Abstract
ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) are a family of multidomain extracellular protease enzymes with 19 members. A growing number of ADAMTS family gene variants have been identified in patients with various hereditary diseases. To understand the genomic landscape and mutational spectrum of ADAMTS family genes, we evaluated all reported variants in the ClinVar database and Human Gene Mutation Database (HGMD), as well as recent literature on Mendelian hereditary disorders associated with ADAMTS family genes. Among 1089 variants in 14 genes reported in public databases, 307 variants previously suggested for pathogenicity in Mendelian diseases were comprehensively re-evaluated using the American College of Medical Genetics and Genomics (ACMG) 2015 guideline. A total of eight autosomal recessive genes were annotated as being strongly associated with specific Mendelian diseases, including two recently discovered genes (ADAMTS9 and ADAMTS19) for their causality in congenital diseases (nephronophthisis-related ciliopathy and nonsyndromic heart valve disease, respectively). Clinical symptoms and affected organs were extremely heterogeneous among hereditary diseases caused by ADAMTS family genes, indicating phenotypic heterogeneity despite their structural and functional similarity. ADAMTS6 was suggested as presenting undiscovered pathogenic mutations responsible for novel Mendelian disorders. Our study is the first to highlight the genomic landscape of ADAMTS family genes, providing an appropriate genetic approach for clinical use.
1. Introduction
ADAMTS proteases, a superfamily of 19 secreted molecules, are zinc metalloendopeptidases; most ADAMTS protease substrates are extracellular matrix components including procollagen, von Willebrand factor, aggrecan, versican, brevican, and neurocan [1]. Since ADAMTS genes share similar structure and catalytic activity, ADAMTS proteins are known to participate in common biological processes, such as skin and cardiac development, connective tissue maintenance, and hemostasis [2]. While various perspectives of ADAMTS family genes have been studied for their clinical significance [3,4,5], a clinical genetic study focusing on their causality in Mendelian disorders is lacking [6].
As high throughput genomic technologies have revolutionized clinical practices, the scope of medical decision making has become broader, with the promise of personalized precision healthcare based on individual genotypes [7]. Genes that were uninvestigated before the next-generation sequencing era appear to possess numerous variants in clinical cases when compared to reference sequences from population databases. Neither underestimation nor overestimation of gene-disease association is helpful for clinical practices or even basic genomic research [8]. Thus, a systematic evaluation with a comprehensive review of up-to-date evidence is essential for accurate and appropriate genetic understanding to enhance the clinical usefulness of genomic studies.
With the help of technological advancements in ADAMTS studies, growing evidence for the association of ADAMTS genes with unique Mendelian disorders raises the need for an accurate and comprehensive evaluation, especially from the genetic perspective. In this study, we evaluate all reported variants in the ClinVar and HGMD mutation databases, as well as recent literature on Mendelian disorders associated with ADAMTS family genes. The information provided in this study demonstrates that the clinical interpretation of all reported mutations in ADAMTS genes requires careful professional curation and application of the latest information to suggest the possibility of ADAMTS gene involvement in novel Mendelian disorders.
2. Materials and Methods
2.1. Collection of Reported ADAMTS Family Gene Variants
All reported ADAMTS family gene variants suggested as being involved in disease were collected from publicly available mutation databases, including the Human Gene Mutation Database (HGMD version 2019.4) [9] and ClinVar (accessed 2019 Dec) [10] (Figure 1). For the HGMD database, variants annotated as “DM” and “DM?” were included in the study since HGMD curators presumably asserted the pathogenicity of these variants. For ClinVar, variants annotated as “pathogenic” or “likely pathogenic”, including all small nucleotide variants and copy number variants, were compiled. The annotation and nomenclature of the variants were confirmed using the Mutalyzer Name Checker tool [11] based on clinically relevant transcripts with the longest transcript and exons in each gene.
