
Unraveling Osteoarthritis: Mechanistic Insights and Emerging Therapies Targeting Pain and Inflammation

 ,
,  , ,
, ,  ,
,
Abstract
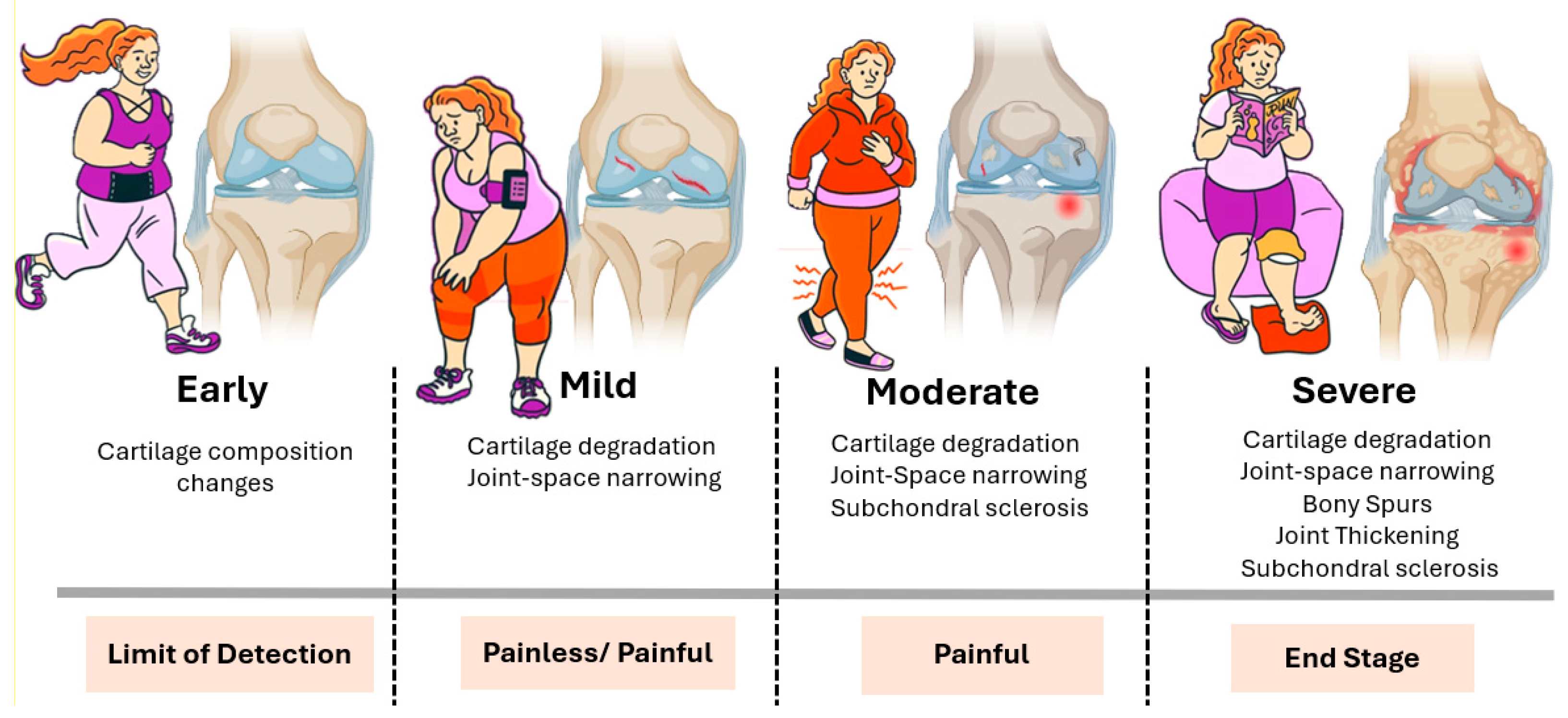
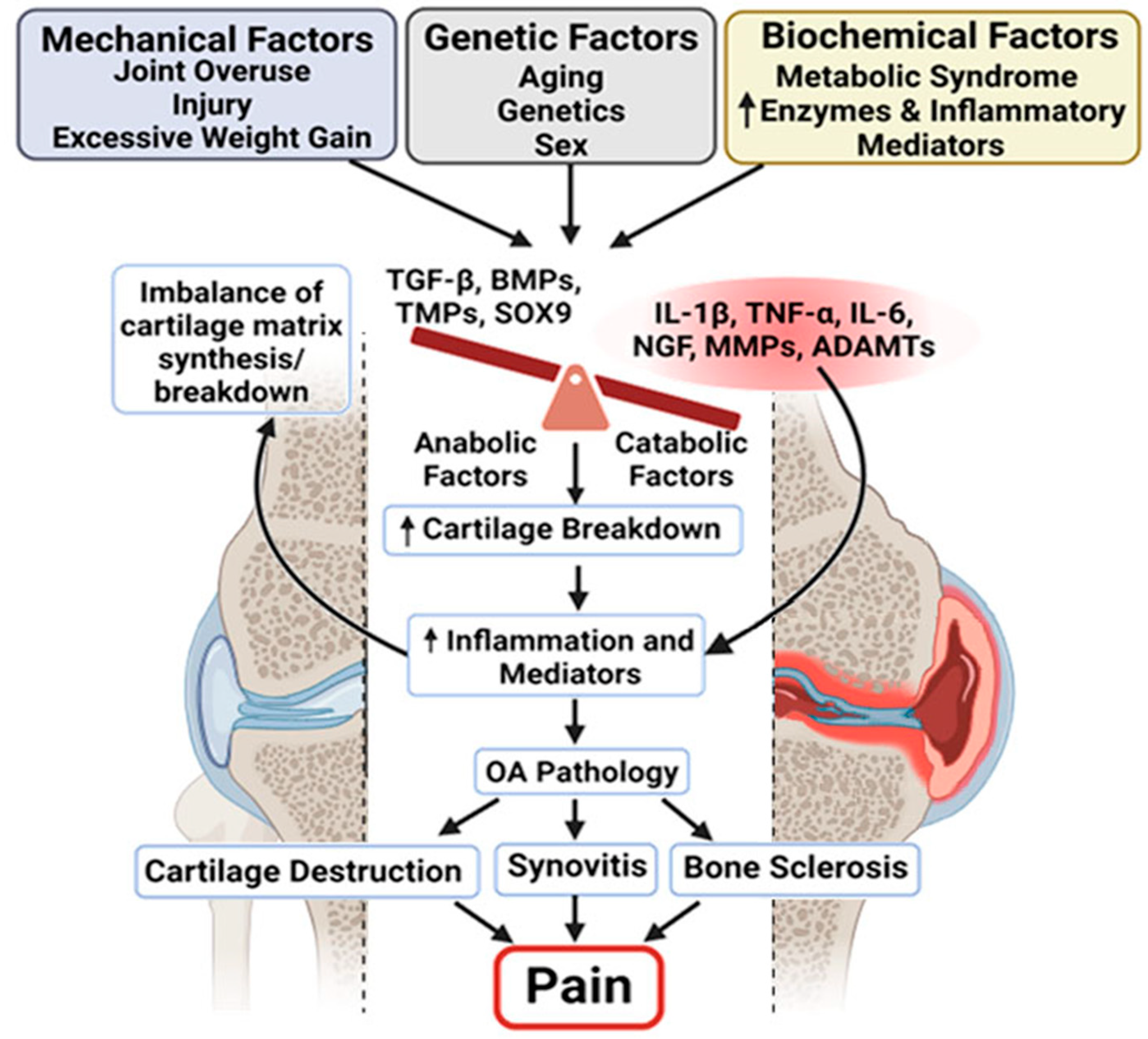
1. Pathophysiology of OA as a Whole-Joint Disease
2. Pain as Both a Sensory and Emotional Experience
3. Exploring Pain in OA: Beyond Sensation to Emotion
3.1. The Role of DAMPs and PRRs in the Pathogenesis of Pain in OA
3.2. Pain Markers in Osteoarthritis
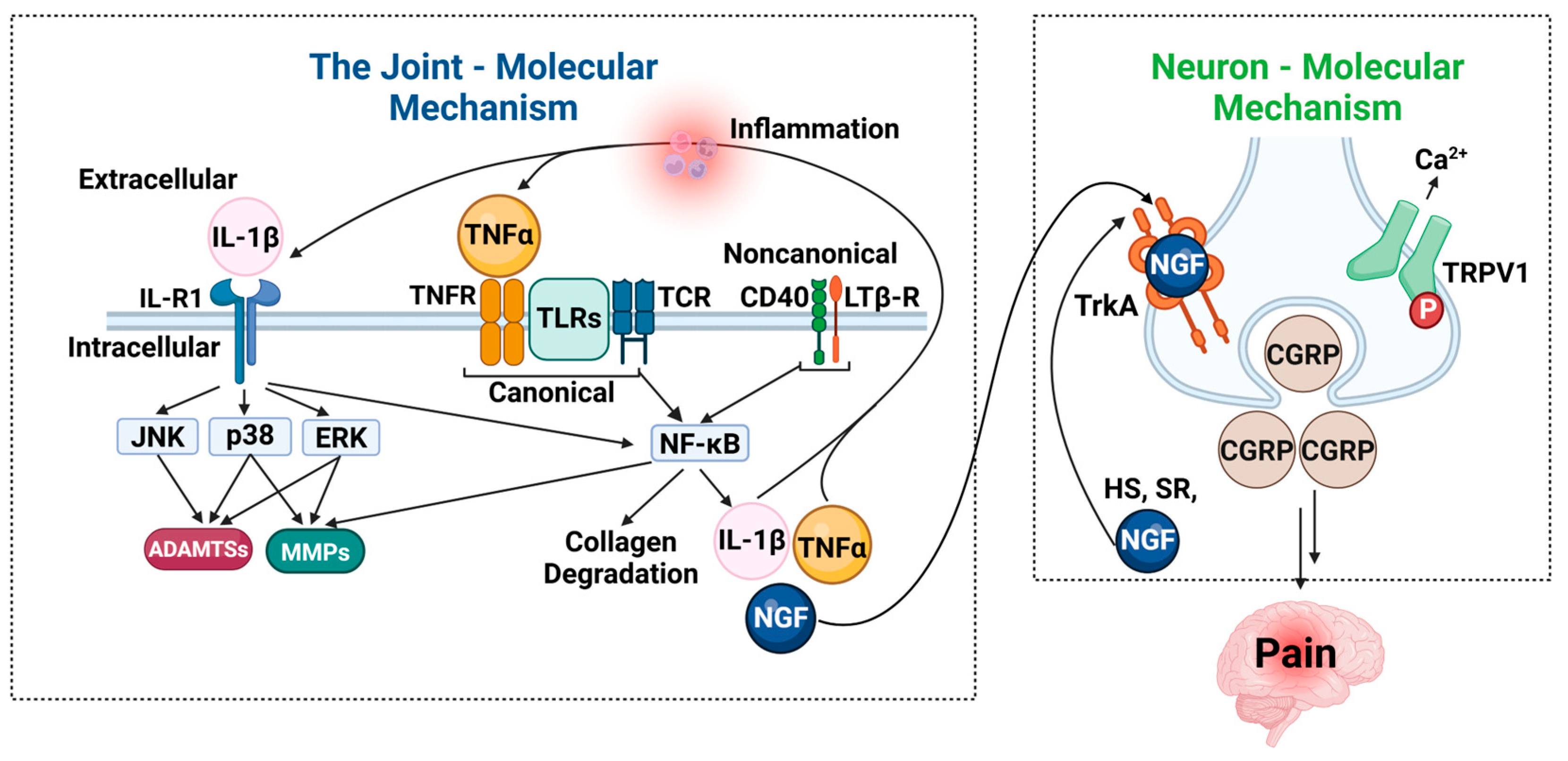
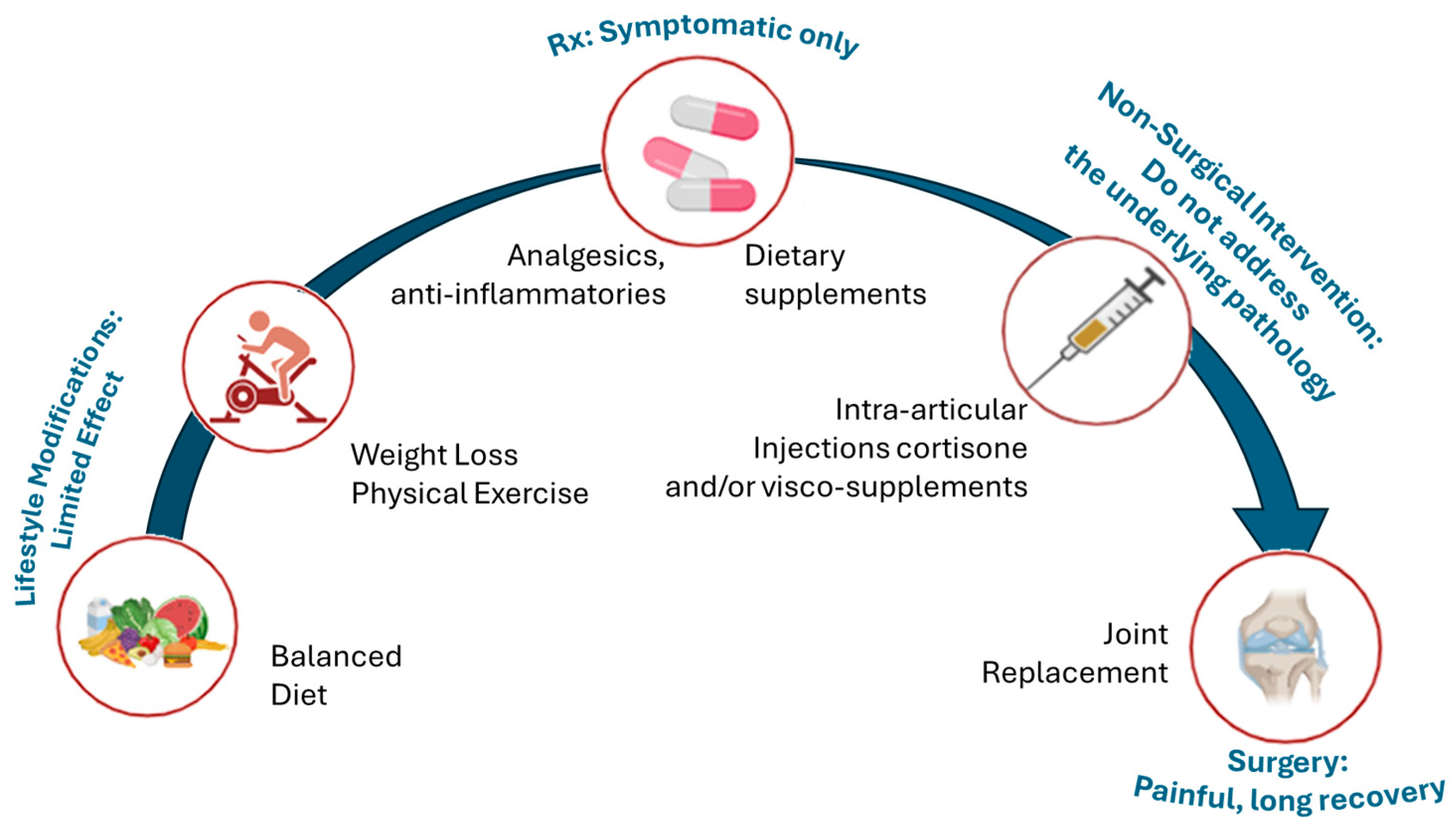
4. Current Treatments for OA
5. Regenerative Therapies Targeting Pain Pathways
5.1. Orthobiologics in Osteoarthritis: PRP, Cell Therapy, and Tissue Engineering Approaches
5.2. Gene Therapies for OA
5.3. Disease-Modifying Osteoarthritis Drugs (DMOADs)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Mode of Action | Target | Benefits |
|---|---|---|---|
| MMP-inhibitor PG-116800 (NCT01919164) | Inhibits cartilage matrix degradation | Cartilage matrix | Limits degradation and slows disease progression |
| Sprifermin (truncated FGF18) | Stimulates chondrocyte proliferation | Cartilage matrix | Improves cartilage thickness [210] |
| BMP-7 or OP-1 (NCT01133613, NCT01111045, NCT00456157) | Promotes chondrogenic differentiation | Cartilage matrix | Enhances cartilage repair and reduces pain [205] |
| AMG 108 (IL-1R1 antibody) (NCT00110942) | Inhibits IL-1β activity | IL-1 receptor | Reduces inflammation and failed to demonstrate significant clinical benefit [211] |
| Adalimumab (TNF inhibitor) (ACTRN 12612000791831) | Blocks TNF-α signaling | TNF-α receptor | Reduces pain and improves physical function |
| Infliximab | Inhibits TNF-alpha | TNFα receptor | Reduced progression of hand OA in recent-onset RA patients [212] |
| Tanezumab (anti-NGF antibody) | Blocks NGF-TrkA interaction | Targets NGF | Improves joint functional and pain scores, safety concerns, and NCT02697773 |
| Trans-capsaicin (CNTX-4975) | Inhibits TRPV1 receptor | TRPV1 | Decreases pain perception |
| Mavatrep (JNJ-39439335) | Inhibits TRPV1 | TRPV1 | Significant pain reduction but dose adjustments needed (EudraCT 2009-010961-21) |
| Selective agonist CR845 | Inhibits opioid receptors | Activates kappa-opioid receptor | Dose-dependent pain reduction, effective in hip OA (NCT02524197 and NCT02944448) |
6. Pain Measurement for Drug Development
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| OA | Osteoarthritis |
| ECM | Extracellular Matrix |
| DRG | Dorsal Root Ganglion |
| ADAMTS | A Disintegrin and Metalloproteinase with Thrombospondin Motifs |
| DAMPs | Damage-Associated Molecular Patterns |
| IL1β | Interleukin 1β |
| IL1R1 | Interleukin 1 Receptor 1 |
| TNF-α | Tumour Necrosis Factor-alpha |
| TrkA | Tropomyosin receptor kinase A |
| LTβ | Lymphotoxin β |
| TLRs | Toll-like receptors |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| NGF | Nerve Growth Factor |
| JAK/STAT | Janus Kinase/Signal Transducer and Activator of Transcription |
| MAPK | Mitogen-Activated Protein Kinase |
| MMP | Matrix Metalloproteinase |
| MSCs | Mesenchymal Stem Cells |
| BMSCs | Bone Marrow-derived Mesenchymal Stem Cells |
| PRP | Platelet-Rich Plasma |
| PRRs | Pattern-Recognition Receptors |
| S100A8/9 | S100 Calcium Binding Proteins A8/A9 |
| IL-1Ra | IL-1 receptor antagonist |
| HMGB1 | High-Mobility Group Box 1 |
| HSPs | Heat Shock Proteins |
| PDGF | Platelet-Derived Growth |
| TGF-β | Transforming Growth Factor Beta |
| HS | Histamine |
| SP | Substance P |
| CSD | Corticosteroid Drugs |
| NRS | Numeric Rating Scale |
| VAS | Visual Analog Scale |
| VDS | Verbal Descriptor Scale |
| MPQ | McGill Pain Questionnaire |
| BPI | Brief Pain Inventory |
| KOOS | Knee Injury and Osteoarthritis Outcome Score |
| HOOS | Hip Disability and Osteoarthritis Outcome Score |
| IKDC | International Knee Documentation Committee |
| CPM | Conditioned Pain Modulation |
| QST | Quantitative Sensory Testing |
References
- Glyn-Jones, S.; Palmer, A.J.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef] [PubMed]
- Cieza, A.; Causey, K.; Kamenov, K.; Hanson, S.W.; Chatterji, S.; Vos, T. Global estimates of the need for rehabilitation based on the Global Burden of Disease study 2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Wu, X.; Tao, C.; Gong, W.; Chen, M.; Qu, M.; Zhong, Y.; He, T.; Chen, S.; Xiao, G. Osteoarthritis: Pathogenic signaling pathways and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 56. [Google Scholar] [CrossRef]
- Bannuru, R.R.; Osani, M.C.; Vaysbrot, E.E.; Arden, N.K.; Bennell, K.; Bierma-Zeinstra, S.M.A.; Kraus, V.B.; Lohmander, L.S.; Abbott, J.H.; Bhandari, M.; et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthr. Cartil. 2019, 27, 1578–1589. [Google Scholar] [CrossRef]
- da Costa, B.R.; Reichenbach, S.; Keller, N.; Nartey, L.; Wandel, S.; Jüni, P.; Trelle, S. Effectiveness of non-steroidal anti-inflammatory drugs for the treatment of pain in knee and hip osteoarthritis: A network meta-analysis. Lancet 2017, 390, E21–E33. [Google Scholar] [CrossRef]
- Zhang, W.; Ouyang, H.; Dass, C.R.; Xu, J. Current research on pharmacologic and regenerative therapies for osteoarthritis. Bone Res. 2016, 4, 15040. [Google Scholar] [CrossRef]
- Pelletier, J.P.; Martel-Pelletier, J.; Rannou, F.; Cooper, C. Efficacy and safety of oral NSAIDs and analgesics in the management of osteoarthritis: Evidence from real-life setting trials and surveys. Semin. Arthritis Rheum. 2016, 45, S22–S27. [Google Scholar] [CrossRef]
- Tong, L.; Yu, H.; Huang, X.; Shen, J.; Xiao, G.; Chen, L.; Wang, H.; Xing, L.; Chen, D. Current understanding of osteoarthritis pathogenesis and relevant new approaches. Bone Res. 2022, 10, 60. [Google Scholar] [CrossRef]
