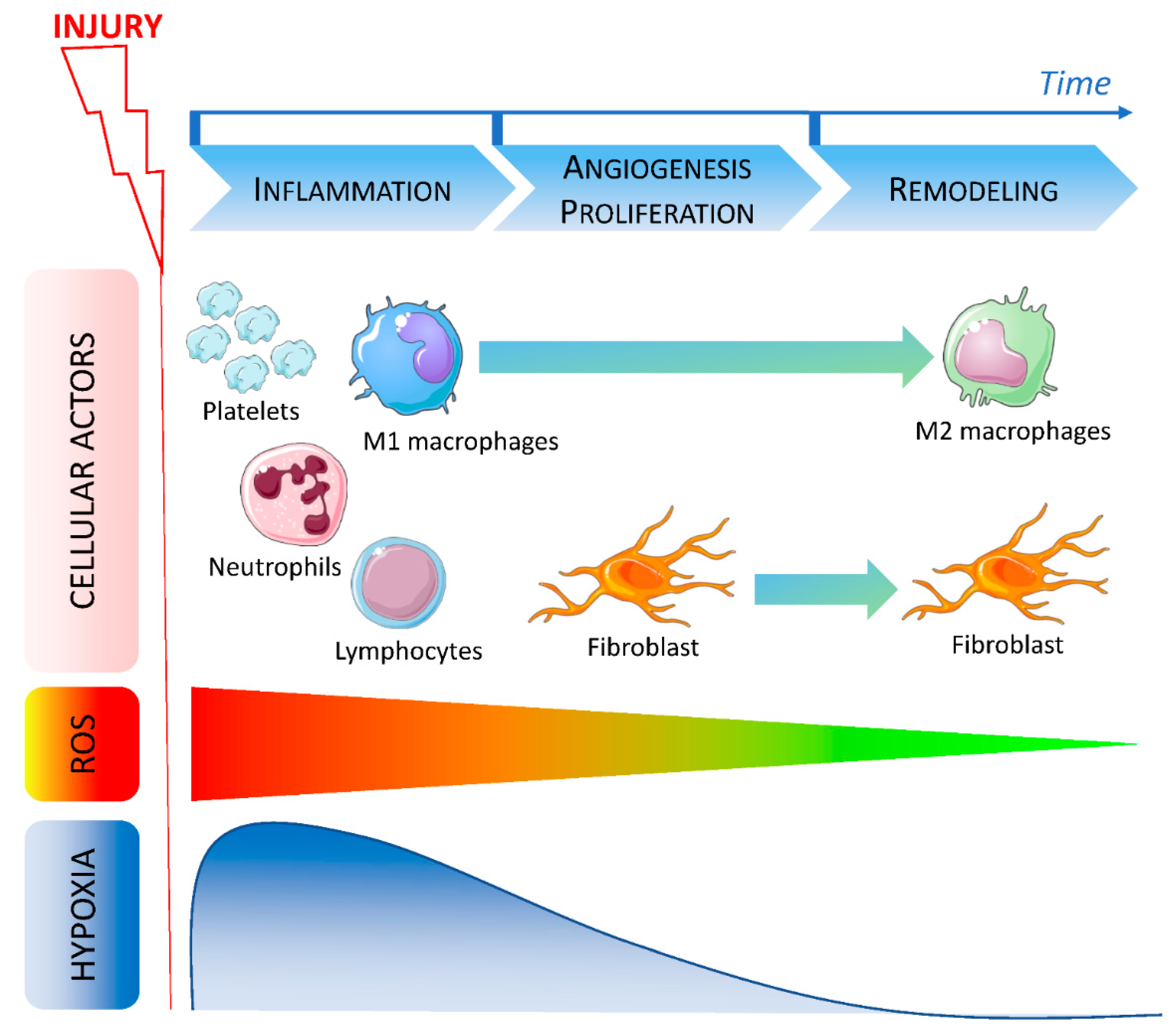
2. Good and Bad Wound Healing: Acute versus Chronic and Chronology of the Cellular Actors
2.1. Macrophages in Wound Healing Process
2.2. ROS in Wound Healing
3. Macrophages Polarization: Role of ROS and NOXs
3.1. ROS in Macrophages Differentiation/Polarization and Function
3.2. NOXs in Macrophages Polarization
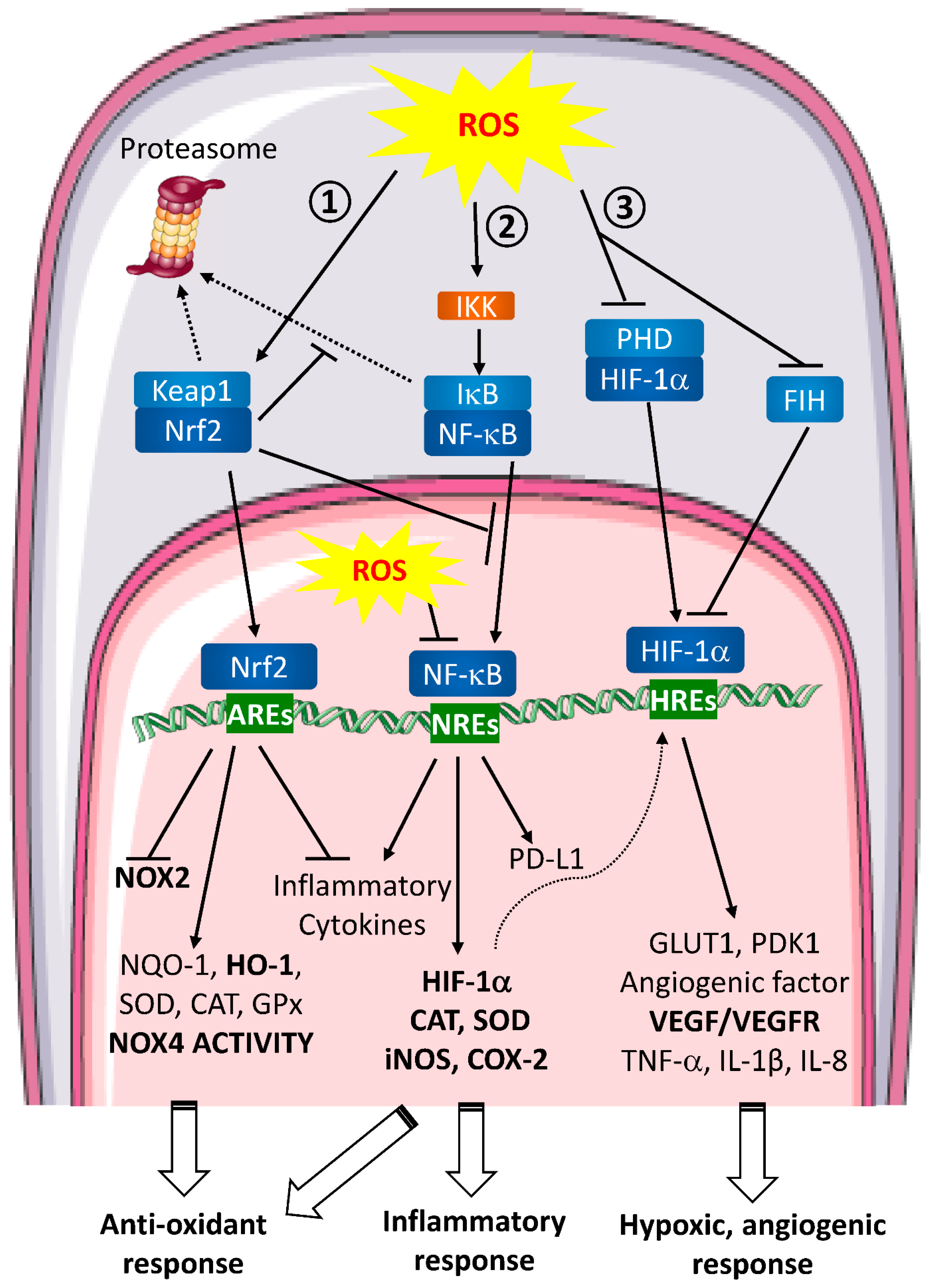
3.3. Molecular Events and Signaling Pathways Involved in Wound Healing
- -
- HIF (Figure 2 ①)
- -
- NF-κB (Figure 2 ②)
- -
- Nrf2 (Figure 2 ③)
4. Metastasis
4.1. Metastasis Hallmarks
4.2. ROS and Metastasis
- -
- Dual effect of ROS
- -
- ROS and angiogenesis
- -
- ROS, EMT and anoikis resistance
- -
- ROS and stemness
4.3. Oxidative Stress and Metastasis: Cellular Actors Involved
- -
- Macrophages
- -
- Fibroblasts
5. Conclusion and Future Perspectives
6. Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Virchow, R. Die Cellularpathologie in Ihrer Begründung Auf Physiologische Und Pathologische Gewebelehre; Hirschwald: Berlin, Germany, 1858; Volume 16. [Google Scholar]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal. Similarities between Tumor Stroma Generation and Wound Healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Singer, A.J.; Clark, R.A. Cutaneous Wound Healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Martin, P. Wound Healing–Aiming for Perfect Skin Regeneration. Science 1997, 276, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Sorg, H.; Tilkorn, D.J.; Hager, S.; Hauser, J.; Mirastschijski, U. Skin Wound Healing: An Update on the Current Knowledge and Concepts. Eur. Surg. Res. 2017, 58, 81–94. [Google Scholar] [CrossRef]
- Gosain, A.; DiPietro, L.A. Aging and Wound Healing. World J. Surg. 2004, 28, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Broughton, G.; Janis, J.E.; Attinger, C.E. The Basic Science of Wound Healing. Plast. Reconstr. Surg. 2006, 117, 12S–34S. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.L.; Groth, A.K.; Branco, A.B. Assessment and Nutritional Aspects of Wound Healing. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 281–288. [Google Scholar] [CrossRef]
- Sindrilaru, A.; Scharffetter-Kochanek, K. Disclosure of the Culprits: Macrophages-Versatile Regulators of Wound Healing. Adv. Wound Care 2013, 2, 357–368. [Google Scholar] [CrossRef]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory Monocytes Recruited after Skeletal Muscle Injury Switch into Antiinflammatory Macrophages to Support Myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef]
- Roy, S.; Khanna, S.; Nallu, K.; Hunt, T.K.; Sen, C.K. Dermal Wound Healing Is Subject to Redox Control. Mol. Ther. 2006, 13, 211–220. [Google Scholar] [CrossRef]
- Bosurgi, L.; Cao, Y.G.; Cabeza-Cabrerizo, M.; Tucci, A.; Hughes, L.D.; Kong, Y.; Weinstein, J.S.; Licona-Limon, P.; Schmid, E.T.; Pelorosso, F.; et al. Macrophage Function in Tissue Repair and Remodeling Requires IL-4 or IL-13 with Apoptotic Cells. Science 2017, 356, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective Depletion of Macrophages Reveals Distinct, Opposing Roles during Liver Injury and Repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, G.S.; Cooper, D.M.; Knighton, D.R.; Margolis, D.J.; Pecoraro, R.E.; Rodeheaver, G.; Robson, M.C. Definitions and Guidelines for Assessment of Wounds and Evaluation of Healing. Arch. Dermatol. 1994, 130, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Gallelli, L.; Butrico, L.; Buffone, G.; Caliò, F.G.; De Caridi, G.; Massara, M.; Barbetta, A.; Amato, B.; Labonia, M.; et al. From Varices to Venous Ulceration: The Story of Chronic Venous Disease Described by Metalloproteinases. Int. Wound J. 2017, 14, 233–240. [Google Scholar] [CrossRef]
- de Franciscis, S.; Gallelli, L.; Amato, B.; Butrico, L.; Rossi, A.; Buffone, G.; Caliò, F.G.; De Caridi, G.; Grande, R.; Serra, R. Plasma MMP and TIMP Evaluation in Patients with Deep Venous Thrombosis: Could They Have a Predictive Role in the Development of Post-Thrombotic Syndrome? Int. Wound J. 2016, 13, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Z.; Yao, B.; Yang, S.; Jiang, L.; Wang, S.; Fan, X.; Yin, H.; Wong, K.; Miyazawa, T.; Chen, J.; et al. CSF-1 Signaling Mediates Recovery from Acute Kidney Injury. J. Clin. Investig. 2012, 122, 4519–4532. [Google Scholar] [CrossRef]