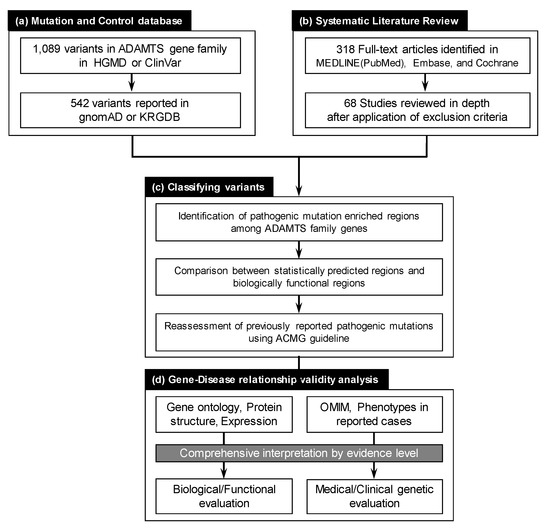
Figure 1.
The overall workflow of the study. (a) ADAMTS family gene variants reported in public mutation and control databases were compiled. A total of 1089 variants were deposited in the Human Gene Mutation Database (HGMD) and ClinVar. Variants reported as “likely pathogenic” or “pathogenic” in ClinVar and as “DM” or “DM?” in HGMD were further examined. Of the 1089 variants, 541 were reported in gnomAD and KRGDB control datasets. (b) Systematic literature review in MEDLINE (PubMed), Embase, and the Cochrane Database of Systematic Reviews electronic databases revealed 318 full-text articles as of January 2020. Only 68 studies fulfilled our criteria and were fully reviewed in depth by two independent authors. (c) Variant classification, according to ACMG guidelines, along with mutation enriched region assessments using two different algorithms, were performed. (d) Evaluation of gene–disease relationships based on comprehensive evidence level interpretation were applied to all ADAMTS family genes.
2.2. Systematic Literature Review for ADAMTS Family Genes on Mendelian Disorders
A systematic online search was performed for publications using MEDLINE (PubMed) (www.ncbi.nlm.nih.gov/pubmed), Embase (www.embase.com), and the Cochrane Database of Systematic Reviews electronic databases (www.cochranelibrary.com). The search was performed from database inception until 31 December 2019. Data for clinical and genomic information, as well as functional studies on variants, were extracted from eligible publications. Two independent authors (J.H.R. and Y.J.C.) assessed articles by title, abstract, and full text. A total of 68 studies that fulfilled the selection criteria of (1) human mutation study (not mouse), (2) germline mutation study with single gene–disease relationship (not association study), and (3) DNA mutation study (not epigenetic or proteomic) were included for further evaluation.
2.3. Evaluating ADAMTS Family Gene Disease Associations
Evidence associating each gene with a specific Mendelian disorder was systematically assessed based on a comprehensive analysis of various aspects, including gene ontology, protein functional domain for mutation location, and expression patterns. The Online Mendelian Inheritance in Man (OMIM) database [12] was used to confirm the currently validated disease associations and inheritance patterns in ADAMTS family genes. The Human Phenotype Ontology (HPO) database [13] was utilized in the interpretation process for organ-specific symptoms of suggested Mendelian disorders. Gene ontology (GO) [14] for ADAMTS family genes was searched using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) [15] and Ensembl Genome Browser [16]. Protein domains and expression patterns among various organs across ADAMTS family genes were compared using UniProt [17] and the Human Protein Atlas (www.proteinatlas.org) [18], respectively.
2.4. Pathogenicity Interpretation for Variant and Gene Evaluation
To evaluate the pathogenic potential of presumably pathogenic ADAMTS family gene variants, we applied the 2015 ACMG guideline for variant classification [19]. To enhance the analysis accuracy, we profiled all aspects of variants, including population allele frequency, prediction algorithm results, and conservation status across species. For gene evaluation in terms of cause-and-effect relationships for hereditary disorders, we applied two recently published prediction algorithms for missense variant burden (i.e., PER (pathogenic mutation enriched regions) viewer [20] and MTR (missense tolerance regions) viewer [21]) in ADAMTS genes. These tools provided a statistical framework to identify gene regions with missense variation intolerance or pathogenic mutation enriched regions. Additionally, oe values (the ratio of observed/expected number of loss-of-function variants in the specific gene) and pLI scores (probability of being loss-of-function intolerant as metrics to measure a transcript’s intolerance to variation) provided by the gnomAD database [22] were incorporated to broaden genomic understanding.