- Farinelli, L.; Riccio, M.; Gigante, A.; De Francesco, F. Pain Management Strategies in Osteoarthritis. Biomedicines 2024, 12, 805. [Google Scholar] [CrossRef]
- Skou, S.T.; Roos, E.M. Good Life with osteoArthritis in Denmark (GLA:D™): Evidence-based education and supervised neuromuscular exercise delivered by certified physiotherapists nationwide. BMC Musculoskelet. Disord. 2017, 18, 72. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.E.V.; Murray, C.M.; Stanton, T.R. Patient perspectives of pain and function after knee replacement: A systematic review and meta-synthesis of qualitative studies. Pain Rep. 2022, 7, e1006. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M.; Newman, S. Trends and developments in hip and knee arthroplasty technology. J. Rehabil. Assist. Technol. Eng. 2021, 8, 2055668320952043. [Google Scholar] [CrossRef]
- Goode, V.M.; Morgan, B.; Muckler, V.C.; Cary, M.P., Jr.; Zdeb, C.E.; Zychowicz, M. Multimodal Pain Management for Major Joint Replacement Surgery. Orthop. Nurs. 2019, 38, 150–156. [Google Scholar] [CrossRef]
- Leifer, V.P.; Katz, J.N.; Losina, E. The burden of OA-health services and economics. Osteoarthr. Cartil. 2022, 30, 10–16. [Google Scholar] [CrossRef]
- Fallon, E.A.; Boring, M.A.; Foster, A.L.; Stowe, E.W.; Lites, T.D.; Odom, E.L.; Seth, P. Prevalence of Diagnosed Arthritis—United States, 2019–2021. Morb. Mortal. Wkly. Rep. 2023, 72, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Yelin, E.; Weinstein, S.; King, T. The burden of musculoskeletal diseases in the United States. Semin. Arthritis Rheum. 2016, 46, 259–260. [Google Scholar] [CrossRef]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef]
- Berenbaum, F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthr. Cartil. 2013, 21, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Coaccioli, S.; Sarzi-Puttini, P.; Zis, P.; Rinonapoli, G.; Varrassi, G. Osteoarthritis: New Insight on Its Pathophysiology. J. Clin. Med. 2022, 11, 6013. [Google Scholar] [CrossRef]
- Abramoff, B.; Caldera, F.E. Osteoarthritis. Med. Clin. N. Am. 2020, 104, 299–311. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, X.; Wang, S.; Jing, Y.; Su, J. Subchondral bone microenvironment in osteoarthritis and pain. Bone Res. 2021, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Bačenková, D.; Trebuňová, M.; Demeterová, J.; Živčák, J. Human Chondrocytes, Metabolism of Articular Cartilage, and Strategies for Application to Tissue Engineering. Int. J. Mol. Sci. 2023, 24, 17096. [Google Scholar] [CrossRef]
- Hodgkinson, T.; Kelly, D.C.; Curtin, C.M.; O’Brien, F.J. Mechanosignalling in cartilage: An emerging target for the treatment of osteoarthritis. Nat. Rev. Rheumatol. 2022, 18, 67–84. [Google Scholar] [CrossRef]
- Heinegård, D.; Saxne, T. The role of the cartilage matrix in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Boileau, C.; Pelletier, J.P.; Roughley, P.J. Cartilage in normal and osteoarthritis conditions. Best Pract. Res. Clin. Rheumatol. 2008, 22, 351–384. [Google Scholar] [CrossRef]
- Burr, D.B.; Gallant, M.A. Bone remodelling in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 665–673. [Google Scholar] [CrossRef]
- Hunter, D.J.; Felson, D.T. Osteoarthritis. BMJ 2006, 332, 639–642. [Google Scholar] [CrossRef]
- Findlay, D.M. Vascular pathology and osteoarthritis. Rheumatology 2007, 46, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R.; Goldring, M.B. Changes in the osteochondral unit during osteoarthritis: Structure, function and cartilage–bone crosstalk. Nat. Rev. Rheumatol. 2016, 12, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Liu, X.; He, Z.; Han, X.; Yan, M.; Qu, X.; Li, X.; Yu, Z. Articular Cartilage Degradation and Aberrant Subchondral Bone Remodeling in Patients with Osteoarthritis and Osteoporosis. J. Bone Miner. Res. 2019, 35, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chan, Y.T.; Yung, P.S.H.; Tuan, R.S.; Jiang, Y. Subchondral Bone Remodeling: A Therapeutic Target for Osteoarthritis. Front. Cell Dev. Biol. 2021, 8, 607764. [Google Scholar] [CrossRef]
- Neogi, T.; Guermazi, A.; Roemer, F.; Nevitt, M.C.; Scholz, J.; Arendt-Nielsen, L.; Woolf, C.; Niu, J.; Bradley, L.A.; Quinn, E.; et al. Association of Joint Inflammation with Pain Sensitization in Knee Osteoarthritis: The Multicenter Osteoarthritis Study. Arthritis Rheumatol. 2016, 68, 654–661. [Google Scholar] [CrossRef]
- Sharma, L.; Song, J.; Felson, D.T.; Cahue, S.; Shamiyeh, E.; Dunlop, D.D. The role of knee alignment in disease progression and functional decline in knee osteoarthritis. JAMA 2001, 286, 188–195. [Google Scholar] [CrossRef]
- Felson, D.T. Clinical practice. Osteoarthritis of the knee. N. Engl. J. Med. 2006, 354, 841–848. [Google Scholar] [CrossRef]
- Diracoglu, D.; Aydin, R.; Baskent, A.; Celik, A. Effects of kinesthesia and balance exercises in knee osteoarthritis. J. Clin. Rheumatol. 2005, 11, 303–310. [Google Scholar] [CrossRef]
- Buckwalter, J.A.; Anderson, D.D.; Brown, T.D.; Tochigi, Y.; Martin, J.A. The Roles of Mechanical Stresses in the Pathogenesis of Osteoarthritis: Implications for Treatment of Joint Injuries. Cartilage 2013, 4, 286–294. [Google Scholar] [CrossRef]
- Bennell, K.L.; Hunt, M.A.; Wrigley, T.V.; Lim, B.W.; Hinman, R.S. Role of muscle in the genesis and management of knee osteoarthritis. Rheum. Dis. Clin. N. Am. 2008, 34, 731–754. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Coras, R.; Torres, A.; Lane, N.E.; Guma, M. Synovial inflammation in osteoarthritis progression. Nat. Rev. Rheumatol. 2022, 18, 258–275. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Barr, A.J.; Cicuttini, F.M.; Conaghan, P.G.; Cooper, C.; Goldring, M.B.; Goldring, S.R.; Jones, G.; Teichtahl, A.J.; Pelletier, J.-P. Osteoarthritis. Nat. Rev. Dis. Primers 2016, 2, 16072. [Google Scholar] [CrossRef] [PubMed]
