
Genetic Diversity, Linkage Disequilibrium and Population Structure of Bulgarian Bread Wheat Assessed by Genome-Wide Distributed SNP Markers: From Old Germplasm to Semi-Dwarf Cultivars

 ,
,  , ,
, ,  , and
, and 
Abstract
1. Introduction
2. Results
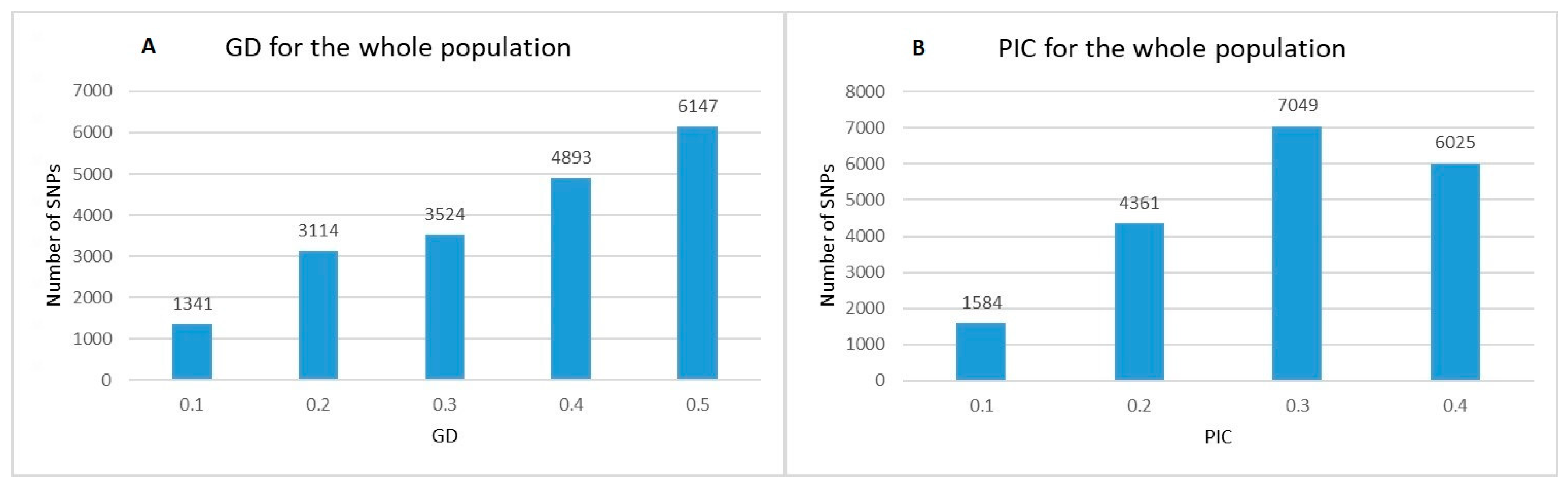
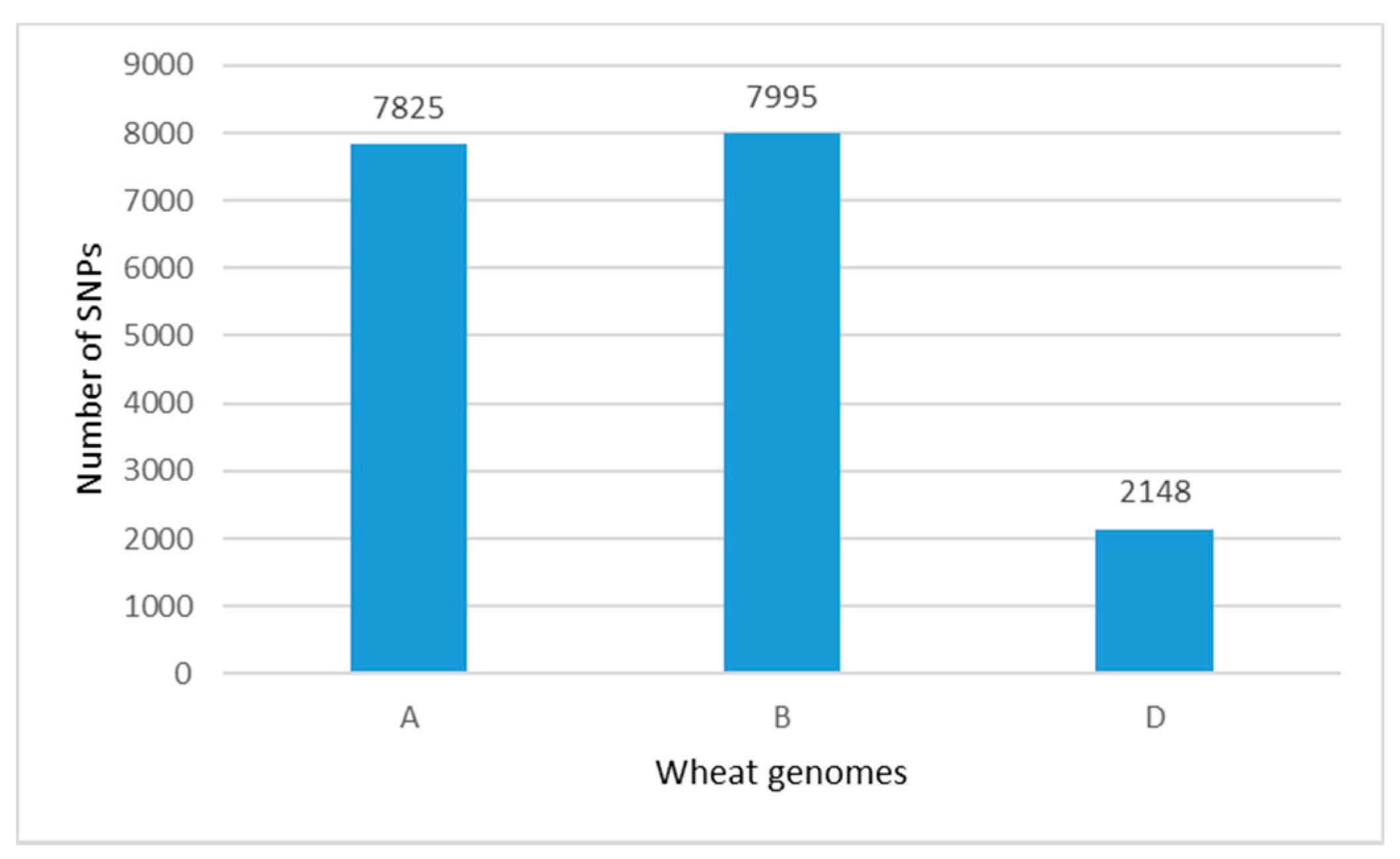
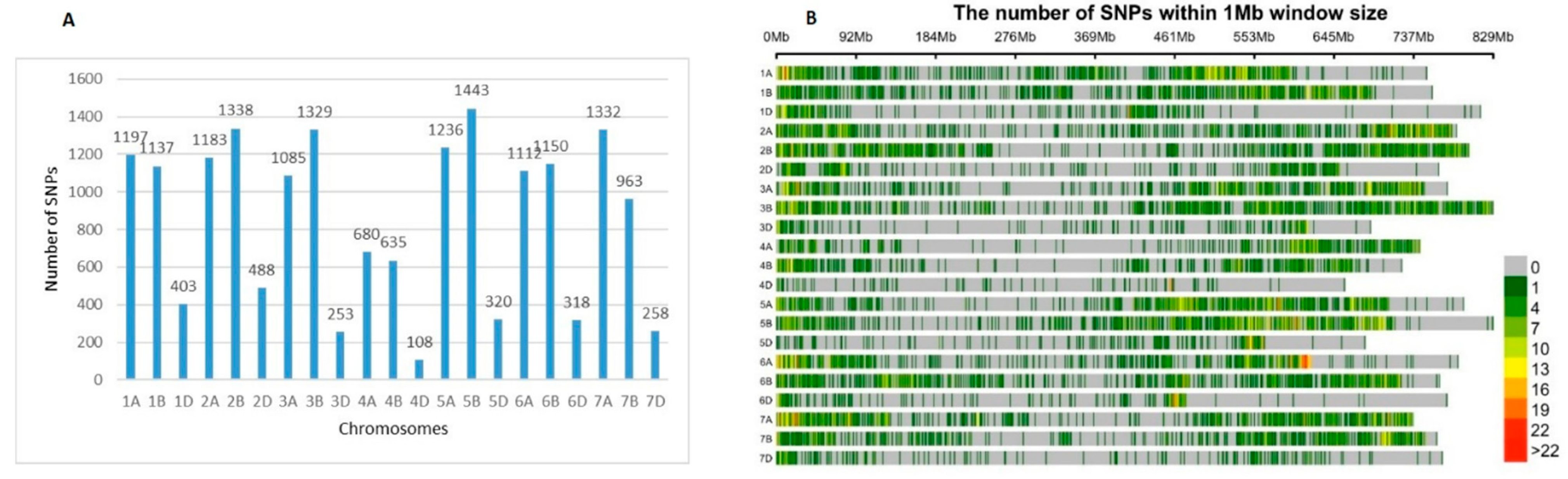
2.1. Genetic Diversity Analysis and SNPs Distribution
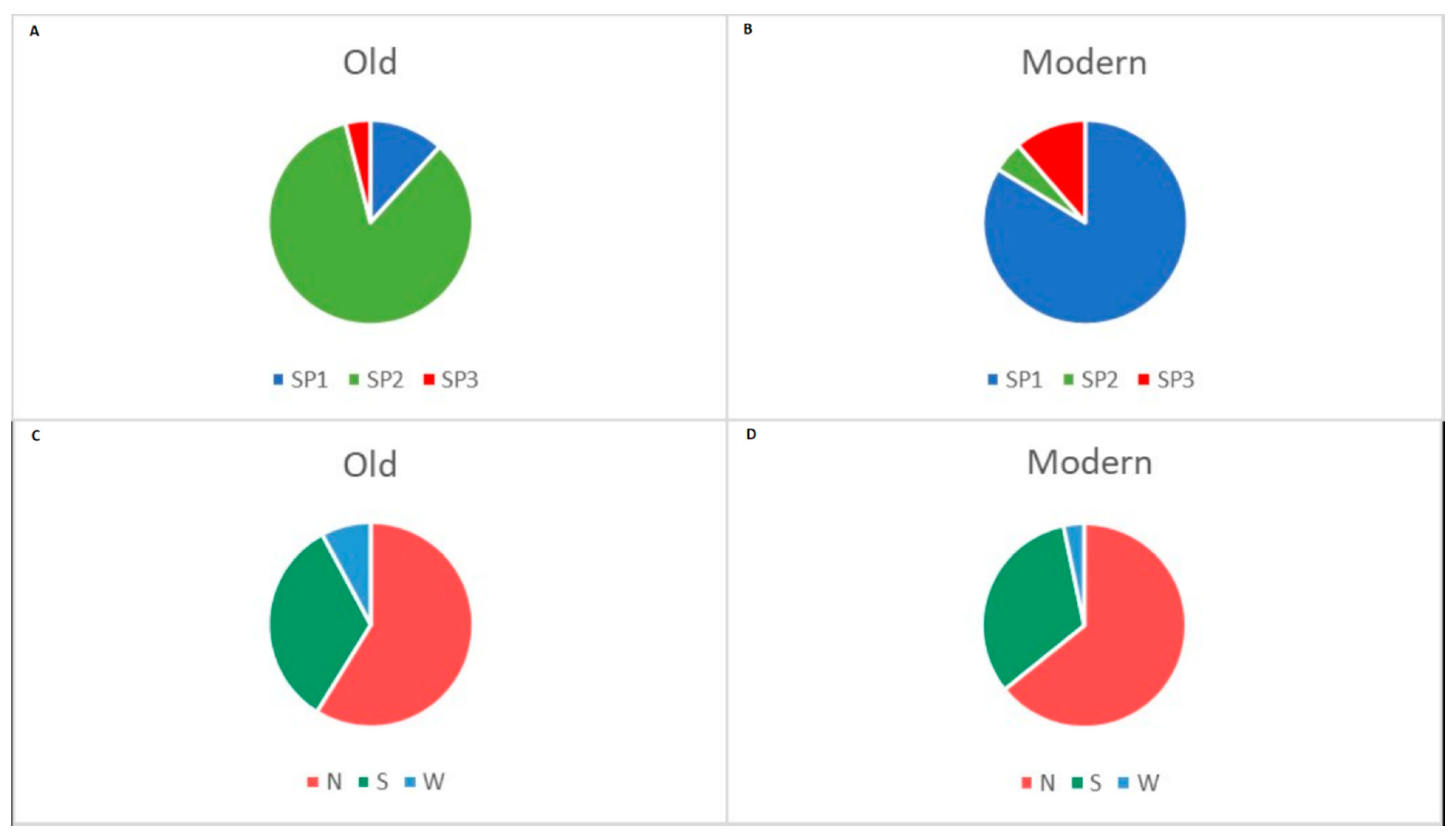
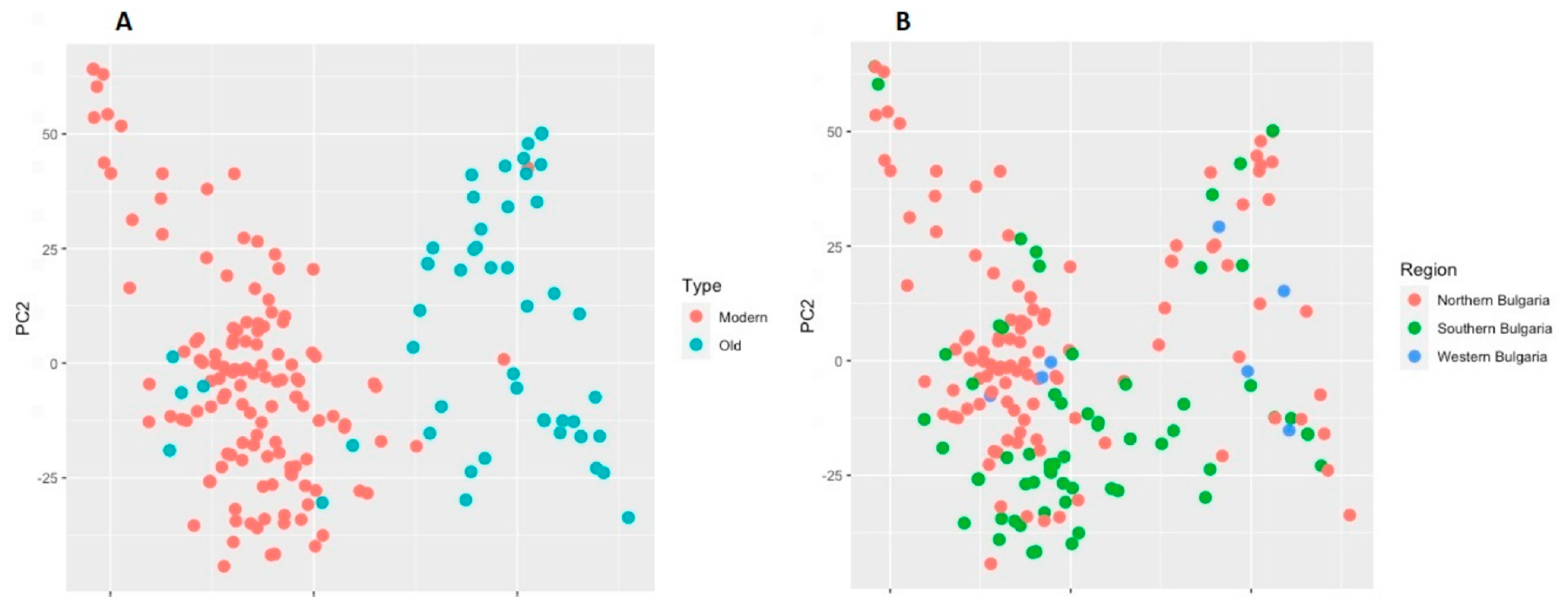
2.2. Differentiation between Old and Modern Germplasm
2.3. Linkage Disequilibrium
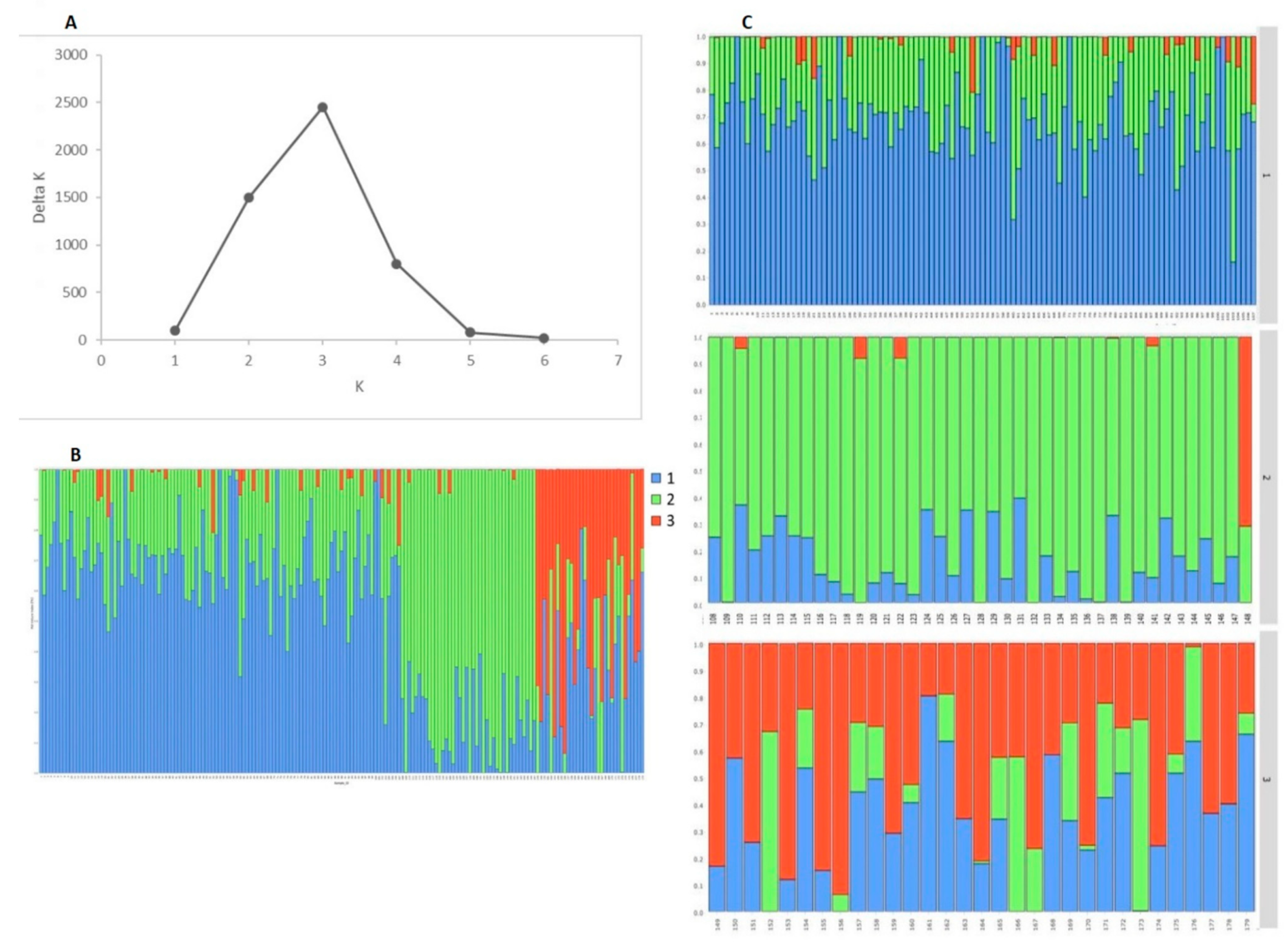
2.4. Population Structure
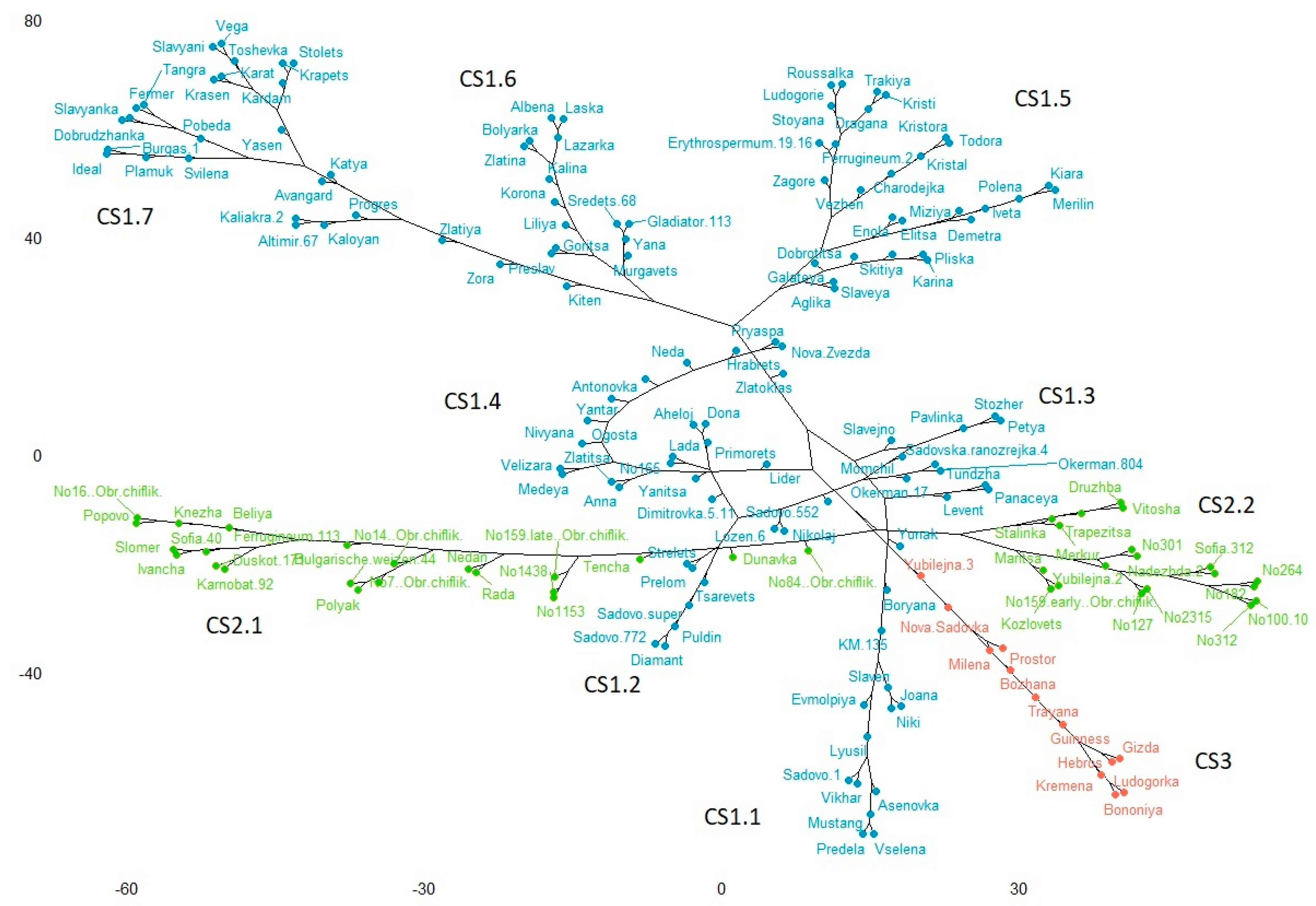
2.5. Cluster Analysis
3. Discussion
3.1. Genetic Diversity and SNPs Distribution
3.2. Linkage Disequilibrium and Population Structure
3.3. Population Differentiation
4. Materials and Methods
4.1. Plant Material
4.2. SNP Genotyping
4.3. Genetic Diversity Analysis
4.4. Population Structure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ganeva, G.; Korzun, V.; Landjeva, S.; Tsenov, N.; Atanasova, M. Identification, distribution and effects of agronomic traits of the semi-dwarfing Rht alleles in Bulgarian bread wheat cultivars. Euphytica 2005, 145, 305–315. [Google Scholar] [CrossRef]
- Kolev, S.; Ganeva, G.; Christov, N.; Belchev, I.; Kostov, K.; Tsenov, N.; Rachovska, G.; Landgeva, S.; Ivanov, M.; Abu-Mhadi, N.; et al. Allele variation in loci for adaptive response and plant height and its effect on grain yield in wheat. Biotechnol. Biotechnol. Equip. 2010, 24, 1807–1813. [Google Scholar] [CrossRef][Green Version]
- Koleva, E.; Alexandrov, V. Drought in the Bulgarian low regions during the 20th century. Theoret. App. Climatol. 2008, 92, 113–120. [Google Scholar] [CrossRef]
- Landjeva, S.; Karceva, T.; Korzun, V.; Ganeva, G. Seedling growth under osmotic stress and agronomic traits in Bulgarian semi-dwarf wheat—comparison of genotypes with Rht8 and/or Rht-B1 genes. Crop Pasture Sci. 2012, 62, 1017–1025. [Google Scholar] [CrossRef]
