Are Inositol Polyphosphates the Missing Link in Dynamic Cullin RING Ligase Regulation by the COP9 Signalosome?


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
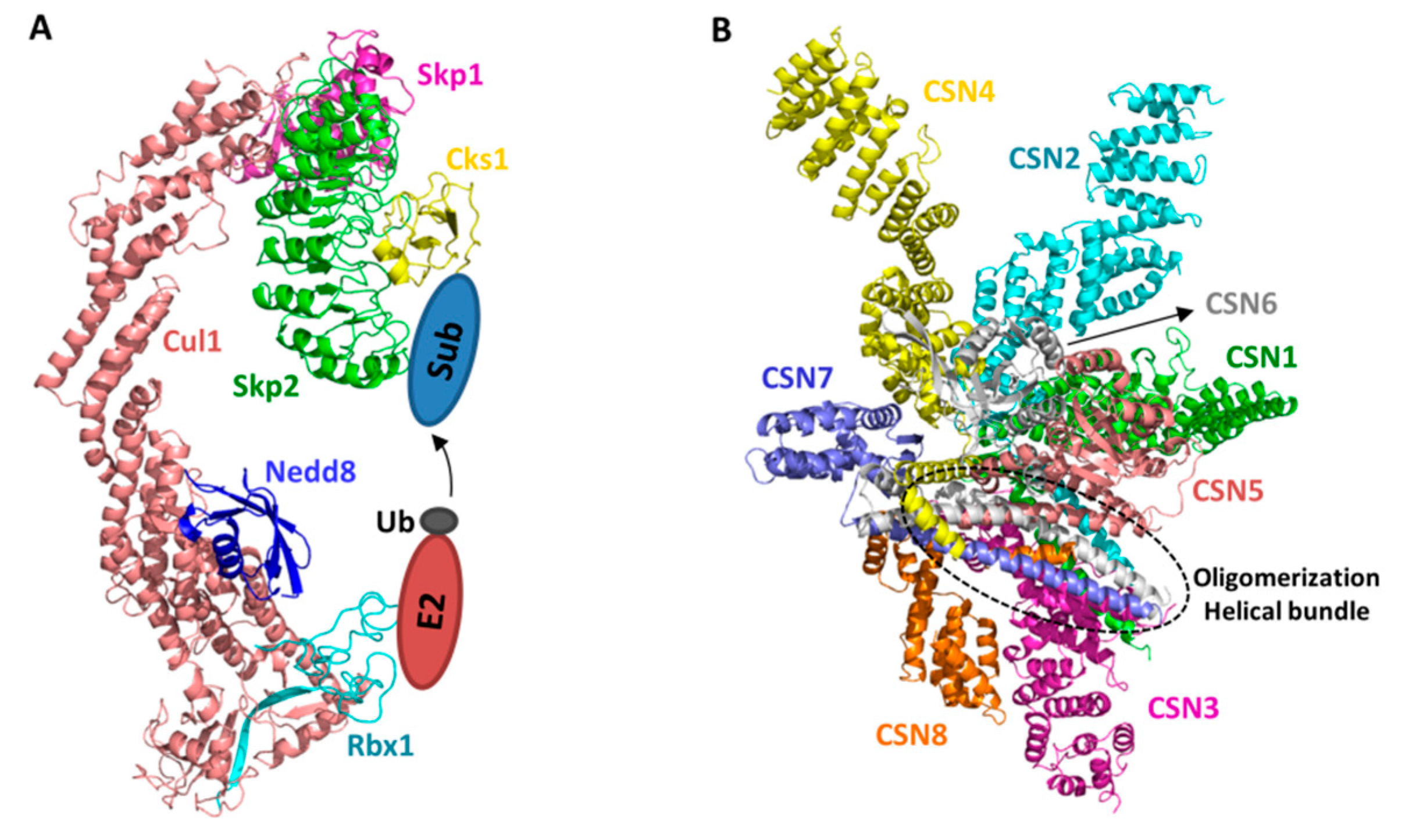
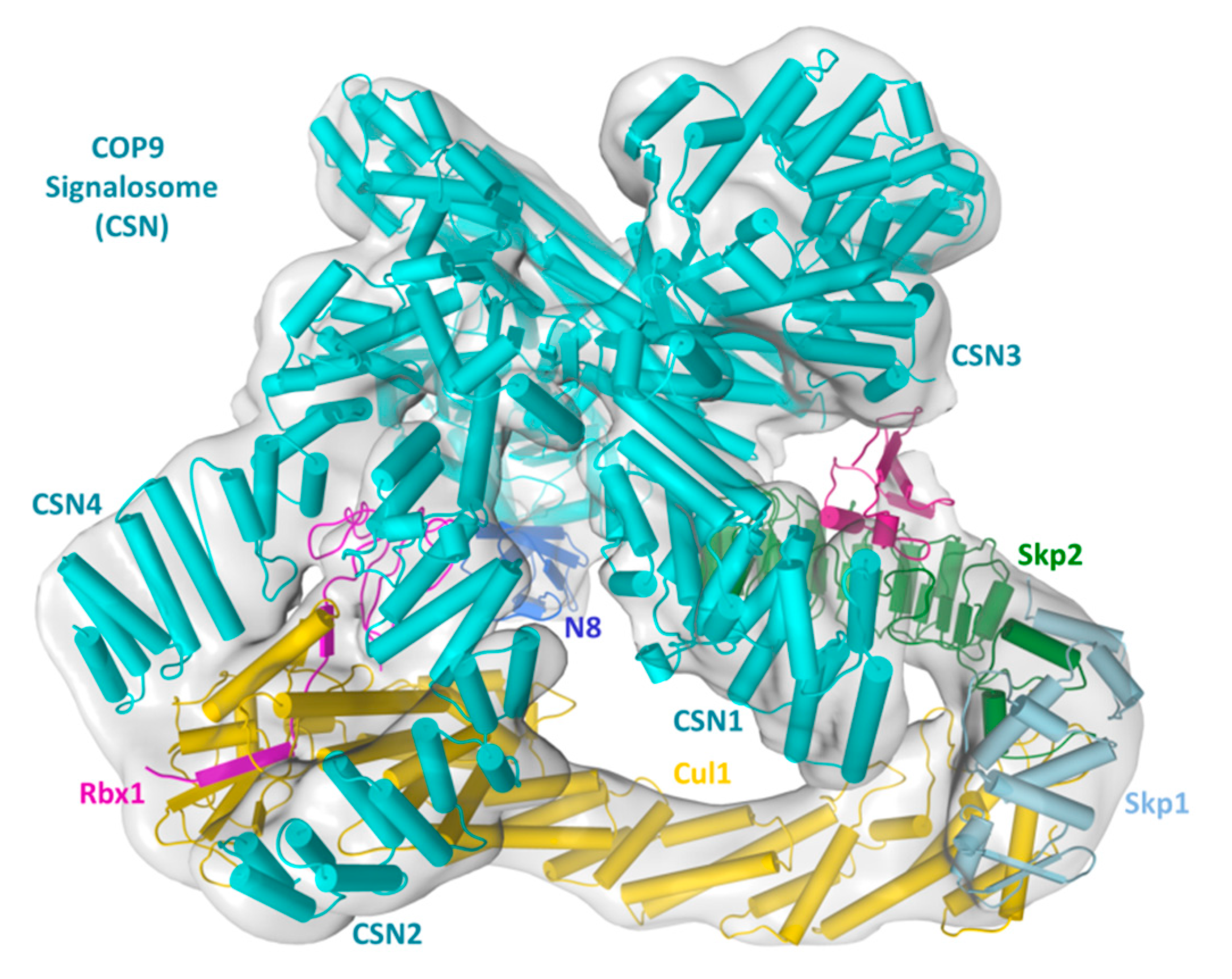
2. Structural Basis of CRL–CSN Supercomplexes
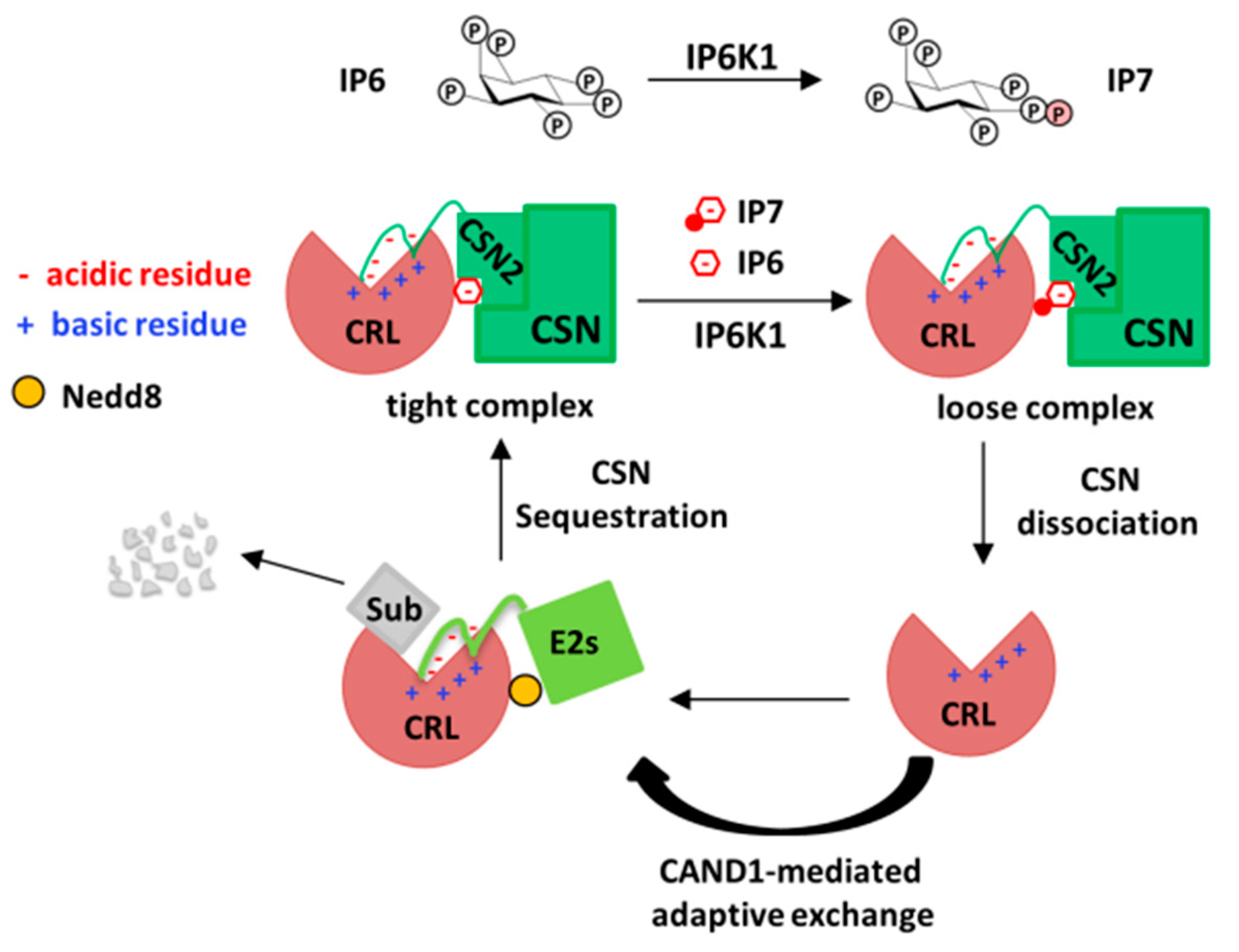
3. The Role of Inositol Polyphosphates and their Synthases in CRL–CSN Complex Dynamics
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wei, N.; Deng, X.W. The COP9 signalosome. Annu. Rev. Cell Dev. Biol. 2003, 19, 261–286. [Google Scholar] [CrossRef] [PubMed]
- Dubiel, D.; Rockel, B.; Naumann, M.; Dubiel, W. Diversity of COP9 signalosome structures and functional consequences. FEBS Lett. 2015, 589, 2507–2513. [Google Scholar] [CrossRef] [PubMed]
- A Chamovitz, D.; Wei, N.; Osterlund, M.T.; Von Arnim, A.G.; Staub, J.M.; Matsui, M.; Deng, X.-W. The COP9 Complex, a Novel Multisubunit Nuclear Regulator Involved in Light Control of a Plant Developmental Switch. Cell 1996, 86, 115–121. [Google Scholar] [CrossRef]
- Wei, N.; Chamovitz, D.A.; Deng, X.-W. Arabidopsis COP9 is a component of a novel signaling complex mediating light control of development. Cell 1994, 78, 117–124. [Google Scholar] [CrossRef]
- Seeger, M.; Kraft, R.; Ferrell, K.; Bech-Otschir, D.; Dumdey, R.; Schade, R.; Gordon, C.; Naumann, M.; Dubiel, W. A novel protein complex involved in signal transduction possessing similarities to 26S proteasome subunits. FASEB J. 1998, 12, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Milic, J.; Tian, Y.; Bernhagen, J. Role of the COP9 Signalosome (CSN) in Cardiovascular Diseases. Biomol. 2019, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Chamovitz, D.A. Role of Cop9 Signalosome Subunits in the Environmental and Hormonal Balance of Plant. Biomolecules 2019, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Xie, L.; Gu, Y.; Li, J.; Xie, J. Roles of Multifunctional COP9 Signalosome Complex in Cell Fate and Implications for Drug Discovery. J. Cell. Physiol. 