- Grässel, S.; Zaucke, F.; Madry, H. Osteoarthritis: Novel Molecular Mechanisms Increase Our Understanding of the Disease Pathology. J. Clin. Med. 2021, 10, 1938. [Google Scholar] [CrossRef]
- Chow, Y.Y.; Chin, K.-Y. The Role of Inflammation in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2020, 2020, 8293921. [Google Scholar] [CrossRef]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Ther. Adv. Musculoskelet. Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef]
- Lotz, M.; Martel-Pelletier, J.; Christiansen, C.; Brandi, M.L.; Bruyère, O.; Chapurlat, R.; Collette, J.; Cooper, C.; Giacovelli, G.; Kanis, J.A.; et al. Value of biomarkers in osteoarthritis: Current status and perspectives. Ann. Rheum. Dis. 2013, 72, 1756–1763. [Google Scholar] [CrossRef]
- Heijink, A.; Gomoll, A.H.; Madry, H.; Drobnič, M.; Filardo, G.; Espregueira-Mendes, J.; Van Dijk, C.N. Biomechanical considerations in the pathogenesis of osteoarthritis of the knee. Knee Surg. Sports Traumatol. Arthrosc. 2012, 20, 423–435. [Google Scholar] [CrossRef]
- Guilak, F. Biomechanical factors in osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2011, 25, 815–823. [Google Scholar] [CrossRef]
- van den Bosch, M.H.J.; Blom, A.B.; van der Kraan, P.M. Inflammation in osteoarthritis: Our view on its presence and involvement in disease development over the years. Osteoarthr. Cartil. 2024, 32, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Lee, K.; Ju, J.H. Recent Updates of Diagnosis, Pathophysiology, and Treatment on Osteoarthritis of the Knee. Int. J. Mol. Sci. 2021, 22, 2619. [Google Scholar] [CrossRef]
- Sellam, J.; Berenbaum, F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 2010, 6, 625–635. [Google Scholar] [CrossRef]
- Mathiessen, A.; Conaghan, P.G. Synovitis in osteoarthritis: Current understanding with therapeutic implications. Arthritis Res. Ther. 2017, 19, 18. [Google Scholar] [CrossRef] [PubMed]
- Ayral, X.; Pickering, E.H.; Woodworth, T.G.; Mackillop, N.; Dougados, M. Synovitis: A potential predictive factor of structural progression of medial tibiofemoral knee osteoarthritis—Results of a 1 year longitudinal arthroscopic study in 422 patients. Osteoarthr. Cartil. 2005, 13, 361–367. [Google Scholar] [CrossRef]
- McKune, C.M. Nociception and pain. In Veterinary Anesthesia and Analgesia: The Fifth Edition of Lumb and Jones; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 584–623. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Herr, M.J. Physiology, Nociception; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Schaible, H.G. Mechanisms of Chronic Pain in Osteoarthritis. Curr. Rheumatol. Rep. 2012, 14, 549–556. [Google Scholar] [CrossRef]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef]
- Wood, M.J.; Miller, R.E.; Malfait, A.-M. The Genesis of Pain in Osteoarthritis: Inflammation as a Mediator of Osteoarthritis Pain. Clin. Geriatr. Med. 2022, 38, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Robbins, S.R.; McDougall, J.J. Osteoarthritis: The genesis of pain. Rheumatology 2017, 57, iv43–iv50. [Google Scholar] [CrossRef]
- Morgan, M.; Nazemian, V.; Harrington, K.; Ivanusic, J.J. Mini review: The role of sensory innervation to subchondral bone in osteoarthritis pain. Front. Endocrinol. 2022, 13, 1047943. [Google Scholar] [CrossRef]
- Yu, H.; Huang, T.; Lu, W.W.; Tong, L.; Chen, D. Osteoarthritis Pain. Int. J. Mol. Sci. 2022, 23, 4642. [Google Scholar] [CrossRef] [PubMed]
- Perrot, S. Osteoarthritis pain. Best Pract. Res. Clin. Rheumatol. 2015, 29, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.P.; Vase, L.; Hooten, W.M. Chronic pain: An update on burden, best practices, and new advances. Lancet 2021, 397, 2082–2097. [Google Scholar] [CrossRef]
- Bechler, C.J. A Categorical Perspective on Attitudes: Implications for Perceived Change, Persuasive Targeting, and the Attitude-Behavior Relationship. Ph.D. Thesis, Stanford University, Stanford, CA, USA, 2021. [Google Scholar]
- Ohashi, Y.; Uchida, K.; Fukushima, K.; Inoue, G.; Takaso, M. Mechanisms of Peripheral and Central Sensitization in Osteoarthritis Pain. Cureus 2023, 15, e35331. [Google Scholar] [CrossRef]
- Wise, B.L.; Niu, J.; Zhang, Y.; Wang, N.; Jordan, J.M.; Choy, E.; Hunter, D.J. Psychological factors and their relation to osteoarthritis pain. Osteoarthr. Cartil. 2010, 18, 883–887. [Google Scholar] [CrossRef]
- Vincent, T.L. Peripheral pain mechanisms in osteoarthritis. PAIN 2020, 161, S138–S146. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.J.; Couto, M.; Sousa, D.M.; Magalhães, A.; Neto, E.; Leitão, L.; Conceição, F.; Monteiro, A.C.; Ribeiro-da-Silva, M.; Lamghari, M. Nociceptive mechanisms driving pain in a post-traumatic osteoarthritis mouse model. Sci. Rep. 2020, 10, 15271. [Google Scholar] [CrossRef]
- Eitner, A.; Hofmann, G.O.; Schaible, H.-G. Mechanisms of Osteoarthritic Pain. Studies in Humans and Experimental Models. Front. Mol. Neurosci. 2017, 10, 349. [Google Scholar] [CrossRef]
- He, Y.; Li, Z.; Alexander, P.G.; Ocasio-Nieves, B.D.; Yocum, L.; Lin, H.; Tuan, R.S. Pathogenesis of Osteoarthritis: Risk Factors, Regulatory Pathways in Chondrocytes, and Experimental Models. Biology 2020, 9, 194. [Google Scholar] [CrossRef]
- Chen, D.; Shen, J.; Zhao, W.; Wang, T.; Han, L.; Hamilton, J.L.; Im, H.-J. Osteoarthritis: Toward a comprehensive understanding of pathological mechanism. Bone Res. 2017, 5, 16044. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kim, J.S.; van Wijnen, A.J.; Im, H.J. Osteoarthritic tissues modulate functional properties of sensory neurons associated with symptomatic OA pain. Mol. Biol. Rep. 2011, 38, 5335–5339. [Google Scholar] [CrossRef] [PubMed]
- Ayobami, O.O.; Goldring, S.R.; Goldring, M.B.; Wright, T.M.; van der Meulen, M.C.H. Contribution of joint tissue properties to load-induced osteoarthritis. Bone Rep. 2022, 17, 101602. [Google Scholar] [CrossRef]