- Ganeva, G.; Petrova, T.; Landjeva, S.; Todorovska, E.; Kolev, S.; Galiba, G.; Szira, F.; Bálint, A. Frost tolerance in winter wheat (Triticum aestivum L.) cultivars: Differential effects of chromosome 5A and association with microsatellite alleles. Biol. Plant. 2013, 57, 184–188. [Google Scholar] [CrossRef]
- Koleva, L.; Landjeva, S.; Tsolova, E.; Ivanov, K. Screening for antixenosis resistance of winter wheat genotypes to cereal leaf beetles (Oulema spp.). In Proceedings of 6th International Scientific Agricultural Symposium “Agrosym 2015”, Jahorina, Bosnia and Herzegovina, 15–18 October 2015; pp. 831–837. [Google Scholar]
- Landjeva, S.; Koutev, V.; Tsenov, N.; Chamurlijski, P.; Trifonova, T.; Nenova, V.; Kartseva, T.; Kocheva, K.; Petrov, P. Productivity and nitrogen use efficiency in bread wheat—Comparative analysis of old and modern Bulgarian cultivars. Sci. Inst. Agric. Karnobat 2014, 3, 267–276. [Google Scholar]
- Zheleva, D.; Todorovska, E.; Christov, N.; Ivanov, P.; Ivanova, I.; Todorov, I. Assessing the genetic variation of Bulgarian bread wheat varieties by biochemical and molecular markers. Biotechnol. Biotechnol. Equip. 2007, 21, 311–321. [Google Scholar] [CrossRef]
- Petrov, P.; Petrova, A.; Dimitrov, I.; Tashev, T.; Olsovska, K.; Brestic, M.; Misheva, S. Relationships between leaf morpho-anatomy, water status and cell membrane stability in leaves of wheat seedlings subjected to severe soil drought. J. Agron. Crop Sci. 2018, 204, 435–441. [Google Scholar] [CrossRef]
- Kolev, S.; Vassilev, D.; Kostov, K.; Todorovska, E. Allele variation in loci for adaptive response in Bulgarian wheat cultivars and landraces and its effect on heading date. Plant Genet. Res. 2011, 9, 251–255. [Google Scholar] [CrossRef]
- Landjeva, S.; Korzun, V.; Ganeva, G. Evaluation of genetic diversity among Bulgarian winter wheat (Triticum aestivum L.) varieties during the period 1925–2003 using microsatellites. Genet. Res. Crop Evol. 2006, 53, 1605–1614. [Google Scholar] [CrossRef]
- Villa, C.T.; Maxted, N.; Scholten, M.; Ford-Lloyd, B. Defining and identifying crop landraces. Plant Genet. Res. Charact. Util. 2005, 3, 373–384. [Google Scholar] [CrossRef]