2017, 232, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Gummlich, L.; Kähne, T.; Naumann, M.; Kilic, E.; Jung, K.; Dubiel, W. New Insights into the Mechanism of COP9 Signalosome–Cullin-RING Ubiquitin-Ligase Pathway Deregulation in Urological Cancers. Int. Rev. Cell Mol. Biol. 2016, 323, 181–229. [Google Scholar]
- Chung, D.; Dellaire, G. The Role of the COP9 Signalosome and Neddylation in DNA Damage Signaling and Repair. Biomolecules 2015, 5, 2388–2416. [Google Scholar] [CrossRef]
- Lee, M.-H.; Zhao, R.; Phan, L.; Yeung, S.-C.J. Roles of COP9 signalosome in cancer. Cell Cycle 2011, 10, 3057–3066. [Google Scholar] [CrossRef] [PubMed]
- Richardson, K.S.; Zundel, W. The Emerging Role of the COP9 Signalosome in Cancer. Mol. Cancer Res. 2005, 3, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Pick, E.; Bramasole, L. Moonlighting and pleiotropy within two regulators of the degradation machinery: the proteasome lid and the CSN. Biochem. Soc. Trans. 2014, 42, 1786–1791. [Google Scholar] [CrossRef] [PubMed]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin–RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Lydeard, J.R.; A Schulman, B.; Harper, J.W. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013, 14, 1050–1061. [Google Scholar] [CrossRef] [PubMed]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Skaar, J.R.; Pagan, J.K.; Pagano, M. SCF ubiquitin ligase targeted therapies. Nat. Rev. Drug Discov. 2014, 13, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, Y. Cullin-RING Ligases as Attractive Anti-cancer Targets. Curr. Pharm. Des. 2013, 19, 3215–3225. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Crews, C.M. Targeted Protein Degradation by Small Molecules. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 107–123. [Google Scholar] [CrossRef]