- Castañeda, S.; Roman-Blas, J.A.; Largo, R.; Herrero-Beaumont, G. Subchondral bone as a key target for osteoarthritis treatment. Biochem. Pharmacol. 2012, 83, 315–323. [Google Scholar] [CrossRef]
- Findlay, D.M.; Kuliwaba, J.S. Bone-cartilage crosstalk: A conversation for understanding osteoarthritis. Bone Res. 2016, 4, 16028. [Google Scholar] [CrossRef]
- Suri, S.; Walsh, D.A. Osteochondral alterations in osteoarthritis. Bone 2012, 51, 204–211. [Google Scholar] [CrossRef]
- Malfait, A.M.; Schnitzer, T.J. Towards a mechanism-based approach to pain management in osteoarthritis. Nat. Rev. Rheumatol. 2013, 9, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, G.; Villano, I.; Ilardi, C.R.; Messina, A.; Monda, V.; Iodice, A.C.; Porro, C.; Panaro, M.A.; Chieffi, S.; Messina, G.; et al. Mechanisms of Transmission and Processing of Pain: A Narrative Review. Int. J. Environ. Res. Public Health 2023, 20, 3064. [Google Scholar] [CrossRef]
- Baral, P.; Udit, S.; Chiu, I.M. Pain and immunity: Implications for host defence. Nat. Rev. Immunol. 2019, 19, 433–447. [Google Scholar] [CrossRef]
- Cao, Y.; Fan, D.; Yin, Y. Pain mechanism in rheumatoid arthritis: From cytokines to central sensitization. Mediat. Inflamm. 2020, 2020, 2076328. [Google Scholar] [CrossRef]
- Miller, R.J.; Malfait, A.M.; Miller, R.E. The innate immune response as a mediator of osteoarthritis pain. Osteoarthr. Cartil. 2020, 28, 562–571. [Google Scholar] [CrossRef]
- Chakrabarti, S. Mechanisms of Peripheral Sensitization in Inflammatory Knee Pain. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2020. [Google Scholar]
- Woolf, C.J. Central sensitization: Implications for the diagnosis and treatment of pain. PAIN 2011, 152, S2–S15. [Google Scholar] [CrossRef]
- Khan, A.; Khan, S.; Kim, Y.S. Insight into Pain Modulation: Nociceptors Sensitization and Therapeutic Targets. Curr. Drug Targets 2019, 20, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Latremoliere, A.; Woolf, C.J. Central sensitization: A generator of pain hypersensitivity by central neural plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef]
- Lluch Girbés, E.; Nijs, J.; Torres-Cueco, R.; López Cubas, C. Pain treatment for patients with osteoarthritis and central sensitization. Phys. Ther. 2013, 93, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Ohtori, S.; Orita, S.; Yamashita, M.; Ishikawa, T.; Ito, T.; Shigemura, T.; Nishiyama, H.; Konno, S.; Ohta, H.; Takaso, M.; et al. Existence of a neuropathic pain component in patients with osteoarthritis of the knee. Yonsei Med. J. 2012, 53, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Bedson, J.; Croft, P.R. The discordance between clinical and radiographic knee osteoarthritis: A systematic search and summary of the literature. BMC Musculoskelet. Disord. 2008, 9, 116. [Google Scholar] [CrossRef]
- Gwilym, S.E.; Filippini, N.; Douaud, G.; Carr, A.J.; Tracey, I. Thalamic atrophy associated with painful osteoarthritis of the hip is reversible after arthroplasty: A longitudinal voxel-based morphometric study. Arthritis Rheum. 2010, 62, 2930–2940. [Google Scholar] [CrossRef]
- Mobasheri, A.; Rayman, M.P.; Gualillo, O.; Sellam, J.; van der Kraan, P.; Fearon, U. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2017, 13, 302–311. [Google Scholar] [CrossRef]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.-P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef]
- Felson, D.T. Osteoarthritis as a disease of mechanics. Osteoarthr. Cartil. 2013, 21, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Berman, J. What Is Osteoarthritis? JAMA 2022, 327, 1300. [Google Scholar] [CrossRef]
- Langworthy, M.; Dasa, V.; Spitzer, A.I. Knee osteoarthritis: Disease burden, available treatments, and emerging options. Ther. Adv. Musculoskelet. Dis. 2024, 16, 1759720X241273009. [Google Scholar] [CrossRef] [PubMed]
- Lambert, C.; Zappia, J.; Sanchez, C.; Florin, A.; Dubuc, J.E.; Henrotin, Y. The Damage-Associated Molecular Patterns (DAMPs) as Potential Targets to Treat Osteoarthritis: Perspectives From a Review of the Literature. Front. Med. 2020, 7, 607186. [Google Scholar] [CrossRef]
- Sengprasert, P.; Kamenkit, O.; Tanavalee, A.; Reantragoon, R. The Immunological Facets of Chondrocytes in Osteoarthritis: A Narrative Review. J. Rheumatol. 2024, 51, 13–24. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, W.; Yong, H.; He, M.; Yang, Y.; Deng, Z.; Li, Y. Macrophages in osteoarthritis: Pathophysiology and therapeutics. Am. J. Transl. Res. 2020, 12, 261–268. [Google Scholar] [PubMed]
- Scanzello, C.R. Role of low-grade inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2017, 29, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef]
- Sofat, N. Analysing the role of endogenous matrix molecules in the development of osteoarthritis. Int. J. Exp. Pathol. 2009, 90, 463–479. [Google Scholar] [CrossRef]
- van Lent, P.L.; Blom, A.B.; Schelbergen, R.F.; Slöetjes, A.; Lafeber, F.P.; Lems, W.F.; Cats, H.; Vogl, T.; Roth, J.; van den Berg, W.B. Active involvement of alarmins S100A8 and S100A9 in the regulation of synovial activation and joint destruction during mouse and human osteoarthritis. Arthritis Rheum. 2012, 64, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Schaefer, L. Complexity of Danger: The Diverse Nature of Damage-associated Molecular Patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef]
- Harris, H.E.; Raucci, A. Alarmin(g) news about danger. EMBO Rep. 2006, 7, 774–778. [Google Scholar] [CrossRef]