- Serpolay, E.; Dawson, J.C.; Chable, V.; Lammerts Van Bueren, E.T.; Osman, A.; Pino, S.; Silveri, D.; Goldringer, I. Diversity of different farmer and modern wheat varieties cultivated in contrasting organic farming conditions in western Europe and implications for European seed and variety legislation. Organ. Agric. 2011, 1, 127–145. [Google Scholar] [CrossRef]
- Newton, A.C.; Akar, T.; Baresel, J.P.; Bebeli, P.J.; Bettencourt, E.; Bladenopoulos, K.V.; Czembor, J.H.; Fasoula, D.A.; Katsiotis, A.; Koutis, K.; et al. Cereal landraces for sustainable agriculture: A review. Agron. Sust. Develop. 2010, 30, 237–269. [Google Scholar] [CrossRef]
- Dvořáček, V.; Dotlačil, L.; Hermuth, J.; Prohasková, A.; Stehno, Z.; Svobodová, L. The utilization of wheat genetic resources in breeding for bread-making quality. Czech J. Genet. Plant Breed. 2011, 47, S71–S76. [Google Scholar] [CrossRef]
- Majdrakov, P. For wheat from Pavlikeni. Seed Prod. 1945, 4, 132–141. (In Bulgarian) [Google Scholar]
- Landjeva, S.; Ganeva, G.; Korzun, V.; Palejev, D.; Chebotar, S.; Kudrjavtsev, A. Genetic diversity of old bread wheat germplasm from the Black Sea region evaluated by microsatellites and agronomic traits. Plant Genet. Res. Charact. Util. 2014, 13, 119–130. [Google Scholar] [CrossRef]
- Börner, A. Preservation of plant genetic resources in the biotechnology era. Biotechnol. J. 2006, 1, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Khlestkina, E.K.; Salina, E.A. SNP markers: Methods of analysis, ways of development, and comparison on an example of common wheat. Rus. J. Genet. 2006, 42, 585–594. [Google Scholar] [CrossRef]
- Ganal, M.W.; Plieske, J.; Hohmeyer, A.; Polley, A.; Röder, M.S. High-throughput genotyping for cereal research and breeding. In Applications of Genetic and Genomic Research in Cereals; Miedaner, T., Korzun, V., Eds.; Woodhead Publishing: Cambridge, UK, 2019; pp. 3–17. [Google Scholar]
- Morgil, H.; Can Gercek, Y.; Tulum, I. Single nucleotide polymorphisms (SNPs) in plant genetics and breeding. In The Recent Topics in Genetic Polymorphisms; Caliskan, M., Ed.; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Hughes, A.R.; Inouye, B.D.; Johnson, M.T.J.; Underwood, N.; Vellend, M. Ecological consequences of genetic diversity. Ecol. Lett. 2008, 11, 609–623. [Google Scholar] [CrossRef]