- Wu, K.; Chen, A.; Pan, Z.-Q. Conjugation of Nedd8 to CUL1 Enhances the Ability of the ROC1-CUL1 Complex to Promote Ubiquitin Polymerization. J. Boil. Chem. 2000, 275, 32317–32324. [Google Scholar] [CrossRef]
- Duda, D.; Schulman, B. Structural Insights into NEDD8 Activation of Cullin-RING Ligases: Conformational Control of Conjugation. Control Conjug. 2008, 134, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Cope, G.A.; Suh, G.S.B.; Aravind, L.; Schwarz, S.E.; Zipursky, S.L.; Koonin, E.V.; Deshaies, R.J. Role of Predicted Metalloprotease Motif of Jab1/Csn5 in Cleavage of Nedd8 from Cul1. Science 2002, 298, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Lyapina, S.; Cope, G.; Shevchenko, A.; Serino, G.; Tsuge, T.; Zhou, C.; Wolf, D.A.; Wei, N.; Deshaies, R.J. Promotion of NEDD8-CUL1 Conjugate Cleavage by COP9 Signalosome. Science 2001, 292, 1382–1385. [Google Scholar] [CrossRef] [PubMed]
- Enchev, R.I.; Scott, D.C.; Da Fonseca, P.C.A.; Schreiber, A.; Monda, J.K.; Schulman, B.A.; Peter, M.; Morris, E.P. Structural basis for a reciprocal regulation between SCF and CSN. Cell Rep. 2012, 2, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Emberley, E.D.; Mosadeghi, R.; Deshaies, R.J. Deconjugation of Nedd8 from Cul1 Is Directly Regulated by Skp1-F-box and Substrate, and the COP9 Signalosome Inhibits Deneddylated SCF by a Noncatalytic Mechanism*. J. Boil. Chem. 2012, 287, 29679–29689. [Google Scholar] [CrossRef] [PubMed]
- A Cope, G.; Deshaies, R.J. Targeted silencing of Jab1/Csn5 in human cells downregulates SCF activity through reduction of F-box protein levels. BMC Biochem. 2006, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Wee, S.; Geyer, R.K.; Toda, T.; Wolf, D.A. CSN facilitates Cullin–RING ubiquitin ligase function by counteracting autocatalytic adapter instability. Nature 2005, 7, 387–391. [Google Scholar] [CrossRef]
- Wolf, D.A.; Zhou, C.; Wee, S. The COP9 signalosome: an assembly and maintenance platform for cullin ubiquitin ligases? Nature 2003, 5, 1029–1033. [Google Scholar] [CrossRef]