- Piccinini, A.M.; Midwood, K.S. DAMPening Inflammation by Modulating TLR Signalling. Mediat. Inflamm. 2010, 2010, 672395. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Burrage, P.S. Matrix Metalloproteinases: Role In Arthritis. Front. Biosci. 2006, 11, 529–543. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, L.M. Immune Proteins in Brain Development and Synaptic Plasticity. Neuron 2009, 64, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Gómez, R.; Villalvilla, A.; Largo, R.; Gualillo, O.; Herrero-Beaumont, G. TLR4 signalling in osteoarthritis—Finding targets for candidate DMOADs. Nat. Rev. Rheumatol. 2015, 11, 159–170. [Google Scholar] [CrossRef]
- Lotz, M.; Loeser, R.F. Effects of aging on articular cartilage homeostasis. Bone 2012, 51, 241–248. [Google Scholar] [CrossRef]
- Kraus, V.B.; Blanco, F.J.; Englund, M.; Karsdal, M.A.; Lohmander, L.S. Call for standardized definitions of osteoarthritis and risk stratification for clinical trials and clinical use. Osteoarthr. Cartil. 2015, 23, 1233–1241. [Google Scholar] [CrossRef]
- Chen, B.; Sun, Y.; Xu, G.; Jiang, J.; Zhang, W.; Wu, C.; Xue, P.; Cui, Z. Role of crosstalk between synovial cells and chondrocytes in osteoarthritis (Review). Exp. Ther. Med. 2024, 27, 201. [Google Scholar] [CrossRef]
- Soares, C.L.R.; Wilairatana, P.; Silva, L.R.; Moreira, P.S.; Vilar Barbosa, N.M.M.; da Silva, P.R.; Coutinho, H.D.M.; de Menezes, I.R.A.; Felipe, C.F.B. Biochemical aspects of the inflammatory process: A narrative review. Biomed. Pharmacother. 2023, 168, 115764. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, M.; Mobasheri, A.; Mozafari, M. Inflammatory mediators in osteoarthritis: A critical review of the state-of-the-art, current prospects, and future challenges. Bone 2016, 85, 81–90. [Google Scholar] [CrossRef]
- Goldring, M.B.; Marcu, K.B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther. 2009, 11, 224. [Google Scholar] [CrossRef]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The Sterile Inflammatory Response. Annu. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef]
- Mukherjee, A.; Das, B. The role of inflammatory mediators and matrix metalloproteinases (MMPs) in the progression of osteoarthritis. Biomater. Biosyst. 2024, 13, 100090. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Van Caam, A.P.M.; Van Der Kraan, P.M. Osteoarthritis year in review 2016: Biology. Osteoarthr. Cartil. 2017, 25, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Knights, A.J.; Redding, S.J.; Maerz, T. Inflammation in osteoarthritis: The latest progress and ongoing challenges. Curr. Opin. Rheumatol. 2023, 35, 128–134. [Google Scholar] [CrossRef]
- Akkiraju, H.; Nohe, A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J. Dev. Biol. 2015, 3, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Deng, Z.; Chen, K.; Jian, S.; Zhou, F.; Yang, Y.; Fu, Z.; Xie, H.; Xiong, J.; Zhu, W. Cartilage tissue engineering: From proinflammatory and anti-inflammatory cytokines to osteoarthritis treatments (Review). Mol. Med. Rep. 2022, 25, 99. [Google Scholar] [CrossRef]
- Maldonado, M.; Nam, J. The role of changes in extracellular matrix of cartilage in the presence of inflammation on the pathology of osteoarthritis. Biomed. Res. Int. 2013, 2013, 284873. [Google Scholar] [CrossRef]
- Mononen, M.E.; Tanska, P.; Isaksson, H.; Korhonen, R.K. A Novel Method to Simulate the Progression of Collagen Degeneration of Cartilage in the Knee: Data from the Osteoarthritis Initiative. Sci. Rep. 2016, 6, 21415. [Google Scholar] [CrossRef]
- Terkawi, M.A.; Ebata, T.; Yokota, S.; Takahashi, D.; Endo, T.; Matsumae, G.; Shimizu, T.; Kadoya, K.; Iwasaki, N. Low-Grade Inflammation in the Pathogenesis of Osteoarthritis: Cellular and Molecular Mechanisms and Strategies for Future Therapeutic Intervention. Biomedicines 2022, 10, 1109. [Google Scholar] [CrossRef]
- Molnar, V.; Matišić, V.; Kodvanj, I.; Bjelica, R.; Jeleč, Ž.; Hudetz, D.; Rod, E.; Čukelj, F.; Vrdoljak, T.; Vidović, D.; et al. Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis. Int. J. Mol. Sci. 2021, 22, 9208. [Google Scholar] [CrossRef]
- Jacques, C.; Gosset, M.; Berenbaum, F.; Gabay, C. The Role of IL-1 and IL-1Ra in Joint Inflammation and Cartilage Degradation. In Vitamins & Hormones; Academic Press: Cambridge, MA, USA, 2006; Volume 74, pp. 371–403. [Google Scholar]
- Arend, W.P.; Malyak, M.; Guthridge, C.J.; Gabay, C. Interleukin-1 receptor antagonist: Role in biology. Annu. Rev. Immunol. 1998, 16, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Brandt, K.D.; Dieppe, P.; Radin, E.L. Etiopathogenesis of Osteoarthritis. Rheum. Dis. Clin. N. Am. 2008, 34, 531–559. [Google Scholar] [CrossRef]
- Pelletier, J.P.; Martel-Pelletier, J.; Abramson, S.B. Osteoarthritis, an inflammatory disease: Potential implication for the selection of new therapeutic targets. Arthritis Rheum. 2001, 44, 1237–1247. [Google Scholar] [CrossRef]
- Bhol, N.K.; Bhanjadeo, M.M.; Singh, A.K.; Dash, U.C.; Ojha, R.R.; Majhi, S.; Duttaroy, A.K.; Jena, A.B. The interplay between cytokines, inflammation, and antioxidants: Mechanistic insights and therapeutic potentials of various antioxidants and anti-cytokine compounds. Biomed. Pharmacother. 2024, 178, 117177. [Google Scholar] [CrossRef]
- Henderson, B.; Pettipher, E.R. Arthritogenic actions of recombinant IL-1 and tumour necrosis factor alpha in the rabbit: Evidence for synergistic interactions between cytokines in vivo. Clin. Exp. Immunol. 1989, 75, 306–310. [Google Scholar]
- Chou, C.-H.; Jain, V.; Gibson, J.; Attarian, D.E.; Haraden, C.A.; Yohn, C.B.; Laberge, R.-M.; Gregory, S.; Kraus, V.B. Synovial cell cross-talk with cartilage plays a major role in the pathogenesis of osteoarthritis. Sci. Rep. 2020, 10, 10868. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Neu, M.; Germershaus, O.; Merkel, O.; Sitterberg, J.; Bakowsky, U.; Kissel, T. Influence of polyethylene glycol chain length on the physicochemical and biological properties of poly(ethylene imine)-graft-poly(ethylene glycol) block copolymer/SiRNA polyplexes. Bioconjug. Chem. 2006, 17, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Erickson, E.A.; Long, D.L. Mitogen-activated protein kinases as therapeutic targets in osteoarthritis. Curr. Opin. Rheumatol. 2008, 20, 581–586. [Google Scholar] [CrossRef]