- Cox, T.S. Deepening the wheat gene pool. J. Crop Prod. 1997, 1, 1–25. [Google Scholar] [CrossRef]
- Lopes, M.S.; El-Basyoni, I.; Baenziger, S.P.; Singh, S.; Royo, C.; Ozbek, K.; Aktas, H.; Ozer, E.; Ozdemir, F.; Manickavelu, A.; et al. Exploiting genetic diversity from landraces in wheat breeding for adaptation to climate change. J. Exp. Bot. 2015, 66, 3477–3486. [Google Scholar] [CrossRef]
- Harlan, J.R. Genetics of disaster. J. Environ. Qual. 1972, 1, 212–215. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Achtar, S.; Moualla, M.Y.; Kalhout, A.; Röder, M.S.; MirAli, N. Assessment of genetic diversity among Syrian durum (Triticum ssp. durum) and bread wheat (Triticum aestivum L.) using SSR markers. Russ. J. Genet. 2010, 46, 1320–1326. [Google Scholar] [CrossRef]
- Zhang, D.D.; Bai, G.H.; Zhu, C.S.; Yu, J.M.; Carver, B.F. Genetic diversity, population structure and linkage disequilibrium in U.S. elite winter wheat. Plant Genome 2010, 3, 117–127. [Google Scholar] [CrossRef]
- Le Couviour, F.; Faure, S.; Poupard, D.; Flodrops, Y.; Dubreuil, P.; Praud, S. Analysis of genetic structure in a panel of elite wheat varieties and relevance for association mapping. Theor. Appl. Genet. 2011, 123, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Min, D.; Yasir, T.A.; Hu, Y.G. Genetic diversity, population structure and linkage disequilibrium in elite Chinese winter wheat investigated with SSR markers. PLoS ONE 2012, 7, e44510. [Google Scholar] [CrossRef]
- Eltaher, S.; Sallam, A.; Belamkar, V.; Emara, H.A.; Nower, A.A.; Salem, K.F.M.; Poland, J.; Baenziger, P.S. Genetic diversity and population structure of F3:6 Nebraska winter wheat genotypes using genotyping-by-sequencing. Front. Genet. 2018, 9, 76. [Google Scholar] [CrossRef]
- Alipour, H.; Bihamta, M.R.; Mohammadi, V.; Peyghambari, S.A.; Bai, G.; Zhang, G. Genotyping-by-sequencing (GBS) revealed molecular genetic diversity of Iranian wheat landraces and cultivars. Front. Plant Sci. 2017, 8, 1293. [Google Scholar] [CrossRef] [PubMed]
- Rufo, R.; Alvaro, F.; Royo, C.; Soriano, J.M. From landraces to improved cultivars: Assessment of genetic diversity and population structure of Mediterranean wheat using SNP markers. PLoS ONE 2019, 14, e0219867. [Google Scholar] [CrossRef]