- Zheng, N.; Schulman, B.A.; Song, L.; Miller, J.J.; Jeffrey, P.D.; Wang, P.; Chu, C.; Koepp, D.M.; Elledge, S.J.; Pagano, M.; et al. Structure of the Cul1–Rbx1–Skp1–F boxSkp2 SCF ubiquitin ligase complex. Nature 2002, 416, 703–709. [Google Scholar] [CrossRef]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1–CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef]
- Schulman, B.A.; Carrano, A.C.; Jeffrey, P.D.; Bowen, Z.; Kinnucan, E.R.E.; Finnin, M.S.; Elledge, S.J.; Harper, J.W.; Pagano, M.; Pavletich, N.P. Insights into SCF ubiquitin ligases from the structure of the Skp1–Skp2 complex. Nature 2000, 408, 381–386. [Google Scholar] [CrossRef]
- Lingaraju, G.M.; Bunker, R.D.; Cavadini, S.; Hess, D.; Hassiepen, U.; Renatus, M.; Fischer, E.S.; Thoma, N.H. Crystal structure of the human COP9 signalosome. Nature 2014, 512, 161–165. [Google Scholar] [CrossRef]
- Lee, J.-H.; Yi, L.; Li, J.; Schweitzer, K.; Borgmann, M.; Naumann, M.; Wu, H. Crystal Structure and Versatile Functional Roles of the COP9 Signalosome Subunit 1. Proc. Natl. Acad. Sci. USA 2013, 110, 11845–11850. [Google Scholar] [CrossRef]
- Echalier, A.; Pan, Y.; Birol, M.; Tavernier, N.; Pintard, L.; Hoh, F.; Ebel, C.; Galophe, N.; Claret, F.X.; Dumas, C. Insights into the regulation of the human COP9 signalosome catalytic subunit, CSN5/Jab1. Proc. Natl. Acad. Sci. USA 2013, 110, 1273–1278. [Google Scholar] [CrossRef]
- Birol, M.; Enchev, R.I.; Padilla, A.; Stengel, F.; Aebersold, R.; Betzi, S.; Yang, Y.; Hoh, F.; Peter, M.; Dumas, C.; et al. Structural and Biochemical Characterization of the Cop9 Signalosome CSN5/CSN6 Heterodimer. PLOS ONE 2014, 9, 105688. [Google Scholar] [CrossRef]
- Dessau, M.; Halimi, Y.; Erez, T.; Chomsky-Hecht, O.; Chamovitz, D.A.; Hirsch, J.A. The Arabidopsis COP9 Signalosome Subunit 7 Is a Model PCI Domain Protein with Subdomains Involved in COP9 Signalosome Assembly[W]. Plant Cell 2008, 20, 2815–2834. [Google Scholar] [CrossRef]