- Prasadam, I.; Crawford, R.; Xiao, Y. Aggravation of ADAMTS and matrix metalloproteinase production and role of ERK1/2 pathway in the interaction of osteoarthritic subchondral bone osteoblasts and articular cartilage chondrocytes—Possible pathogenic role in osteoarthritis. J. Rheumatol. 2012, 39, 621–634. [Google Scholar] [CrossRef]
- Poole, A.R.; Kobayashi, M.; Yasuda, T.; Laverty, S.; Mwale, F.; Kojima, T.; Sakai, T.; Wahl, C.; El-Maadawy, S.; Webb, G.; et al. Type II collagen degradation and its regulation in articular cartilage in osteoarthritis. Ann. Rheum. Dis. 2002, 61 (Suppl. 2), II78–II81. [Google Scholar] [CrossRef]
- Marcu, K.B.; Otero, M.; Olivotto, E.; Borzi, R.M.; Goldring, M.B. NF-kappaB signaling: Multiple angles to target OA. Curr. Drug Targets 2010, 11, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.; Aguda, B.D.; Rath, B.; Agarwal, S. Biomechanical thresholds regulate inflammation through the NF-kappaB pathway: Experiments and modeling. PLoS ONE 2009, 4, e5262. [Google Scholar] [CrossRef]
- Gu, R.; Liu, N.; Luo, S.; Huang, W.; Zha, Z.; Yang, J. MicroRNA-9 regulates the development of knee osteoarthritis through the NF-kappaB1 pathway in chondrocytes. Medicine 2016, 95, e4315. [Google Scholar] [CrossRef] [PubMed]
- Roman-Blas, J.A.; Jimenez, S.A. NF-kappaB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthr. Cartil. 2006, 14, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Rigoglou, S.; Papavassiliou, A.G. The NF-κB signalling pathway in osteoarthritis. Int. J. Biochem. Cell Biol. 2013, 45, 2580–2584. [Google Scholar] [CrossRef]
- Choi, M.C.; Jo, J.; Park, J.; Kang, H.K.; Park, Y. NF-κB Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells 2019, 8, 734. [Google Scholar] [CrossRef]
- Malfait, A.M.; Miller, R.E.; Block, J.A. Targeting neurotrophic factors: Novel approaches to musculoskeletal pain. Pharmacol. Ther. 2020, 211, 107553. [Google Scholar] [CrossRef]
- Malfait, A.M.; Miller, R.E.; Miller, R.J. Basic Mechanisms of Pain in Osteoarthritis: Experimental Observations and New Perspectives. Rheum. Dis. Clin. N. Am. 2021, 47, 165–180. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, J.; McNaughton, P.A. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 2005, 24, 4211–4223. [Google Scholar] [CrossRef]
- Schaefer, I.; Prato, V.; Arcourt, A.; Taberner, F.J.; Lechner, S.G. Differential modulation of voltage-gated sodium channels by nerve growth factor in three major subsets of TrkA-expressing nociceptors. Mol. Pain 2018, 14, 1744806918814640. [Google Scholar] [CrossRef]
- Dyck, P.J.; Peroutka, S.; Rask, C.; Burton, E.; Baker, M.K.; Lehman, K.A.; Gillen, D.A.; Hokanson, J.L.; O’Brien, P.C. Intradermal recombinant human nerve growth factor induces pressure allodynia and lowered heat-pain threshold in humans. Neurology 1997, 48, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Uchida, K.; Fukushima, K.; Satoh, M.; Koyama, T.; Tsuchiya, M.; Saito, H.; Takahira, N.; Inoue, G.; Takaso, M. NGF Expression and Elevation in Hip Osteoarthritis Patients with Pain and Central Sensitization. Biomed. Res. Int. 2021, 2021, 9212585. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Wang, Z.; Tao, H. Mechanism and therapeutic effectiveness of nerve growth factor in osteoarthritis pain. Ther. Clin. Risk Manag. 2017, 13, 951–956. [Google Scholar] [CrossRef]
- Lisowska, B.; Lisowski, A.; Siewruk, K. Substance P and Chronic Pain in Patients with Chronic Inflammation of Connective Tissue. PLoS ONE 2015, 10, e0139206. [Google Scholar] [CrossRef]
- Semenistaja, S.; Skuja, S.; Kadisa, A.; Groma, V. Healthy and Osteoarthritis-Affected Joints Facing the Cellular Crosstalk. Int. J. Mol. Sci. 2023, 24, 4120. [Google Scholar] [CrossRef]
- Yin, X.; Wang, Q.; Tang, Y.; Wang, T.; Zhang, Y.; Yu, T. Research progress on macrophage polarization during osteoarthritis disease progression: A review. J. Orthop. Surg. Res. 2024, 19, 584. [Google Scholar] [CrossRef]
- Mushenkova, N.V.; Nikiforov, N.G.; Shakhpazyan, N.K.; Orekhova, V.A.; Sadykhov, N.K.; Orekhov, A.N. Phenotype Diversity of Macrophages in Osteoarthritis: Implications for Development of Macrophage Modulating Therapies. Int. J. Mol. Sci. 2022, 23, 8381. [Google Scholar] [CrossRef] [PubMed]
- Mai, L.; Liu, Q.; Huang, F.; He, H.; Fan, W. Involvement of Mast Cells in the Pathophysiology of Pain. Front. Cell. Neurosci. 2021, 15, 665066. [Google Scholar] [CrossRef]
- Hao, G.; Han, S.; Xiao, Z.; Shen, J.; Zhao, Y.; Hao, Q. Synovial mast cells and osteoarthritis: Current understandings and future perspectives. Heliyon 2024, 10, e41003. [Google Scholar] [CrossRef]
- Jiang, P.; Hu, K.; Jin, L.; Luo, Z. A brief review of current treatment options for osteoarthritis including disease-modifying osteoarthritis drugs (DMOADs) and novel therapeutics. Ann. Med. Surg. 2024, 86, 4042–4048. [Google Scholar] [CrossRef]
- Allen, K.D.; Ambrose, K.R.; Booker, S.Q.; Buck, A.N.; Huffman, K.F. Non-Pharmacological Pain Management for Osteoarthritis: Review Update. Curr. Rheumatol. Rep. 2025, 27, 19. [Google Scholar] [CrossRef] [PubMed]
- Hermann, W.; Lambova, S.; Muller-Ladner, U. Current Treatment Options for Osteoarthritis. Curr. Rheumatol. Rev. 2018, 14, 108–116. [Google Scholar] [CrossRef] [PubMed]
- van Laar, M.; Pergolizzi, J.V., Jr.; Mellinghoff, H.U.; Merchante, I.M.; Nalamachu, S.; O’Brien, J.; Perrot, S.; Raffa, R.B. Pain treatment in arthritis-related pain: Beyond NSAIDs. Open Rheumatol. J. 2012, 6, 320–330. [Google Scholar] [CrossRef]
- Wongrakpanich, S.; Wongrakpanich, A.; Melhado, K.; Rangaswami, J. A Comprehensive Review of Non-Steroidal Anti-Inflammatory Drug Use in The Elderly. Aging Dis. 2018, 9, 143–150. [Google Scholar] [CrossRef]