- Alemu, A.; Feyissa, T.; Letta, T.; Abeyo, B. Genetic diversity and population structure analysis based on the high-density SNP markers in Ethiopian durum wheat (Triticum turgidum ssp. durum). BMC Genetics 2020, 21, 18. [Google Scholar] [CrossRef]
- Rimbert, H.; Darrier, B.; Navarro, J.; Kitt, J.; Choulet, F.; Leveugle, M.; Paux, E. High throughput SNP discovery and genotyping in hexaploid wheat. PLoS ONE 2018, 13, e0186329. [Google Scholar] [CrossRef]
- Beil, C.T.; Manmathan, H.K.; Anderson, V.A.; Morgounov, A.; Haley, S.D. Population structure and genetic diversity analysis of germplasm from the winter wheat Eastern European regional yield trial (WWEERYT). Crop Sci. 2017, 57, 812–820. [Google Scholar] [CrossRef]
- Bhatta, M.; Shamanin, V.; Shepelev, S.; Baenziger, P.S.; Pozherukova, V.; Pototskaya, I.; Morgounov, A. Genetic diversity and population structure analysis of synthetic and bread wheat accessions in Western Siberia. J. App. Genet. 2019, 60, 283–289. [Google Scholar] [CrossRef]
- Bhatta, M.; Morgounov, A.; Belamkar, V.; Poland, J.; Baenziger, P.S. Unlocking the novel genetic diversity and population structure of synthetic Hexaploid wheat. BMC Genom. 2018, 19, 591. [Google Scholar] [CrossRef]
- El Baidouri, M.; Murat, F.; Veyssiere, M.; Molinier, M.; Flores, R.; Burlot, L.; Salse, J. Reconciling the evolutionary origin of bread wheat (Triticum aestivum). New Phytol. 2016, 213, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R. Linkage disequilibrium and association mapping. Meth. Mol. Biol. 2007, 376, 1–15. [Google Scholar] [CrossRef]
- Chao, S.; Dubcovsky, J.; Dvorak, J.; Luo, M.-C.; Baenziger, S.P.; Matnyazov, R.; Akhunov, E.D. Population- and genome-specific patterns of linkage disequilibrium and SNP variation in spring and winter wheat (Triticum aestivum L.). BMC Genom. 2010, 11, 727. [Google Scholar] [CrossRef] [PubMed]
- Tsenov, N.; Kostov, K.; Todorov, I.; Panayotov, I.; Stoeva, I.; Atanassova, D.; Mankovsky, I.; Chamurliysky, P. Problems, achievements and prospects in breeding for grain productivity of winter wheat. Field Crop. Stud. 2009, 2, 261–273. [Google Scholar]