- Hofmann, K.; Bucher, P. The PCI domain: a common theme in three multiprotein complexes. Trends Biochem. Sci. 1998, 23, 204–205. [Google Scholar] [CrossRef]
- Cavadini, S.; Fischer, E.S.; Bunker, R.D.; Potenza, A.; Lingaraju, G.M.; Goldie, K.N.; Mohamed, W.I.; Faty, M.; Petzold, G.; Beckwith, R.E.J.; et al. Cullin–RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature 2016, 531, 598–603. [Google Scholar] [CrossRef]
- Mosadeghi, R.; Reichermeier, K.M.; Winkler, M.; Schreiber, A.; Reitsma, J.M.; Zhang, Y.; Stengel, F.; Cao, J.; Kim, M.; Sweredoski, M.J.; et al. Structural and kinetic analysis of the COP9-Signalosome activation and the cullin-RING ubiquitin ligase deneddylation cycle. eLife 2016, 5, e12102. [Google Scholar] [CrossRef]
- Kleiger, G.; Saha, A.; Lewis, S.; Kuhlman, B.; Deshaies, R.J. Rapid E2-E3 assembly and disassembly enable processive ubiquitylation of cullin-RING ubiquitin ligase substrates. Cell 2009, 139, 957–968. [Google Scholar] [CrossRef]
- Fischer, E.S.; Scrima, A.; Böhm, K.; Matsumoto, S.; Lingaraju, G.M.; Faty, M.; Yasuda, T.; Cavadini, S.; Wakasugi, M.; Hanaoka, F.; et al. The Molecular Basis of CRL4DDB2/CSA Ubiquitin Ligase Architecture, Targeting, and Activation. Cell 2011, 147, 1024–1039. [Google Scholar] [CrossRef]
- Bornstein, G.; Ganoth, D.; Hershko, A. Regulation of neddylation and deneddylation of cullin1 in SCFSkp2 ubiquitin ligase by F-box protein and substrate. Proc. Natl. Acad. Sci. USA 2006, 103, 11515–11520. [Google Scholar] [CrossRef]
- Besten, W.D.; Verma, R.; Kleiger, G.; Oania, R.S.; Deshaies, R.J. NEDD8 links Cullin–Ring ubiquitin Ligase function to the p97 pathway. Nat. Struct. Mol. Boil. 2012, 19, 511–516. [Google Scholar] [CrossRef]
- Liu, X.; Reitsma, J.M.; Mamrosh, J.L.; Zhang, Y.; Straube, R.; Deshaies, R.J. Cand1-Mediated Adaptive Exchange Mechanism Enables Variation in F-Box Protein Expression. Mol. Cell 2018, 69, 773–786.e6. [Google Scholar] [CrossRef]