- Fuggle, N.R.; Cooper, C.; Oreffo, R.O.C.; Price, A.J.; Kaux, J.F.; Maheu, E.; Cutolo, M.; Honvo, G.; Conaghan, P.G.; Berenbaum, F.; et al. Alternative and complementary therapies in osteoarthritis and cartilage repair. Aging Clin. Exp. Res. 2020, 32, 547–560. [Google Scholar] [CrossRef]
- Primorac, D.; Molnar, V.; Matišić, V.; Hudetz, D.; Jeleč, Ž.; Rod, E.; Čukelj, F.; Vidović, D.; Vrdoljak, T.; Dobričić, B.; et al. Comprehensive Review of Knee Osteoarthritis Pharmacological Treatment and the Latest Professional Societies’ Guidelines. Pharmaceuticals 2021, 14, 205. [Google Scholar] [CrossRef] [PubMed]
- Guermazi, A.; Neogi, T.; Katz, J.N.; Kwoh, C.K.; Conaghan, P.G.; Felson, D.T.; Roemer, F.W. Intra-articular Corticosteroid Injections for the Treatment of Hip and Knee Osteoarthritis-related Pain: Considerations and Controversies with a Focus on Imaging-Radiology Scientific Expert Panel. Radiology 2020, 297, 503–512. [Google Scholar] [CrossRef]
- Kompel, A.J.; Roemer, F.W.; Murakami, A.M.; Diaz, L.E.; Crema, M.D.; Guermazi, A. Intra-articular Corticosteroid Injections in the Hip and Knee: Perhaps Not as Safe as We Thought? Radiology 2019, 293, 656–663. [Google Scholar] [CrossRef]
- Boutin, R.D.; Pai, J.; Meehan, J.P.; Newman, J.S.; Yao, L. Rapidly progressive idiopathic arthritis of the hip: Incidence and risk factors in a controlled cohort study of 1471 patients after intra-articular corticosteroid injection. Skelet. Radiol. 2021, 50, 2449–2457. [Google Scholar] [CrossRef]
- Peck, J.; Slovek, A.; Miro, P.; Vij, N.; Traube, B.; Lee, C.; Berger, A.A.; Kassem, H.; Kaye, A.D.; Sherman, W.F.; et al. A Comprehensive Review of Viscosupplementation in Osteoarthritis of the Knee. Orthop. Rev. (Pavia) 2021, 13, 25549. [Google Scholar] [CrossRef]
- Tapasvi, S.; Mohanty, S.S.; Vedavyasa Acharya, K.K.; Bhattacharya, K.; Easwaran, R.; Charugulla, S.N. Viscosupplementation for Management of Knee Osteoarthritis from an Indian Perspective: An Expert Consensus Report. Pain Ther. 2019, 8, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Madry, H. Surgical therapy in osteoarthritis. Osteoarthr. Cartil. 2022, 30, 1019–1034. [Google Scholar] [CrossRef]
- Hawker, G.A.; Wright, J.G.; Coyte, P.C.; Williams, J.I.; Harvey, B.; Glazier, R.; Badley, E.M. Differences between men and women in the rate of use of hip and knee arthroplasty. N. Engl. J. Med. 2000, 342, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Malfait, A.M. Modelling pain in post-traumatic osteoarthritis of the knee. PAIN 2012, 153, 257–258. [Google Scholar] [CrossRef]
- Roseti, L.; Desando, G.; Cavallo, C.; Petretta, M.; Grigolo, B. Articular Cartilage Regeneration in Osteoarthritis. Cells 2019, 8, 1305. [Google Scholar] [CrossRef]
- Primorac, D.; Molnar, V.; Tsoukas, D.; Uzieliene, I.; Tremolada, C.; Brlek, P.; Klarić, E.; Vidović, D.; Zekušić, M.; Pachaleva, J.; et al. Tissue engineering and future directions in regenerative medicine for knee cartilage repair: A comprehensive review. Croat. Med. J. 2024, 65, 268–287. [Google Scholar] [CrossRef] [PubMed]
- Luyten, F.P.; Vanlauwe, J. Tissue engineering approaches for osteoarthritis. Bone 2012, 51, 289–296. [Google Scholar] [CrossRef]
- Zhu, C.; Wu, W.; Qu, X. Mesenchymal stem cells in osteoarthritis therapy: A review. Am. J. Transl. Res. 2021, 13, 448–461. [Google Scholar]
- Ganguly, P.; Birch, M. Biomaterial Scaffolds for Mesenchymal Stem Cell Based Therapy Aimed at Tissue Engineering Application for Osteoarthritis. Recent Pat. Nanomed. 2015, 5, 19–32. [Google Scholar] [CrossRef]
- Mavrogenis, A.F.; Karampikas, V.; Zikopoulos, A.; Sioutis, S.; Mastrokalos, D.; Koulalis, D.; Scarlat, M.M.; Hernigou, P. Orthobiologics: A review. Int. Orthop. 2023, 47, 1645–1662. [Google Scholar] [CrossRef]
- Everts, P.; Onishi, K.; Jayaram, P.; Lana, J.F.; Mautner, K. Platelet-Rich Plasma: New Performance Understandings and Therapeutic Considerations in 2020. Int. J. Mol. Sci. 2020, 21, 7794. [Google Scholar] [CrossRef]
- Liang, J.; Liu, P.; Yang, X.; Liu, L.; Zhang, Y.; Wang, Q.; Zhao, H. Biomaterial-based scaffolds in promotion of cartilage regeneration: Recent advances and emerging applications. J. Orthop. Transl. 2023, 41, 54–62. [Google Scholar] [CrossRef]
- Ip, H.L.; Nath, D.K.; Sawleh, S.H.; Kabir, M.H.; Jahan, N. Regenerative Medicine for Knee Osteoarthritis—The Efficacy and Safety of Intra-Articular Platelet-Rich Plasma and Mesenchymal Stem Cells Injections: A Literature Review. Cureus 2020, 12, e10575. [Google Scholar] [CrossRef] [PubMed]
- Tjandra, K.C.; Novriansyah, R.; Sudiasa, I.N.S.; Ar, A.; Rahmawati, N.A.D.; Dilogo, I.H. Modified Mesenchymal stem cell, platelet-rich plasma, and hyaluronic acid intervention in early stage osteoarthritis: A systematic review, meta-analysis, and meta-regression of arthroscopic-guided intra-articular approaches. PLoS ONE 2024, 19, e0295876. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Xiang, X.-N.; Yu, X.; Liu, Y.; Jiang, H.-Y.; Peng, J.-L.; He, C.-Q.; He, H.-C. Mechanisms and applications of the regenerative capacity of platelets-based therapy in knee osteoarthritis. Biomed. Pharmacother. 2024, 178, 117226. [Google Scholar] [CrossRef]
- Filardo, G.; Di Matteo, B.; Kon, E.; Merli, G.; Marcacci, M. Platelet-rich plasma in tendon-related disorders: Results and indications. Knee Surg. Sports Traumatol. Arthrosc. 2018, 26, 1984–1999. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy Reddy, S.H.; Reddy, R.; Babu, N.C.; Ashok, G.N. Stem-cell therapy and platelet-rich plasma in regenerative medicines: A review on pros and cons of the technologies. J. Oral Maxillofac. Pathol. 2018, 22, 367–374. [Google Scholar] [CrossRef]