3. Results
3.1. Gene-Disease Association of ADAMTS Family Genes Based on Pathogenic Mutations
In order to identify definitive gene–disease associations among mutated ADAMTS family genes in Mendelian disorders, we reviewed all publications archived from mutation databases and systematic literature review. A total of eight ADAMTS family genes were revealed to have strong causality for various Mendelian disorders if mutated (Table 1). Although several recently updated reviews have already briefly covered six ADAMTS genes [5], our evidence suggested two additional ADAMTS genes with a high probability of involvement in different diseases [23,24]. Except for ADAMTS3 and ADAMTS10, six genes presented a unique association with specific disease phenotypes, indicating low genetic heterogeneity for unique Mendelian disorders caused by corresponding ADAMTS gene mutations. Regarding inheritance modes, all ADAMTS mutations acted recessively due to the nature of enzymes since enzymes are mostly haplo-sufficient to alleviate heterozygote loss of function mutation. This phenomenon could also be related to the dominance of nonsense, frameshift, and splice-site mutations in terms of the mutational spectrum in most ADAMTS genes. While ADAMTS13 possessed more than 200 pathogenic mutations with clinical validations, mutations in seven other ADAMTS genes were rarely reported, suggesting a very low prevalence of these ultra-rare diseases (Table 1).

Table 1.
ADAMTS family genes responsible for Mendelian disorders with strong evidence as of 2020.
When all clinical cases were reviewed for organ-specific phenotypes, HPO analysis revealed phenotypic heterogeneity among ADAMTS family genes (Figure 2). While ocular symptoms were the most common, a broad spectrum of eye diseases were caused by ADAMTS10, ADAMTS17, and ADAMTS18 [25,26,27,28]. Since ADAMTS10 and ADAMTS17 consisted of common domains, including PLAC, clinical presentations in Weill–Marchesani syndrome caused by ADAMTS10 mutations and Weill–Marchesani-like syndrome caused by ADAMTS17 mutations shared similar ocular and skeletal phenotypes [26]. In contrast, phenotypes in Ehlers–Danlos syndrome and Hennekam syndrome patients completely differed and involved different organs, although ADAMTS2 and ADAMTS3 possess the same procollagen N-propeptidase domains [29,30]. Furthermore, ADAMTS9 with a unique GON domain presented neural, hearing, and renal phenotypes if mutated, all of which are specifically caused by ciliary dysfunction [23].
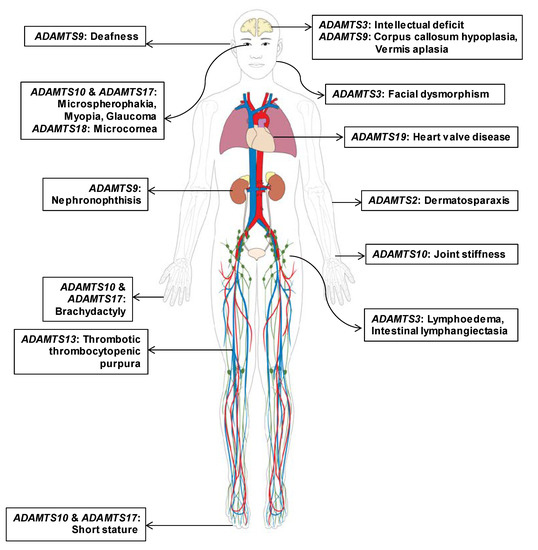
Figure 2.
Clinical findings and distribution of affected organs in individuals with Mendelian disorders caused by pathogenic mutations in different ADAMTS family genes. Predominant and recurrently reported clinical presentations of eight ADAMTS genes with a strong causal relationship to Mendelian disorders are marked according to organ. Heterogeneous distribution of affected organ types indicates the phenotypic heterogeneity among hereditary disorders caused by pathogenic germline mutations in ADAMTS genes.