- Reitsma, J.M.; Liu, X.; Reichermeier, K.M.; Moradian, A.; Sweredoski, M.J.; Hess, S.; Deshaies, R.J. Composition and regulation of the cellular repertoire of SCF ubiquitin ligases. Cell 2017, 171, 1326–1339.e14. [Google Scholar] [CrossRef]
- Goldenberg, S.J.; Cascio, T.C.; Shumway, S.D.; Garbutt, K.C.; Liu, J.; Xiong, Y.; Zheng, N. Structure of the Cand1-Cul1-Roc1 Complex Reveals Regulatory Mechanisms for the Assembly of the Multisubunit Cullin-Dependent Ubiquitin Ligases. Cell 2004, 119, 517–528. [Google Scholar] [CrossRef]
- Hwang, H.J.; Jang, H.J.; Cocco, L.; Suh, P.G. The regulation of insulin secretion via phosphoinositide-specific phospholipase Cbeta signaling. Adv. Biol. Regul. 2019, 71, 10–18. [Google Scholar] [CrossRef]
- Verbsky, J.W.; Chang, S.C.; Wilson, M.P.; Mochizuki, Y.; Majerus, P.W. The pathway for the production of inositol hexakisphosphate in human cells. J. Biol. Chem. 2005, 280, 1911–1920. [Google Scholar] [CrossRef]
- Shears, S.B. Intimate connections: Inositol pyrophosphates at the interface of metabolic regulation and cell signaling. J. Cell Physiol. 2018, 233, 1897–1912. [Google Scholar] [CrossRef]
- Wilson, M.P.; Sun, Y.; Cao, L.; Majerus, P.W. Inositol 1,3,4-Trisphosphate 5/6-Kinase Is a Protein Kinase That Phosphorylates the Transcription Factors c-Jun and ATF-2. J. Boil. Chem. 2001, 276, 40998–41004. [Google Scholar] [CrossRef]
- Sun, Y.; Wilson, M.P.; Majerus, P.W. Inositol 1,3,4-Trisphosphate 5/6-Kinase Associates with the COP9 Signalosome by Binding to CSN1. J. Boil. Chem. 2002, 277, 45759–45764. [Google Scholar] [CrossRef]
- Uhle, S.; Medalia, O.; Waldron, R.; Dumdey, R.; Henklein, P.; Bech-Otschir, D.; Huang, X.; Berse, M.; Sperling, J.; Schade, R.; et al. Protein kinase CK2 and protein kinase D are associated with the COP9 signalosome. EMBO J. 2003, 22, 1302–1312. [Google Scholar] [CrossRef]