- Panayotov, I. Strategy of wheat breeding in Bulgaria. Bulg. J. Agric. Sci. 2000, 6, 513–523. [Google Scholar]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- International Wheat Genome Sequencing Consortium (IWGSC); Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Khurana, J.P. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, 7191. [Google Scholar] [CrossRef]
- Ganal, M.W.; Roeder, M.S. Microsatellite and SNP markers in wheat breeding. In Genomics Assisted Crop Improvement, Genomics Applications in Crops; Varshney, R.K., Tuberosa, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; Volume 2, pp. 1–24. [Google Scholar]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom. Proteom. Bioinform. 2021, in press. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef]
- Jost, L. GST and its relatives do not measure differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [CrossRef] [PubMed]
- Meirmans, P.G. Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 2006, 60, 2399–2402. [Google Scholar] [CrossRef]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodöhl, P.A. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Meth. Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef]
- Culley, T.M.; Wallace, L.E.; Gengler-Nowak, K.M.; Crawford, D.J. A comparison of two methods of calculating GST, a genetic measure of population differentiation. Am. J. Bot. 2002, 89, 460–465. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, W.S. Robust locally weighted regression and smoothing scatterplots. J. Am. Stat. Assoc. 1979, 74, 829–836. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Cons. Genet. Res. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 18. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome/ Genome | Old Germplasm | Modern Cultivars | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | GD | PIC | LD (r2) | LD (%) | LD Decay | SNP | GD | PIC | LD (r2) | LD (%) | LD Decay | |
| 1A | 1115 | 0.40 * | 0.31 * | 0.67 * | 23 | 6 | 1120 | 0.35 | 0.28 | 0.45 | 30 | 3 |
| 1B | 1068 | 0.37 * | 0.29 * | 0.58 * | 22 | 3 | 1102 | 0.33 | 0.27 | 0.44 | 45 | 3 |
| 1D | 393 | 0.39 | 0.31 | 0.64 * | 18 | 3 | 399 | 0.39 | 0.31 | 0.51 | 31 | 3 |