- Minutti, C.M.; Knipper, J.A.; Allen, J.E.; Zaiss, D.M.W. Tissue-Specific Contribution of Macrophages to Wound Healing. Semin. Cell Dev. Biol. 2017, 61, 3–11. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth Factors and Cytokines in Wound Healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Resolution of Liver Fibrosis: Basic Mechanisms and Clinical Relevance. Semin. Liver. Dis. 2015, 35, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Rodero, M.P.; Khosrotehrani, K. Skin Wound Healing Modulation by Macrophages. Int. J. Clin. Exp. Pathol 2010, 3, 643–653. [Google Scholar] [PubMed]
- Novak, M.L.; Koh, T.J. Phenotypic Transitions of Macrophages Orchestrate Tissue Repair. Am. J. Pathol. 2013, 183, 1352–1363. [Google Scholar] [CrossRef]
- Lech, M.; Anders, H.-J. Macrophages and Fibrosis: How Resident and Infiltrating Mononuclear Phagocytes Orchestrate All Phases of Tissue Injury and Repair. Biochim. Biophys. Acta 2013, 1832, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.E.; García, A.J. Macrophage Phenotypes in Tissue Repair and the Foreign Body Response: Implications for Biomaterial-Based Regenerative Medicine Strategies. Acta Biomater. 2021, 133, 4–16. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Nair, M.G. Macrophages in Wound Healing: Activation and Plasticity. Immunol. Cell Biol. 2019, 97, 258–267. [Google Scholar] [CrossRef]
- Sen, C.K.; Roy, S. Redox Signals in Wound Healing. Biochim. Biophys. Acta 2008, 1780, 1348–1361. [Google Scholar] [CrossRef]
- Ojha, N.; Roy, S.; He, G.; Biswas, S.; Velayutham, M.; Khanna, S.; Kuppusamy, P.; Zweier, J.L.; Sen, C.K. Assessment of Wound-Site Redox Environment and the Significance of Rac2 in Cutaneous Healing. Free Radic. Biol. Med. 2008, 44, 682–691. [Google Scholar] [CrossRef]
- Hattori, H.; Subramanian, K.K.; Sakai, J.; Jia, Y.; Li, Y.; Porter, T.F.; Loison, F.; Sarraj, B.; Kasorn, A.; Jo, H.; et al. Small-Molecule Screen Identifies Reactive Oxygen Species as Key Regulators of Neutrophil Chemotaxis. Proc. Natl. Acad. Sci. USA 2010, 107, 3546–3551. [Google Scholar] [CrossRef]
- Schafer, M.; Werner, S. Oxidative Stress in Normal and Impaired Wound Repair. Pharmacol. Res. 2008, 58, 165–171. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Hakami, N.Y.; Dusting, G.J.; Chan, E.C.; Shah, M.H.; Peshavariya, H.M. Wound Healing After Alkali Burn Injury of the Cornea Involves Nox4-Type NADPH Oxidase. Investig. Ophthalmol. Vis. Sci 2020, 61, 20. [Google Scholar] [CrossRef]
- Sen, C.K.; Khanna, S.; Babior, B.M.; Hunt, T.K.; Ellison, E.C.; Roy, S. Oxidant-Induced Vascular Endothelial Growth Factor Expression in Human Keratinocytes and Cutaneous Wound Healing. J. Biol. Chem. 2002, 277, 33284–33290. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, C.K.; Trocme, S.D.; Ansari, N.H. Acceleration of Corneal Wound Healing in Diabetic Rats by the Antioxidant Trolox. Res. Commun. Mol. Pathol. Pharmacol. 1996, 93, 3–12. [Google Scholar] [PubMed]
- Shukla, A.; Rasik, A.M.; Patnaik, G.K. Depletion of Reduced Glutathione, Ascorbic Acid, Vitamin E and Antioxidant Defence Enzymes in a Healing Cutaneous Wound. Free Radic. Res. 1997, 26, 93–101. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, D.H.; Ash, K.; Lord, J.; Newman, J.; Zukowski, M. Accelerated Laser Resurfacing Wound Healing Using a Triad of Topical Antioxidants. Dermatol. Surg. 1998, 24, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Bilgen, F.; Ural, A.; Kurutas, E.B.; Bekerecioglu, M. The Effect of Oxidative Stress and Raftlin Levels on Wound Healing. Int. Wound J. 2019, 16, 1178–1184. [Google Scholar] [CrossRef]
- Feng, C.; Yu, B.; Song, C.; Wang, J.; Zhang, L.; Ji, X.; Wang, Y.; Fang, Y.; Liao, Z.; Wei, M.; et al. Static Magnetic Fields Reduce Oxidative Stress to Improve Wound Healing and Alleviate Diabetic Complications. Cells 2022, 11, 443. [Google Scholar] [CrossRef]
- Schilrreff, P.; Alexiev, U. Chronic Inflammation in Non-Healing Skin Wounds and Promising Natural Bioactive Compounds Treatment. Int. J. Mol. Sci. 2022, 23, 4928. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, L.; Xiong, Y.; Panayi, A.C.; Abududilibaier, A.; Hu, Y.; Yu, C.; Zhou, W.; Sun, Y.; Liu, M.; et al. Antioxidant Therapy and Antioxidant-Related Bionanomaterials in Diabetic Wound Healing. Front. Bioeng. Biotechnol. 2021, 9, 707479. [Google Scholar] [CrossRef] [PubMed]