3.2. Updated ADAMTS Family Genes Responsible for Mendelian Disorders; ADAMTS9 and ADAMTS19
Our group recently found ADAMTS9 mutations are a cause of nephronophthisis-related ciliopathies (NPHP–RCs) [23]. NPHP–RCs are a group of inherited diseases associated with defects in primary cilium structure and function. In this study, homozygosity mapping and whole-exome sequencing identified two cases with homozygous mutations in ADAMTS9. A novel homozygous frameshift truncating mutation (c.4575_4576del; p.Gln1525Hisfs*60) in ADAMTS9 was identified in an European patient with NPHP and early-onset end-stage renal disease (ESRD) with the Joubert syndrome phenotype, including symptoms of vermis aplasia and corpus callosum hypoplasia. In addition, the patient presented with proteinuria, deafness, atrial septal defects, coloboma, and short stature. An Arabic child from consanguineous parents also had ESRD from NPHP, resulting from an amino acid substitution (c.194C>G; p.Thr65Arg) at Thr65, a highly conserved residue for protein function. The patient exhibited proteinuria, deafness, hepatosplenomegaly, anemia, thrombocytopenia, short stature, and osteopenia. While ADAMTS9 is known to be a secreted extracellular metalloproteinase, an in vitro study showed that ADAMTS9-loss resulted in shortened cilia and defective sonic hedgehog signaling. Knockdown of adamts9 in zebrafish recapitulated NPHP–RC phenotypes, including renal cysts and hydrocephalus. These findings suggest that the identified mutations in ADAMTS9 cause NPHP–RC and that ADAMTS9 is required for the formation and function of primary cilia.
Another study on ADAMTS19 recently identified pathogenic mutations as the cause of non-syndromic heart valve disease [24]. Exome sequencing of four affected individuals in two consanguineous families showed novel homozygous truncating mutations in ADAMTS19 (homozygous deletion involving exons 1 to 8 and homozygous nonsense mutation (c.1984C > T; p.Arg662*)). The authors suggested that a unique feature results from these ADAMTS19 mutations, causing only heart valve disorders without affecting any other organs; this indicates non-syndromic features.
3.3. Evidence-Based Evaluation for Gene-Disease Relationships in ADAMTS Genes
Various aspects of gene evaluation, including GO, protein domains, expression databases, and functional assay availability, were reviewed (Table 2). When commonly shared biological process and molecular function GO terms were evaluated in all ADAMTS family genes, diverse GO terms appeared across various groups of ADAMTS genes. While “proteolysis” and “collagen processes” were dominantly annotated for biological processes, “metalloendopeptidase”, “metal ion binding”, and “zinc ion binding” were reported in more than half of the ADAMTS family genes. As phenotypic variability was characteristic in the affected organ spectrum, expression patterns of ADAMTS family genes were heterogeneous. However, in vitro functional assays were only available for seven genes despite the commonly shared functions among ADAMTS family genes.
Table 2.
Evidence-based evaluation of the clinical validity of ADAMTS family genes in gene–disease relationships.
3.4. Mutational Spectrum of ADAMTS Family Genes in Current Databases
When mutation databases (HGMD and ClinVar) were searched for ADAMTS gene variants, a total of 286 SNVs and 21 CNVs were collected. While eight genes with a strong causality in specific Mendelian disorders presented 34 SNVs on average, other ADAMTS genes with currently deficient clinical evidence possessed up to four SNVs (Table 3). Percentages of frameshift/nonsense/splicing variants were dominant in most causative ADAMTS genes, although missense variants were more predominant in ADAMTS18.
Table 3.
Mutational spectrum of ADAMTS family genes in Mendelian hereditary disorders in current databases and literature.