- Tan, X.; Calderón-Villalobos, L.I.A.; Sharon, M.; Zheng, C.; Robinson, C.V.; Estelle, M.; Zheng, N. Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 2007, 446, 640–645. [Google Scholar] [CrossRef]
- Rao, F.; Xu, J.; Khan, A.B.; Gadalla, M.M.; Cha, J.Y.; Xu, R.; Tyagi, R.; Dang, Y.; Chakraborty, A.; Snyder, S.H. Inositol hexakisphosphate kinase-1 mediates assembly/disassembly of the CRL4–signalosome complex to regulate DNA repair and cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 16005–16010. [Google Scholar] [CrossRef]
- Scrima, A.; Fischer, E.S.; Lingaraju, G.M.; Böhm, K.; Cavadini, S.; Thoma, N.H. Detecting UV-lesions in the genome: The modular CRL4 ubiquitin ligase does it best! FEBS Lett. 2011, 585, 2818–2825. [Google Scholar] [CrossRef]
- Scherer, P.C.; Ding, Y.; Liu, Z.; Xu, J.; Mao, H.; Barrow, J.C.; Wei, N.; Zheng, N.; Snyder, S.H.; Rao, F. Inositol hexakisphosphate (IP6) generated by IP5K mediates cullin-COP9 signalosome interactions and CRL function. Proc. Natl. Acad. Sci. USA 2016, 113, 3503–3508. [Google Scholar] [CrossRef]
- Kleiger, G.; Hao, B.; Mohl, D.A.; Deshaies, R.J. The Acidic Tail of the Cdc34 Ubiquitin-conjugating Enzyme Functions in Both Binding to and Catalysis with Ubiquitin Ligase SCFCdc4*. J. Boil. Chem. 2009, 284, 36012–36023. [Google Scholar] [CrossRef]
- Shears, S.B. Inositol pyrophosphates: why so many phosphates? Adv. Biol. Regul. 2015, 57, 203–216. [Google Scholar] [CrossRef]
- Castellana, M.; Wilson, M.Z.; Xu, Y.; Joshi, P.; Cristea, I.M.; Rabinowitz, J.D.; Gitai, Z.; Wingreen, N.S. Enzyme clustering accelerates processing of intermediates through metabolic channeling. Nat. Biotechnol. 2014, 32, 1011–1018. [Google Scholar] [CrossRef]
- Li, L.; Wang, M.; Yu, G.; Chen, P.; Li, H.; Wei, D.; Zhu, J.; Xie, L.; Jia, H.; Shi, J.; et al. Overactivated Neddylation Pathway as a Therapeutic Target in Lung Cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Sekeres, M.A.; Fram, R.J.; Hua, Z.; Ades, L. Phase 3 study of first line pevonedistat (PEV) + azacitidine (AZA) versus single-agent AZA in patients with higher-risk myelodysplastic syndromes (HR MDS), chronic myelomonocytic leukemia (CMML) or low-blast acute myelogenous leukemia (AML). J. Clin. Oncol. 2018, TPS7077. [Google Scholar] [CrossRef]