- Aviello, G.; Knaus, U.G. ROS in Gastrointestinal Inflammation: Rescue Or Sabotage? Br. J. Pharmacol. 2017, 174, 1704–1718. [Google Scholar] [CrossRef] [PubMed]
- Rasik, A.M.; Shukla, A. Antioxidant Status in Delayed Healing Type of Wounds: Delayed Healing Wounds and Antioxidants. Int. J. Exp. Pathol. 2001, 81, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Dworzański, J.; Strycharz-Dudziak, M.; Kliszczewska, E.; Kiełczykowska, M.; Dworzańska, A.; Drop, B.; Polz-Dacewicz, M. Glutathione Peroxidase (GPx) and Superoxide Dismutase (SOD) Activity in Patients with Diabetes Mellitus Type 2 Infected with Epstein-Barr Virus. PLoS ONE 2020, 15, e0230374. [Google Scholar] [CrossRef]
- Prasad, A.; Manoharan, R.R.; Sedlářová, M.; Pospíšil, P. Free Radical-Mediated Protein Radical Formation in Differentiating Monocytes. Int. J. Mol. Sci 2021, 22, 9963. [Google Scholar] [CrossRef]
- Helfinger, V.; Palfi, K.; Weigert, A.; Schröder, K. The NADPH Oxidase Nox4 Controls Macrophage Polarization in an NFκB-Dependent Manner. Oxid. Med. Cell. Longev. 2019, 2019, 3264858. [Google Scholar] [CrossRef]
- Lee, C.F.; Qiao, M.; Schröder, K.; Zhao, Q.; Asmis, R. Nox4 Is a Novel Inducible Source of Reactive Oxygen Species in Monocytes and Macrophages and Mediates Oxidized Low Density Lipoprotein-Induced Macrophage Death. Circ. Res. 2010, 106, 1489–1497. [Google Scholar] [CrossRef]
- Xu, Q.; Choksi, S.; Qu, J.; Jang, J.; Choe, M.; Banfi, B.; Engelhardt, J.F.; Liu, Z.-G. NADPH Oxidases Are Essential for Macrophage Differentiation. J. Biol. Chem. 2016, 291, 20030–20041. [Google Scholar] [CrossRef]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.-G. ROS Play a Critical Role in the Differentiation of Alternatively Activated Macrophages and the Occurrence of Tumor-Associated Macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef]
- Griess, B.; Mir, S.; Datta, K.; Teoh-Fitzgerald, M. Scavenging Reactive Oxygen Species Selectively Inhibits M2 Macrophage Polarization and Their Pro-Tumorigenic Function in Part, via Stat3 Suppression. Free Radic. Biol. Med. 2020, 147, 48–60. [Google Scholar] [CrossRef]
- Tan, H.-Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 2795090. [Google Scholar] [CrossRef] [PubMed]
- Nassif, R.M.; Chalhoub, E.; Chedid, P.; Hurtado-Nedelec, M.; Raya, E.; Dang, P.M.-C.; Marie, J.-C.; El-Benna, J. Metformin Inhibits ROS Production by Human M2 Macrophages via the Activation of AMPK. Biomedicines 2022, 10, 319. [Google Scholar] [CrossRef] [PubMed]
- Sanmun, D.; Witasp, E.; Jitkaew, S.; Tyurina, Y.Y.; Kagan, V.E.; Åhlin, A.; Palmblad, J.; Fadeel, B. Involvement of a Functional NADPH Oxidase in Neutrophils and Macrophages during Programmed Cell Clearance: Implications for Chronic Granulomatous Disease. Am. J. Physiol.-Cell Physiol. 2009, 297, C621–C631. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Christenson, K.; Karlsson, A.; Dahlgren, C.; Bylund, J. Divergent Effects on Phagocytosis by Macrophage-Derived Oxygen Radicals. J. Innate Immun. 2009, 1, 592–598. [Google Scholar] [CrossRef]
- Kumar, A.; Barrett, J.P.; Alvarez-Croda, D.-M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. NOX2 Drives M1-like Microglial/Macrophage Activation and Neurodegeneration Following Experimental Traumatic Brain Injury. Brain Behav. Immun. 2016, 58, 291–309. [Google Scholar] [CrossRef]
- Balce, D.R.; Li, B.; Allan, E.R.O.; Rybicka, J.M.; Krohn, R.M.; Yates, R.M. Alternative Activation of Macrophages by IL-4 Enhances the Proteolytic Capacity of Their Phagosomes through Synergistic Mechanisms. Blood 2011, 118, 4199–4208. [Google Scholar] [CrossRef]
- Han, C.; Sheng, Y.; Wang, J.; Zhou, X.; Li, W.; Zhang, C.; Guo, L.; Yang, Y. NOX4 Promotes Mucosal Barrier Injury in Inflammatory Bowel Disease by Mediating Macrophages M1 Polarization through ROS. Int. Immunopharmacol. 2022, 104, 108361. [Google Scholar] [CrossRef]
- Lokmic, Z.; Musyoka, J.; Hewitson, T.D.; Darby, I.A. Hypoxia and Hypoxia Signaling in Tissue Repair and Fibrosis. Int. Rev. Cell Mol. Biol. 2012, 296, 139–185. [Google Scholar] [CrossRef]