| 2A | 1156 | 0.37 * | 0.30 * | 0.67 * | 17 | 2 | 1153 | 0.35 | 0.28 | 0.53 | 25 | 2 |
| 2B | 1273 | 0.37 * | 0.29 | 0.69 * | 13 | 2 | 1193 | 0.36 | 0.29 | 0.39 | 32 | 3 |
| 2D | 450 | 0.37 | 0.30 | 0.71 * | 15 | 5 | 464 | 0.37 | 0.29 | 0.54 | 33 | 8 |
| 3A | 1024 | 0.35 * | 0.28 | 0.60 * | 14 | 5 | 1064 | 0.36 | 0.28 | 0.41 | 29 | 4 |
| 3B | 1264 | 0.37 * | 0.30 * | 0.56 * | 23 | 5 | 1303 | 0.35 | 0.28 | 0.4 | 43 | 4 |
| 3D | 241 | 0.38 | 0.30 | 0.71 * | 9 | 3 | 243 | 0.37 | 0.29 | 0.49 | 16 | 3 |
| 4A | 657 | 0.36 * | 0.29 * | 0.69 * | 6 | 3 | 648 | 0.33 | 0.27 | 0.36 | 28 | 3 |
| 4B | 591 | 0.34 | 0.28 | 0.71 * | 25 | 6 | 612 | 0.35 | 0.28 | 0.45 | 39 | 6 |
| 4D | 95 | 0.33 | 0.27 | 0.67 * | 8 | 6 | 105 | 0.35 | 0.28 | 0.34 | 14 | 8 |
| 5A | 1164 | 0.35 | 0.28 | 0.70 * | 12 | 4 | 1217 | 0.35 | 0.28 | 0.42 | 37 | 6 |
| 5B | 1344 | 0.36 * | 0.29 * | 0.55 * | 17 | 3 | 1427 | 0.35 | 0.28 | 0.39 | 40 | 5 |
| 5D | 311 | 0.38 * | 0.30 * | 0.60 * | 17 | 2 | 311 | 0.32 | 0.27 | 0.49 | 23 | 3 |
| 6A | 964 | 0.34 | 0.28 * | 0.49 * | 27 | 1 | 1093 | 0.34 | 0.27 | 0.44 | 32 | 2 |
| 6B | 1093 | 0.38 * | 0.30 * | 0.68 * | 13 | 3 | 1129 | 0.36 | 0.29 | 0.40 | 40 | 6 |
| 6D | 303 | 0.36 | 0.29 | 0.71 * | 13 | 1 | 308 | 0.35 | 0.28 | 0.56 | 21 | 3 |
| 7A | 1275 | 0.36 | 0.29 | 0.71 * | 11 | 2 | 1286 | 0.36 | 0.29 | 0.41 | 30 | 3 |
| 7B | 918 | 0.36 * | 0.29 * | 0.67 * | 13 | 3 | 933 | 0.35 | 0.28 | 0.36 | 34 | 4 |
| 7D | 243 | 0.35 | 0.28 | 0.45 * | 7 | 2 | 256 | 0.34 | 0.28 | 0.35 | 10 | 4 |
| A genome | 7355 | 0.36 * | 0.29 * | 0.65 * | 19 | 3.3 | 7581 | 0.35 | 0.28 | 0.43 | 30 | 3.3 |
| B genome | 7551 | 0.36 * | 0.29 * | 0.63 * | 18 | 3.6 | 7699 | 0.35 | 0.28 | 0.4 | 39 | 4.4 |
| D genome | 2036 | 0.37 * | 0.29 | 0.64 * | 12 | 3.1 | 2086 | 0.36 | 0.29 | 0.47 | 21 | 4.6 |
| Total/Average | 16,942 | 0.36 * | 0.29 * | 0.64 * | 16 | 3.3 | 17,366 | 0.35 | 0.28 | 0.43 | 30 | 4.1 |
| Subpopulation Region/Type | SP1 | SP2 | SP3 | Total | |||
|---|---|---|---|---|---|---|---|
| Old | Modern | Old | Modern | Old | Modern | ||
| Northern Bulgaria | 4 | 68 | 26 | 3 | 0 | 8 | 109 |
| Southern Bulgaria | 2 | 31 | 13 | 3 | 2 | 6 | 57 |