- Rozen, S.; -Levi, M.G.F.; Ben-Nissan, G.; Mizrachi, L.; Gabashvili, A.; Levin, Y.; Ben-Dor, S.; Eisenstein, M.; Sharon, M. CSNAP is a stoichiometric subunit of the COP9 signalosome. Cell Rep. 2015, 13, 585–598. [Google Scholar] [CrossRef]
- Duda, D.M.; Olszewski, J.L.; Tron, A.E.; Hammel, M.; Lambert, L.J.; Waddell, M.B.; Mittag, T.; DeCaprio, J.A.; Schulman, B.A. Structure of a Glomulin-RBX1-CUL1 complex: inhibition of a RING E3 ligase through masking of its E2-binding surface. Mol. Cell 2012, 47, 371–382. [Google Scholar] [CrossRef]
- Xu, G.-P.; Zhang, Z.-L.; Xiao, S.; Zhuang, L.-K.; Xia, D.; Zou, Q.-P.; Jia, P.-M.; Tong, J.-H. Rig-G negatively regulates SCF-E3 ligase activities by disrupting the assembly of COP9 signalosome complex. Biochem. Biophys. Res. Commun. 2013, 432, 425–430. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Rao, F. Are Inositol Polyphosphates the Missing Link in Dynamic Cullin RING Ligase Regulation by the COP9 Signalosome? Biomolecules 2019, 9, 349. https://doi.org/10.3390/biom9080349
Zhang X, Rao F. Are Inositol Polyphosphates the Missing Link in Dynamic Cullin RING Ligase Regulation by the COP9 Signalosome? Biomolecules. 2019; 9(8):349. https://doi.org/10.3390/biom9080349
Chicago/Turabian StyleZhang, Xiaozhe, and Feng Rao. 2019. "Are Inositol Polyphosphates the Missing Link in Dynamic Cullin RING Ligase Regulation by the COP9 Signalosome?" Biomolecules 9, no. 8: 349. https://doi.org/10.3390/biom9080349
APA StyleZhang, X., & Rao, F. (2019). Are Inositol Polyphosphates the Missing Link in Dynamic Cullin RING Ligase Regulation by the COP9 Signalosome? Biomolecules, 9(8), 349. https://doi.org/10.3390/biom9080349