- Smith, K.A.; Waypa, G.B.; Schumacker, P.T. Redox Signaling during Hypoxia in Mammalian Cells. Redox. Biol. 2017, 13, 228–234. [Google Scholar] [CrossRef]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Finkel, T. Signal Transduction by Mitochondrial Oxidants. J. Biol. Chem. 2012, 287, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 and Mechanisms of Hypoxia Sensing. Curr. Opin. Cell Biol. 2001, 13, 167–171. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive Oxygen Species Generated at Mitochondrial Complex III Stabilize Hypoxia-Inducible Factor-1α during Hypoxia. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Wang, D.; Malo, D.; Hekimi, S. Elevated Mitochondrial Reactive Oxygen Species Generation Affects the Immune Response via Hypoxia-Inducible Factor-1alpha in Long-Lived Mclk1+/− Mouse Mutants. J. Immunol. 2010, 184, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Nishi, K.; Oda, T.; Takabuchi, S.; Oda, S.; Fukuda, K.; Adachi, T.; Semenza, G.L.; Shingu, K.; Hirota, K. LPS Induces Hypoxia-Inducible Factor 1 Activation in Macrophage-Differentiated Cells in a Reactive Oxygen Species-Dependent Manner. Antioxid. Redox. Signal. 2008, 10, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Y.; Li, Y.; Yu, Q.; Jin, X.; Wang, X.; Jia, A.; Hu, Y.; Han, L.; Wang, J.; et al. HIF1α-Dependent Glycolysis Promotes Macrophage Functional Activities in Protecting against Bacterial and Fungal Infection. Sci. Rep. 2018, 8, 3603. [Google Scholar] [CrossRef] [PubMed]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1α Is Essential for Myeloid Cell-Mediated Inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef]
- Kerber, E.L.; Padberg, C.; Koll, N.; Schuetzhold, V.; Fandrey, J.; Winning, S. The Importance of Hypoxia-Inducible Factors (HIF-1 and HIF-2) for the Pathophysiology of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2020, 21, 8551. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared Principles in NF-KappaB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Lawrence, T.; Fong, C. The Resolution of Inflammation: Anti-Inflammatory Roles for NF-KappaB. Int. J. Biochem. Cell Biol. 2010, 42, 519–523. [Google Scholar] [CrossRef]
- Müller, C.W.; Harrison, S.C. The Structure of the NF-Kappa B P50:DNA-Complex: A Starting Point for Analyzing the Rel Family. FEBS Lett. 1995, 369, 113–117. [Google Scholar] [CrossRef]
- Bonizzi, G.; Karin, M. The Two NF-KappaB Activation Pathways and Their Role in Innate and Adaptive Immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Roux, C.; Jafari, S.M.; Shinde, R.; Duncan, G.; Cescon, D.W.; Silvester, J.; Chu, M.F.; Hodgson, K.; Berger, T.; Wakeham, A.; et al. Reactive Oxygen Species Modulate Macrophage Immunosuppressive Phenotype through the Up-Regulation of PD-L1. Proc. Natl. Acad. Sci. USA 2019, 116, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Formentini, L.; Santacatterina, F.; Núñez de Arenas, C.; Stamatakis, K.; López-Martínez, D.; Logan, A.; Fresno, M.; Smits, R.; Murphy, M.P.; Cuezva, J.M. Mitochondrial ROS Production Protects the Intestine from Inflammation through Functional M2 Macrophage Polarization. Cell Rep. 2017, 19, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Guo, Y.; Cheng, Y.; Zhao, J.; Wang, Y.; Rong, J. Natural Product Celastrol Suppressed Macrophage M1 Polarization against Inflammation in Diet-Induced Obese Mice via Regulating Nrf2/HO-1, MAP Kinase and NF-ΚB Pathways. Aging 2017, 9, 2069–2082. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Li, W.; Kong, A.-N. Molecular Mechanisms of Nrf2-Mediated Antioxidant Response. Mol. Carcinog. 2009, 48, 91–104. [Google Scholar] [CrossRef]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural Basis of Keap1 Interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef]
- Piotrowska, M.; Swierczynski, M.; Fichna, J.; Piechota-Polanczyk, A. The Nrf2 in the Pathophysiology of the Intestine: Molecular Mechanisms and Therapeutic Implications for Inflammatory Bowel Diseases. Pharmacol. Res. 2021, 163, 105243. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 Regulates ROS Production by Mitochondria and NADPH Oxidase. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 794–801. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 Suppresses Macrophage Inflammatory Response by Blocking Proinflammatory Cytokine Transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Li, C.; Cheng, N.; Cui, X.; Xu, X.; Zhou, G. Redox Regulation in Cancer Stem Cells. Oxid. Med. Cell. Longev. 2015, 2015, 750798. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, H.; Pan, H.; Zhou, X.; Ruan, Q.; Kong, D.; Chu, Z.; Li, H.; Huang, J.; Huang, X.; et al. Nrf2 Suppression Delays Diabetic Wound Healing Through Sustained Oxidative Stress and Inflammation. Front. Pharmacol. 2019, 10, 1099. [Google Scholar] [CrossRef] [PubMed]