To predict the possibility of a missense mutational burden at “hot spots” on ADAMTS genes, two recently validated prediction tools (i.e., PER-viewer [20] and MTR-viewer [21]) were utilized. As PER-viewer considers the positional conservation status among paralogs, each ADAMTS gene was compared to other genes within the ADAMTS family. Four families were incorporated using PER-viewer, and this algorithm suggested that there were no definitive pathogenic mutation enriched regions for missense mutations in any ADAMTS gene, according to the suggested threshold of Bonferroni-adjusted p-value below 0.05. In contrast, ADAMTS6, ADAMTS9, ADAMTS10, and ADAMTS14 appeared to have missense mutation enriched regions according to MTR-viewer based on the criterion of MTR score under 0.6 and FDR-adjusted p-value under 0.1, as defined by algorithm developers. As missense mutations in ADAMTS9 and ADAMTS10 are validated for their pathogenicity in association with Mendelian disorders, ADAMTS6 and ADAMTS14 are expected to be responsible for currently undiscovered Mendelian disorders with pathogenic missense mutations. Furthermore, oe value for missense variants in ADAMTS6 was relatively low among ADAMTS family genes, indicating the possibility of missense mutations to be elucidated in the future. Another oe value for loss of function (LoF) mutations provided by the gnomAD database, along with the pLI score from a previous version of gnomAD, suggested ADAMTS2 and ADAMTS6 as LoF genes if pathogenic mutations caused Mendelian disorders. As all reported ADAMTS2 and ADAMTS6 variants did not satisfy the pathogenicity grades, pathogenic LoF mutations with distinctive dysfunctions might cause severe Mendelian disorders or even embryo lethality.
3.5. Reassessment of Previously Reported Pathogenic Mutations in ADAMTS Genes
The ACMG variant interpretation guideline [19] was applied for previously reported pathogenic mutations (Table 4). The mutational spectrum of ADAMTS13 pathogenic mutations, which was well-reviewed in recently published literatures, revealed a heterogeneous pattern across mutation positions and variant types (Table S1). Among the diverse evidence in the ACMG guideline, allele frequency in the control database highlighted as predicting the prevalence of Mendelian disorders were ultra-rare. A total of 45 mutations were validated for their suggested pathogenicity (Table 4), while another 12 presumably annotated pathogenic variants provided insufficient evidence for pathogenicity interpretation (Table S2). Positional distribution of pathogenic and likely pathogenic mutations were dispersed among seven genes (Figure 3).
Table 4.
Reassessment of previously reported pathogenic mutations in ADAMTS family genes using the American College of Medical Genetics and Genomics (ACMG) variant interpretation guideline.
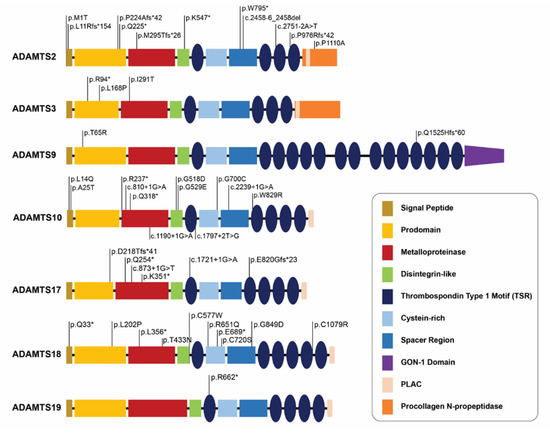
Figure 3.
Comparative analysis of pathogenic mutation loci across ADAMTS genes with strong relationship with specific Mendelian disorders. Positional annotations of pathogenic mutations in ADAMTS genes by protein functional domains indicate the wide distribution of mutations and absence of hot spots. “*” (asterisk) indicates a translation termination codon.
4. Discussion
In this study, we identified a subset of ADAMTS family genes with strong evidence of being causative genes for specific Mendelian disorders. As a growing number of variants are detected by up-to-date sequencing technologies [7], ADAMTS family genes are expected to be highlighted for their association with human disorders, especially hereditary diseases [3,4,5]. Understanding the genomic landscape and mutational spectrum of ADAMTS family genes in clinical cases will not only benefit further characterization of ADAMTS family proteins in biology but the future development and application of therapeutics for Mendelian disorders caused by ADAMTS gene mutations.