| Western Bulgaria | 0 | 4 | 4 | 0 | 0 | 0 | 8 |
| Q means | 0.719 | 0.707 | 0.830 | 0.632 | 0.739 | 0.721 | |
| SP | No of Accessions | HT | HS | DST | GST | Nm |
|---|---|---|---|---|---|---|
| Total | 179 | 0.3586 | 0.3481 | 0.0242 | 0.0675 | 6.91 |
| SP1 | 109 | 0.3336 | - | - | - | - |
| SP2 | 49 | 0.3419 | - | - | - | - |
| SP3 | 16 | 0.3289 | - | |||
| Admixed | 5 | - | - | - | - | - |
| Old | 51 | 0.3654 | 0.3419 | 0.0536 | 0.1467 | 2.91 |
| Modern | 128 | 0.3533 | 0.3313 | 0.0493 | 0.1395 | 3.08 |
| SP1-SP2 | 158 | 0.3608 | 0.3376 | 0.0525 | 0.1455 | 2.94 |
| SP1-SP3 | 125 | 0.3370 | 0.3313 | 0.0128 | 0.0380 | 12.66 |
| SP2-SP3 | 65 | 0.3582 | 0.3353 | 0.0517 | 0.1443 | 2.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aleksandrov, V.; Kartseva, T.; Alqudah, A.M.; Kocheva, K.; Tasheva, K.; Börner, A.; Misheva, S. Genetic Diversity, Linkage Disequilibrium and Population Structure of Bulgarian Bread Wheat Assessed by Genome-Wide Distributed SNP Markers: From Old Germplasm to Semi-Dwarf Cultivars. Plants 2021, 10, 1116. https://doi.org/10.3390/plants10061116
Aleksandrov V, Kartseva T, Alqudah AM, Kocheva K, Tasheva K, Börner A, Misheva S. Genetic Diversity, Linkage Disequilibrium and Population Structure of Bulgarian Bread Wheat Assessed by Genome-Wide Distributed SNP Markers: From Old Germplasm to Semi-Dwarf Cultivars. Plants. 2021; 10(6):1116. https://doi.org/10.3390/plants10061116
Chicago/Turabian StyleAleksandrov, Vladimir, Tania Kartseva, Ahmad M. Alqudah, Konstantina Kocheva, Krasimira Tasheva, Andreas Börner, and Svetlana Misheva. 2021. "Genetic Diversity, Linkage Disequilibrium and Population Structure of Bulgarian Bread Wheat Assessed by Genome-Wide Distributed SNP Markers: From Old Germplasm to Semi-Dwarf Cultivars" Plants 10, no. 6: 1116. https://doi.org/10.3390/plants10061116
APA StyleAleksandrov, V., Kartseva, T., Alqudah, A. M., Kocheva, K., Tasheva, K., Börner, A., & Misheva, S. (2021). Genetic Diversity, Linkage Disequilibrium and Population Structure of Bulgarian Bread Wheat Assessed by Genome-Wide Distributed SNP Markers: From Old Germplasm to Semi-Dwarf Cultivars. Plants, 10(6), 1116. https://doi.org/10.3390/plants10061116