- Weis, N.; Weigert, A.; von Knethen, A.; Brüne, B. Heme Oxygenase-1 Contributes to an Alternative Macrophage Activation Profile Induced by Apoptotic Cell Supernatants. MBoC 2009, 20, 1280–1288. [Google Scholar] [CrossRef]
- Khor, T.O.; Huang, M.-T.; Kwon, K.H.; Chan, J.Y.; Reddy, B.S.; Kong, A.-N. Nrf2-Deficient Mice Have an Increased Susceptibility to Dextran Sulfate Sodium-Induced Colitis. Cancer Res. 2006, 66, 11580–11584. [Google Scholar] [CrossRef]
- Osburn, W.O.; Karim, B.; Dolan, P.M.; Liu, G.; Yamamoto, M.; Huso, D.L.; Kensler, T.W. Increased Colonic Inflammatory Injury and Formation of Aberrant Crypt Foci in Nrf2-Deficient Mice upon Dextran Sulfate Treatment. Int. J. Cancer 2007, 121, 1883–1891. [Google Scholar] [CrossRef]
- Feng, R.; Morine, Y.; Ikemoto, T.; Imura, S.; Iwahashi, S.; Saito, Y.; Shimada, M. Nrf2 Activation Drive Macrophages Polarization and Cancer Cell Epithelial-Mesenchymal Transition during Interaction. Cell Commun. Signal. 2018, 16, 54. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Mukherjee, S.; Choudhury, S.; Gupta, P.; Adhikary, A.; Baral, R.; Chattopadhyay, S. Reactive Oxygen Species in the Tumor Niche Triggers Altered Activation of Macrophages and Immunosuppression: Role of Fluoxetine. Cell Signal. 2015, 27, 1398–1412. [Google Scholar] [CrossRef]
- Zhuyan, J.; Chen, M.; Zhu, T.; Bao, X.; Zhen, T.; Xing, K.; Wang, Q.; Zhu, S. Critical Steps to Tumor Metastasis: Alterations of Tumor Microenvironment and Extracellular Matrix in the Formation of Pre-Metastatic and Metastatic Niche. Cell Biosci. 2020, 10, 89. [Google Scholar] [CrossRef]
- Catalano, V.; Turdo, A.; Di Franco, S.; Dieli, F.; Todaro, M.; Stassi, G. Tumor and Its Microenvironment: A Synergistic Interplay. Semin. Cancer Biol. 2013, 23, 522–532. [Google Scholar] [CrossRef]
- Kennel, K.B.; Greten, F.R. Immune Cell–Produced ROS and Their Impact on Tumor Growth and Metastasis. Redox Biol. 2021, 42, 101891. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Lotfi, R.; Eisenbacher, J.; Solgi, G.; Fuchs, K.; Yildiz, T.; Nienhaus, C.; Rojewski, M.T.; Schrezenmeier, H. Human Mesenchymal Stem Cells Respond to Native but Not Oxidized Damage Associated Molecular Pattern Molecules from Necrotic (Tumor) Material. Eur. J. Immunol. 2011, 41, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- van Beijnum, J.R.; Nowak-Sliwinska, P.; van den Boezem, E.; Hautvast, P.; Buurman, W.A.; Griffioen, A.W. Tumor Angiogenesis Is Enforced by Autocrine Regulation of High-Mobility Group Box 1. Oncogene 2013, 32, 363–374. [Google Scholar] [CrossRef]
- Patidar, A.; Selvaraj, S.; Sarode, A.; Chauhan, P.; Chattopadhyay, D.; Saha, B. DAMP-TLR-Cytokine Axis Dictates the Fate of Tumor. Cytokine 2018, 104, 114–123. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the Extracellular Matrix: Drivers of Tumour Metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef]
- Sakamoto, S.; Kyprianou, N. Targeting Anoikis Resistance in Prostate Cancer Metastasis. Mol. Aspects. Med. 2010, 31, 205–214. [Google Scholar] [CrossRef]
- Alizadeh, A.M.; Shiri, S.; Farsinejad, S. Metastasis Review: From Bench to Bedside. Tumour. Biol. 2014, 35, 8483–8523. [Google Scholar] [CrossRef]
- Wang, S.-S.; Jiang, J.; Liang, X.-H.; Tang, Y.-L. Links between Cancer Stem Cells and Epithelial-Mesenchymal Transition. Onco Targets Ther. 2015, 8, 2973–2980. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Sorolla, M.A.; Hidalgo, I.; Sorolla, A.; Montal, R.; Pallisé, O.; Salud, A.; Parisi, E. Microenvironmental Reactive Oxygen Species in Colorectal Cancer: Involved Processes and Therapeutic Opportunities. Cancers 2021, 13, 5037. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G. Targeting Extracellular ROS Signaling of Tumor Cells. Anticancer Res. 2014, 34, 1467–1482. [Google Scholar] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive Oxygen Species in Cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Assi, M. The Differential Role of Reactive Oxygen Species in Early and Late Stages of Cancer. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R646–R653. [Google Scholar] [CrossRef]