A total of eight ADAMTS family genes are currently known to be responsible for Mendelian disorders. Since all ADAMTS proteins are metalloproteases, the autosomal recessive modes of all genes are compatible with the characteristics of ADAMTS enzymes [1]. Furthermore, the mutational spectrum of nonsense and frameshift mutations could be explained by the LoF mechanism in most genes, as expected. While ADAMTS13 mutations are well studied for heterogenous mutations across the gene in more than 200 congenital thrombotic thrombocytopenic purpura patients [31,32], only small numbers of patients and mutations have been reported in the other seven genes. Although the prevalence of ultra-rare Mendelian diseases caused by ADAMTS genes is expected [5], it is important to broaden the genetic spectrum of these genes as more patients with pathogenic ADAMTS gene mutations will present in the future. It is noteworthy that two recently discovered Mendelian disorders arising from ADAMTS9 and ADAMTS19 broadened the spectrum of affected organs (renal and cardiac diseases, respectively) that have never been associated with six previously identified ADAMTS genes responsible for Mendelian disorders.
Clinical symptoms and affected organs are extremely heterogeneous among hereditary diseases caused by ADAMTS genes. Phenotypic heterogeneity in these Mendelian disorders, despite shared enzymatic functions and similar functional defective features of germline mutations among ADAMTS family genes, is noticeable and it is alarming that paralogs within the ADAMTS gene family might function differently in different organs. Redundancy or compensation by other normal ADAMTS genes instead of affected ADAMTS genes is less likely to be anticipated based on the diverse symptoms among these disorders. Furthermore, subgrouping of ADAMTS genes according to shared unique protein domains did not fully correlate with the clinical presentations in affected patients, suggesting that organ-specific functions among individual ADAMTS genes are essential to their physiological roles.
To evaluate the possibility of yet-to-be-discovered Mendelian disorders caused by other ADAMTS genes, we applied two recently validated prediction algorithms [20,21] for missense burden estimation by regions across genes. Although only one algorithm predicted that four genes (ADAMTS6, ADAMTS9, ADAMTS10, ADAMTS14) possessed missense intolerance regions, pathogenic missense mutations in ADAMTS9 and ADAMTS10 indeed account for relatively high proportions among mutation types. Therefore, we suggest that ADAMTS6 and ADAMTS14, which are not currently considered causative genes for any Mendelian disorders, might be responsible for novel hereditary disorders caused by pathogenic missense mutations. Furthermore, ADAMTS6 and ADAMTS2 also presented high pLI scores and low oe values for LoF using the gnomAD database. As nonsense and frameshift mutations in ADAMTS2 are responsible for the Ehlers–Danlos syndrome, LoF mutations in ADAMTS6 are expected to cause novel disease entities with diverse organotypic symptoms. Altogether, it will be interesting to examine whether any other ADAMTS proteins are involved in different forms of Mendelian diseases in the future.
When we applied the 2015 ACMG guideline [19] for pathogenicity interpretation in all reported ADAMTS family gene variants and compiled all published information on functional assays to evaluate the clinical validity of gene-disease relationships, the importance of an appropriate functional evaluation on diverse ADAMTS gene variants was once again confirmed in terms of accurately interpreting pathogenicity. Although ADAMTS1 and ADAMTS16 variants were suggested to be responsible for congenital mandibular prognathism and inherited hypertension, respectively, insufficient evidence for defective functions in ADAMTS proteins caused by variants and the scarcity of clinical reports did not allow strongly valid annotations for either the gene–disease relationship or variant pathogenicity. While the conventional genetic assessment of ADAMTS gene family variants should be considered in the context of prediction algorithm results, conservation status across species, and population allele frequencies, functional assays for clear defects appear to be the most important factor in the process of ACMG guideline application to establish a strong link between a pathogenic mutation and an associated Mendelian disorder.
5. Conclusions
In conclusion, we evaluated the genomic landscape and mutational spectrum of ADAMTS family genes in Mendelian disorders based on a gene evidence review of variants using publicly available databases and systematic literature reviews. Although eight ADAMTS family genes have a strong causal relationship with diverse Mendelian diseases in an autosomal recessive manner, there are additional possibilities for other ADAMTS family genes, such as ADAMTS6, to have a high potential in causing novel hereditary disorders based on our analysis. Despite an ultra-rare prevalence of pathogenic germline ADAMTS mutations responsible for genetic diseases (with the exception of ADAMTS13), it is important to accurately assess variants for their pathogenicity, together with metalloproteinase-specific functional assays for ADAMTS family proteins.
Supplementary Materials
The following are available online at https://www.mdpi.com/2218-273X/10/3/449/s1,. Table S1: Reassessment of variants in ADAMTS family genes with strong evidence for Mendelian disorders previously suggested for association but disqualified for pathogenicity using the ACMG variant interpretation guideline. Table S2: List of 202 pathogenic mutations in ADAMTS13 (NM_139025.4) with strong evidence for hereditary thrombotic thrombocytopenic purpura or Upshaw–Schulman syndrome.
Author Contributions
J.H.R. and Y.J.C. analyzed and processed public genomic data and literature on ADAMTS gene family. H.Y.G. conceived of and directed the entire project and wrote the manuscript with help of J.H.R. and Y.J.C. All authors critically reviewed the paper. All authors have read and agreed to the published version of the manuscript.
Funding
H.Y.G. was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning [2018R1A2A3074572].
Acknowledgments
We thank MID (Medical Illustration and Design) for providing support with the medical illustrations.
Conflicts of Interest
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Apte, S.S. Adamts proteins: Concepts, challenges, and prospects. Methods Mol. Biol. 2020, 2043, 1–12. [Google Scholar] [PubMed]
- Apte, S.S. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (adamts) superfamily: Functions and mechanisms. J. Biol. Chem. 2009, 284, 31493–31497. [Google Scholar] [CrossRef] [PubMed]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The adamts (a disintegrin and metalloproteinase with thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef] [PubMed]
- Dubail, J.; Apte, S.S. Insights on adamts proteases and adamts-like proteins from mammalian genetics. Matrix Biol. 2015, 44, 24–37. [Google Scholar] [CrossRef]
- Mead, T.J.; Apte, S.S. Adamts proteins in human disorders. Matrix Biol. 2018, 71, 225–239. [Google Scholar] [CrossRef]
- Le Goff, C.; Cormier-Daire, V. The adamts(l) family and human genetic disorders. Hum. Mol. Genet. 2011, 20, R163–R167. [Google Scholar] [CrossRef]
- Adams, D.R.; Eng, C.M. Next-generation sequencing to diagnose suspected genetic disorders. New Engl. J. Med. 2018, 379, 1353–1362. [Google Scholar] [CrossRef]
- Grant, A.R.; Cushman, B.J.; Cave, H.; Dillon, M.W.; Gelb, B.D.; Gripp, K.W.; Lee, J.A.; Mason-Suares, H.; Rauen, K.A.; Tartaglia, M.; et al. Assessing the gene-disease association of 19 genes with the rasopathies using the clingen gene curation framework. Hum. Mutat. 2018, 39, 1485–1493. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The human gene mutation database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. Clinvar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Wildeman, M.; van Ophuizen, E.; den Dunnen, J.T.; Taschner, P.E. Improving sequence variant descriptions in mutation databases and literature using the mutalyzer sequence variation nomenclature checker. Hum. Mutat. 2008, 29, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. Omim.Org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef] [PubMed]
- Kohler, S.; Carmody, L.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.P.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; McMurry, J.A.; et al. Expansion of the human phenotype ontology (hpo) knowledge base and resources. Nucleic Acids Res. 2019, 47, D1018–D1027. [Google Scholar] [CrossRef] [PubMed]
- The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 2019, 47, D330–D338. [CrossRef]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2019, 47, D745–D751. [Google Scholar] [CrossRef]