- Spitz, D.R.; Sim, J.E.; Ridnour, L.A.; Galoforo, S.S.; Lee, Y.J. Glucose Deprivation-Induced Oxidative Stress in Human Tumor Cells. A Fundamental Defect in Metabolism? Ann. N. Y. Acad. Sci. 2000, 899, 349–362. [Google Scholar] [CrossRef]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of Vascular Endothelial Growth Factor Gene Transcription by Hypoxia-Inducible Factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef]
- Han, X.; Sun, S.; Zhao, M.; Cheng, X.; Chen, G.; Lin, S.; Guan, Y.; Yu, X. Celastrol Stimulates Hypoxia-Inducible Factor-1 Activity in Tumor Cells by Initiating the ROS/Akt/P70S6K Signaling Pathway and Enhancing Hypoxia-Inducible Factor-1α Protein Synthesis. PLoS ONE 2014, 9, e112470. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/MTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef]
- Xia, C.; Meng, Q.; Liu, L.-Z.; Rojanasakul, Y.; Wang, X.-R.; Jiang, B.-H. Reactive Oxygen Species Regulate Angiogenesis and Tumor Growth through Vascular Endothelial Growth Factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef]
- Liu, L.-Z.; Hu, X.-W.; Xia, C.; He, J.; Zhou, Q.; Shi, X.; Fang, J.; Jiang, B.-H. Reactive Oxygen Species Regulate Epidermal Growth Factor-Induced Vascular Endothelial Growth Factor and Hypoxia-Inducible Factor-1alpha Expression through Activation of AKT and P70S6K1 in Human Ovarian Cancer Cells. Free Radic. Biol. Med. 2006, 41, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- West, X.Z.; Malinin, N.L.; Merkulova, A.A.; Tischenko, M.; Kerr, B.A.; Borden, E.C.; Podrez, E.A.; Salomon, R.G.; Byzova, T.V. Oxidative Stress Induces Angiogenesis by Activating TLR2 with Novel Endogenous Ligands. Nature 2010, 467, 972–976. [Google Scholar] [CrossRef]
- Wartenberg, M.; Budde, P.; De Mareés, M.; Grünheck, F.; Tsang, S.Y.; Huang, Y.; Chen, Z.-Y.; Hescheler, J.; Sauer, H. Inhibition of Tumor-Induced Angiogenesis and Matrix-Metalloproteinase Expression in Confrontation Cultures of Embryoid Bodies and Tumor Spheroids by Plant Ingredients Used in Traditional Chinese Medicine. Lab. Investig. 2003, 83, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, H.E.; Casterline, B.W.; Rada, B.; Korzeniowska, A.; Leto, T.L. Nox4 Involvement in TGF-Beta and SMAD3-Driven Induction of the Epithelial-to-Mesenchymal Transition and Migration of Breast Epithelial Cells. Free Radic. Biol. Med. 2012, 53, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, K.; Matsuno, Y.; Yoshida, K.; Sherpa, M.; Nakajima, M.; Matsuyama, M.; Kiwamoto, T.; Morishima, Y.; Ishii, Y.; Hizawa, N. ROS-Nrf2 Pathway Mediates the Development of TGF-Β1-Induced Epithelial-Mesenchymal Transition through the Activation of Notch Signaling. Eur. J. Cell Biol. 2021, 100, 151181. [Google Scholar] [CrossRef]
- Padua, D.; Massagué, J. Roles of TGFbeta in Metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Diebold, I.; Petry, A.; Hess, J.; Görlach, A. The NADPH Oxidase Subunit NOX4 Is a New Target Gene of the Hypoxia-Inducible Factor-1. Mol. Biol. Cell 2010, 21, 2087–2096. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, Z.; Hu, X. Inhibiting Cancer Metastasis via Targeting NAPDH Oxidase 4. Biochem. Pharmacol. 2013, 86, 253–266. [Google Scholar] [CrossRef]
- Du, S.; Miao, J.; Zhu, Z.; Xu, E.; Shi, L.; Ai, S.; Wang, F.; Kang, X.; Chen, H.; Lu, X.; et al. NADPH Oxidase 4 Regulates Anoikis Resistance of Gastric Cancer Cells through the Generation of Reactive Oxygen Species and the Induction of EGFR. Cell Death Dis. 2018, 9, 948. [Google Scholar] [CrossRef]
- Kim, H.; Sung, J.Y.; Park, E.-K.; Kho, S.; Koo, K.H.; Park, S.-Y.; Goh, S.-H.; Jeon, Y.K.; Oh, S.; Park, B.-K.; et al. Regulation of Anoikis Resistance by NADPH Oxidase 4 and Epidermal Growth Factor Receptor. Br. J. Cancer 2017, 116, 370–381. [Google Scholar] [CrossRef]
- Binker, M.G.; Binker-Cosen, A.A.; Richards, D.; Oliver, B.; Cosen-Binker, L.I. EGF Promotes Invasion by PANC-1 Cells through Rac1/ROS-Dependent Secretion and Activation of MMP-2. Biochem. Biophys. Res. Commun. 2009, 379, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Tobar, N.; Villar, V.; Santibanez, J.F. ROS-NFkappaB Mediates TGF-Beta1-Induced Expression of Urokinase-Type Plasminogen Activator, Matrix Metalloproteinase-9 and Cell Invasion. Mol. Cell. Biochem. 2010, 340, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Basak, D.; Uddin, M.N.; Hancock, J. The Role of Oxidative Stress and Its Counteractive Utility in Colorectal Cancer (CRC). Cancers 2020, 12, 3336. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer Stem Cell Metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Dando, I.; Cordani, M.; Dalla Pozza, E.; Biondani, G.; Donadelli, M.; Palmieri, M. Antioxidant Mechanisms and ROS-Related MicroRNAs in Cancer Stem Cells. Oxid. Med. Cell. Longev. 2015, 2015, 425708. [Google Scholar] [CrossRef]
- Sato, A.; Okada, M.; Shibuya, K.; Watanabe, E.; Seino, S.; Narita, Y.; Shibui, S.; Kayama, T.; Kitanaka, C. Pivotal Role for ROS Activation of P38 MAPK in the Control of Differentiation and Tumor-Initiating Capacity of Glioma-Initiating Cells. Stem Cell Res. 2014, 12, 119–131. [Google Scholar] [CrossRef]
- Cao, Y.; Fang, Y.; Cai, J.; Li, X.; Xu, F.; Yuan, N.; Zhang, S.; Wang, J. ROS Functions as an Upstream Trigger for Autophagy to Drive Hematopoietic Stem Cell Differentiation. Hematology 2016, 21, 613–618. [Google Scholar] [CrossRef]
- Singel, K.L.; Segal, B.H. Neutrophils in the Tumor Microenvironment: Trying to Heal the Wound That Cannot Heal. Immunol. Rev. 2016, 273, 329–343. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A. Macrophages, Innate Immunity and Cancer: Balance, Tolerance, and Diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef]
- Kotsafti, A.; Scarpa, M.; Castagliuolo, I.; Scarpa, M. Reactive Oxygen Species and Antitumor Immunity-From Surveillance to Evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef]
- Wang, H.; Tian, T.; Zhang, J. Tumor-Associated Macrophages (TAMs) in Colorectal Cancer (CRC): From Mechanism to Therapy and Prognosis. Int. J. Mol. Sci 2021, 22, 8470. [Google Scholar] [CrossRef] [PubMed]
- Ryder, M.; Ghossein, R.A.; Ricarte-Filho, J.C.M.; Knauf, J.A.; Fagin, J.A. Increased Density of Tumor-Associated Macrophages Is Associated with Decreased Survival in Advanced Thyroid Cancer. Endocr. Relat. Cancer 2008, 15, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-Associated Microglia/Macrophages Predict Poor Prognosis in High-Grade Gliomas and Correlate with an Aggressive Tumour Subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Hwang, I.; Kang, S.H.; Shin, H.C.; Kwon, S.Y. Tumor-Associated Macrophages as Potential Prognostic Biomarkers of Invasive Breast Cancer. J. Breast Cancer 2019, 22, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Que, K.-T.; Zhang, Z.; Yi, Z.J.; Zhao, P.X.; You, Y.; Gong, J.-P.; Liu, Z.-J. Iron Overloaded Polarizes Macrophage to Proinflammation Phenotype through ROS/Acetyl-P53 Pathway. Cancer Med. 2018, 7, 4012–4022. [Google Scholar] [CrossRef]
- Wu, Q.; Allouch, A.; Paoletti, A.; Leteur, C.; Mirjolet, C.; Martins, I.; Voisin, L.; Law, F.; Dakhli, H.; Mintet, E.; et al. NOX2-Dependent ATM Kinase Activation Dictates pro-Inflammatory Macrophage Phenotype and Improves Effectiveness to Radiation Therapy. Cell Death Differ. 2017, 24, 1632–1644. [Google Scholar] [CrossRef]
- Mantovani, A.; Locati, M. Tumor-Associated Macrophages as a Paradigm of Macrophage Plasticity, Diversity, and Polarization: Lessons and Open Questions. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1478–1483. [Google Scholar] [CrossRef]
- Chen, L.-M.; Tseng, H.-Y.; Chen, Y.-A.; Al Haq, A.T.; Hwang, P.-A.; Hsu, H.-L. Oligo-Fucoidan Prevents M2 Macrophage Differentiation and HCT116 Tumor Progression. Cancers 2020, 12, 421. [Google Scholar] [CrossRef]
- Salpeter, S.J.; Pozniak, Y.; Merquiol, E.; Ben-Nun, Y.; Geiger, T.; Blum, G. A Novel Cysteine Cathepsin Inhibitor Yields Macrophage Cell Death and Mammary Tumor Regression. Oncogene 2015, 34, 6066–6078. [Google Scholar] [CrossRef]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of Tumor Associated Macrophages in Tumor Angiogenesis and Lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef]