- UniProt Consortium. Uniprot: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Perez-Palma, E.; May, P.; Iqbal, S.; Niestroj, L.M.; Du, J.; Heyne, H.O.; Castrillon, J.A.; O’Donnell-Luria, A.; Nurnberg, P.; Palotie, A.; et al. Identification of pathogenic variant enriched regions across genes and gene families. Genome Res. 2020, 30, 62–71. [Google Scholar] [CrossRef]
- Silk, M.; Petrovski, S.; Ascher, D.B. Mtr-viewer: Identifying regions within genes under purifying selection. Nucleic Acids Res. 2019, 47, W121–W126. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv 2019, 531210. [Google Scholar] [CrossRef]
- Choi, Y.J.; Halbritter, J.; Braun, D.A.; Schueler, M.; Schapiro, D.; Rim, J.H.; Nandadasa, S.; Choi, W.I.; Widmeier, E.; Shril, S.; et al. Mutations of adamts9 cause nephronophthisis-related ciliopathy. Am. J. Hum. Genet. 2019, 104, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Wunnemann, F.; Ta-Shma, A.; Preuss, C.; Leclerc, S.; van Vliet, P.P.; Oneglia, A.; Thibeault, M.; Nordquist, E.; Lincoln, J.; Scharfenberg, F.; et al. Loss of adamts19 causes progressive non-syndromic heart valve disease. Nat. Genet. 2020, 52, 40–47. [Google Scholar] [CrossRef]
- Dagoneau, N.; Benoist-Lasselin, C.; Huber, C.; Faivre, L.; Megarbane, A.; Alswaid, A.; Dollfus, H.; Alembik, Y.; Munnich, A.; Legeai-Mallet, L.; et al. Adamts10 mutations in autosomal recessive weill-marchesani syndrome. Am. J. Hum. Genet. 2004, 75, 801–806. [Google Scholar] [CrossRef]
- Morales, J.; Al-Sharif, L.; Khalil, D.S.; Shinwari, J.M.; Bavi, P.; Al-Mahrouqi, R.A.; Al-Rajhi, A.; Alkuraya, F.S.; Meyer, B.F.; Al Tassan, N. Homozygous mutations in adamts10 and adamts17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am. J. Hum. Genet. 2009, 85, 558–568. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Alshammari, M.J.; Khan, A.O.; Mohamed, J.Y.; Alhabib, F.A.; Alkuraya, F.S. The syndrome of microcornea, myopic chorioretinal atrophy, and telecanthus (mmcat) is caused by mutations in adamts18. Hum. Mutat. 2013, 34, 1195–1199. [Google Scholar] [CrossRef]
- Chandra, A.; Arno, G.; Williamson, K.; Sergouniotis, P.I.; Preising, M.N.; Charteris, D.G.; Thompson, D.A.; Holder, G.E.; Borman, A.D.; Davagnanam, I.; et al. Expansion of ocular phenotypic features associated with mutations in adamts18. JAMA Ophthalmol. 2014, 132, 996–1001. [Google Scholar] [CrossRef][Green Version]
- Van Damme, T.; Colige, A.; Syx, D.; Giunta, C.; Lindert, U.; Rohrbach, M.; Aryani, O.; Alanay, Y.; Simsek-Kiper, P.O.; Kroes, H.Y.; et al. Expanding the clinical and mutational spectrum of the ehlers-danlos syndrome, dermatosparaxis type. Genet. Med. 2016, 18, 882–891. [Google Scholar] [CrossRef]
- Brouillard, P.; Dupont, L.; Helaers, R.; Coulie, R.; Tiller, G.E.; Peeden, J.; Colige, A.; Vikkula, M. Loss of adamts3 activity causes hennekam lymphangiectasia-lymphedema syndrome 3. Hum. Mol. Genet. 2017, 26, 4095–4104. [Google Scholar] [CrossRef]
- Alwan, F.; Vendramin, C.; Liesner, R.; Clark, A.; Lester, W.; Dutt, T.; Thomas, W.; Gooding, R.; Biss, T.; Watson, H.G.; et al. Characterization and treatment of congenital thrombotic thrombocytopenic purpura. Blood 2019, 133, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- van Dorland, H.A.; Taleghani, M.M.; Sakai, K.; Friedman, K.D.; George, J.N.; Hrachovinova, I.; Knobl, P.N.; von Krogh, A.S.; Schneppenheim, R.; Aebi-Huber, I.; et al. The international hereditary thrombotic thrombocytopenic purpura registry: Key findings at enrollment until 2017. Haematologica 2019, 104, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).