- Luput, L.; Licarete, E.; Sesarman, A.; Patras, L.; Alupei, M.C.; Banciu, M. Tumor-Associated Macrophages Favor C26 Murine Colon Carcinoma Cell Proliferation in an Oxidative Stress-Dependent Manner. Oncol. Rep. 2017, 37, 2472–2480. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.S.K.; Tan, M.J.; Sng, M.K.; Teo, Z.; Phua, T.; Choo, C.C.; Li, L.; Zhu, P.; Tan, N.S. Cancer-Associated Fibroblasts Enact Field Cancerization by Promoting Extratumoral Oxidative Stress. Cell Death Dis. 2017, 8, e2562. [Google Scholar] [CrossRef] [PubMed]
- Onfroy-Roy, L.; Hamel, D.; Malaquin, L.; Ferrand, A. Colon Fibroblasts and Inflammation: Sparring Partners in Colorectal Cancer Initiation? Cancers 2021, 13, 1749. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative Stress in Cancer Associated Fibroblasts Drives Tumor-Stroma Co-Evolution: A New Paradigm for Understanding Tumor Metabolism, the Field Effect and Genomic Instability in Cancer Cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef]
- Policastro, L.L.; Ibañez, I.L.; Notcovich, C.; Duran, H.A.; Podhajcer, O.L. The Tumor Microenvironment: Characterization, Redox Considerations, and Novel Approaches for Reactive Oxygen Species-Targeted Gene Therapy. Antioxid. Redox Signal. 2013, 19, 854–895. [Google Scholar] [CrossRef]
- Giannoni, E.; Bianchini, F.; Calorini, L.; Chiarugi, P. Cancer Associated Fibroblasts Exploit Reactive Oxygen Species through a Proinflammatory Signature Leading to Epithelial Mesenchymal Transition and Stemness. Antioxid. Redox Signal. 2011, 14, 2361–2371. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-KappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Zhao, H.; Huang, J.; Li, Y.; Lv, X.; Zhou, H.; Wang, H.; Xu, Y.; Wang, C.; Wang, J.; Liu, Z. ROS-Scavenging Hydrogel to Promote Healing of Bacteria Infected Diabetic Wounds. Biomaterials 2020, 258, 120286. [Google Scholar] [CrossRef]
- Konaté, M.M.; Antony, S.; Doroshow, J.H. Inhibiting the Activity of NADPH Oxidase in Cancer. Antioxid. Redox Signal. 2020, 33, 435–454. [Google Scholar] [CrossRef]
- Hadi, T.; Douhard, R.; Dias, A.M.M.; Wendremaire, M.; Pezzè, M.; Bardou, M.; Sagot, P.; Garrido, C.; Lirussi, F. Beta3 Adrenergic Receptor Stimulation in Human Macrophages Inhibits NADPHoxidase Activity and Induces Catalase Expression via PPARγ Activation. Biochim. Biophys. Acta 2017, 1864, 1769–1784. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.T.B.; Araújo-Filho, H.G.; Barreto, A.S.; Quintans-Júnior, L.J.; Quintans, J.S.S.; Barreto, R.S.S. Wound Healing Properties of Flavonoids: A Systematic Review Highlighting the Mechanisms of Action. Phytomedicine 2021, 90, 153636. [Google Scholar] [CrossRef] [PubMed]
- Magrì, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chilà, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S.; et al. High-Dose Vitamin C Enhances Cancer Immunotherapy. Sci. Transl. Med. 2020, 12, eaay8707. [Google Scholar] [CrossRef] [PubMed]
- Luchtel, R.A.; Bhagat, T.; Pradhan, K.; Jacobs, W.R.; Levine, M.; Verma, A.; Shenoy, N. High-Dose Ascorbic Acid Synergizes with Anti-PD1 in a Lymphoma Mouse Model. Proc. Natl. Acad. Sci. USA 2020, 117, 1666–1677. [Google Scholar] [CrossRef] [PubMed]
- Dunnill, C.; Patton, T.; Brennan, J.; Barrett, J.; Dryden, M.; Cooke, J.; Leaper, D.; Georgopoulos, N.T. Reactive Oxygen Species (ROS) and Wound Healing: The Functional Role of ROS and Emerging ROS-Modulating Technologies for Augmentation of the Healing Process. Int. Wound J. 2017, 14, 89–96. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez, T.; Wendremaire, M.; Lagarde, J.; Duquet, O.; Alibert, L.; Paquette, B.; Garrido, C.; Lirussi, F. Wound Healing versus Metastasis: Role of Oxidative Stress. Biomedicines 2022, 10, 2784. https://doi.org/10.3390/biomedicines10112784
Lopez T, Wendremaire M, Lagarde J, Duquet O, Alibert L, Paquette B, Garrido C, Lirussi F. Wound Healing versus Metastasis: Role of Oxidative Stress. Biomedicines. 2022; 10(11):2784. https://doi.org/10.3390/biomedicines10112784
Chicago/Turabian StyleLopez, Tatiana, Maeva Wendremaire, Jimmy Lagarde, Oriane Duquet, Line Alibert, Brice Paquette, Carmen Garrido, and Frédéric Lirussi. 2022. "Wound Healing versus Metastasis: Role of Oxidative Stress" Biomedicines 10, no. 11: 2784. https://doi.org/10.3390/biomedicines10112784
APA StyleLopez, T., Wendremaire, M., Lagarde, J., Duquet, O., Alibert, L., Paquette, B., Garrido, C., & Lirussi, F. (2022). Wound Healing versus Metastasis: Role of Oxidative Stress. Biomedicines, 10(11), 2784. https://doi.org/10.3390/biomedicines10112784