- Seong, G.J.; Hong, S.; Jung, S.A.; Lee, J.J.; Lim, E.; Kim, S.J.; Lee, J.H. TGF-beta-induced interleukin-6 participates in transdifferentiation of human Tenon’s fibroblasts to myofibroblasts. Mol. Vis. 2009, 15, 2123–2128. [Google Scholar]
- Dhillon, R.S.; Schwarz, E.M.; Maloney, M.D. Platelet-rich plasma therapy—Future or trend? Arthritis Res. Ther. 2012, 14, 219. [Google Scholar] [CrossRef]
- Simental-Mendía, M.; Ortega-Mata, D.; Tamez-Mata, Y.; Olivo, C.A.A.; Vilchez-Cavazos, F. Comparison of the clinical effectiveness of activated and non-activated platelet-rich plasma in the treatment of knee osteoarthritis: A systematic review and meta-analysis. Clin. Rheumatol. 2023, 42, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Bensa, A.; Previtali, D.; Sangiorgio, A.; Boffa, A.; Salerno, M.; Filardo, G. PRP Injections for the Treatment of Knee Osteoarthritis: The Improvement Is Clinically Significant and Influenced by Platelet Concentration: A Meta-analysis of Randomized Controlled Trials. Am. J. Sports Med. 2025, 53, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.I.; Whitney, K.; Evans, T.; LaPrade, R.F. Platelet-Rich Plasma and Cartilage Repair. Curr. Rev. Musculoskelet. Med. 2018, 11, 573–582. [Google Scholar] [CrossRef]
- GBD 2017 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 359 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1859–1922. [Google Scholar] [CrossRef] [PubMed]
- Wang-Saegusa, A.; Cugat, R.; Ares, O.; Seijas, R.; Cuscó, X.; Garcia-Balletbó, M. Infiltration of plasma rich in growth factors for osteoarthritis of the knee short-term effects on function and quality of life. Arch. Orthop. Trauma Surg. 2011, 131, 311–317. [Google Scholar] [CrossRef]
- Bansal, H.; Leon, J.; Pont, J.L.; Wilson, D.A.; Bansal, A.; Agarwal, D.; Preoteasa, I. Platelet-rich plasma (PRP) in osteoarthritis (OA) knee: Correct dose critical for long term clinical efficacy. Sci. Rep. 2021, 11, 3971. [Google Scholar] [CrossRef]
- Lin, J.; Huang, J.; Jiao, Z.; Nian, M.; Li, C.; Dai, Y.; Jia, S.; Zhang, X. Mesenchymal stem cells for osteoarthritis: Recent advances in related cell therapy. Bioeng. Transl. Med. 2025, 10, e10701. [Google Scholar] [CrossRef]
- Thoene, M.; Bejer-Olenska, E.; Wojtkiewicz, J. The Current State of Osteoarthritis Treatment Options Using Stem Cells for Regenerative Therapy: A Review. Int. J. Mol. Sci. 2023, 24, 8925. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, R.-J.; Wu, Y.; Huang, D.; Li, Y.; Liu, Y. Advances in Stem Cell-Based Therapies in the Treatment of Osteoarthritis. Int. J. Mol. Sci. 2024, 25, 394. [Google Scholar] [CrossRef]
- Tian, R.; Su, S.; Yu, Y.; Liang, S.; Ma, C.; Jiao, Y.; Xing, W.; Tian, Z.; Jiang, T.; Wang, J. Revolutionizing osteoarthritis treatment: How mesenchymal stem cells hold the key. Biomed. Pharmacother. 2024, 173, 116458. [Google Scholar] [CrossRef]
- Maleitzke, T.; Elazaly, H.; Festbaum, C.; Eder, C.; Karczewski, D.; Perka, C.; Duda, G.N.; Winkler, T. Mesenchymal Stromal Cell-Based Therapy-An Alternative to Arthroplasty for the Treatment of Osteoarthritis? A State of the Art Review of Clinical Trials. J. Clin. Med. 2020, 9, 2062. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, J.A.; Jones, I.A.; Han, B.; Vangsness, C.T., Jr. Intra-articular Mesenchymal Stem Cell Therapy for the Human Joint: A Systematic Review. Am. J. Sports Med. 2018, 46, 3550–3563. [Google Scholar] [CrossRef]
- Krawczenko, A.; Klimczak, A. Adipose Tissue-Derived Mesenchymal Stem/Stromal Cells and Their Contribution to Angiogenic Processes in Tissue Regeneration. Int. J. Mol. Sci. 2022, 23, 2425. [Google Scholar] [CrossRef]
- Stratton, S.; Shelke, N.B.; Hoshino, K.; Rudraiah, S.; Kumbar, S.G. Bioactive polymeric scaffolds for tissue engineering. Bioact. Mater. 2016, 1, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Stamnitz, S.; Klimczak, A. Mesenchymal Stem Cells, Bioactive Factors, and Scaffolds in Bone Repair: From Research Perspectives to Clinical Practice. Cells 2021, 10, 1925. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Chen, Y.Y.; Spencer, M.J. Successes and challenges in clinical gene therapy. Gene Ther. 2023, 30, 738–746. [Google Scholar] [CrossRef]
- Evans, C.H.; Kraus, V.B.; Setton, L.A. Progress in intra-articular therapy. Nat. Rev. Rheumatol. 2014, 10, 11–22. [Google Scholar] [CrossRef]
- Vlashi, R.; Zhang, X.; Li, H.; Chen, G. Potential therapeutic strategies for osteoarthritis via CRISPR/Cas9 mediated gene editing. Rev. Endocr. Metab. Disord. 2024, 25, 339–367. [Google Scholar] [CrossRef]
- Vickram, A.S.; Manikandan, S.; Richard, T.; Lakshmi, S.V.; Chopra, H. Targeted Gene Therapy: Promises and Challenges in Disease Management. J. Bio-X Res. 2024, 07, 81–89. [Google Scholar] [CrossRef]
- Cho, Y.; Jeong, S.; Kim, H.; Kang, D.; Lee, J.; Kang, S.-B.; Kim, J.-H. Disease-modifying therapeutic strategies in osteoarthritis: Current status and future directions. Exp. Mol. Med. 2021, 53, 1689–1696. [Google Scholar] [CrossRef]
- Kim, H.; Seo, J.; Lee, Y.; Park, K.; Perry, T.A.; Arden, N.K.; Mobasheri, A.; Choi, H. The current state of the osteoarthritis drug development pipeline: A comprehensive narrative review of the present challenges and future opportunities. Ther. Adv. Musculoskelet. Dis. 2022, 14, 1759720x221085952. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Michaelis, M.; Ladel, C.; Siebuhr, A.S.; Bihlet, A.R.; Andersen, J.R.; Guehring, H.; Christiansen, C.; Bay-Jensen, A.C.; Kraus, V.B. Disease-modifying treatments for osteoarthritis (DMOADs) of the knee and hip: Lessons learned from failures and opportunities for the future. Osteoarthr. Cartil. 2016, 24, 2013–2021. [Google Scholar] [CrossRef]
- Cohen, S.B.; Proudman, S.; Kivitz, A.J.; Burch, F.X.; Donohue, J.P.; Burstein, D.; Sun, Y.N.; Banfield, C.; Vincent, M.S.; Ni, L.; et al. A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee. Arthritis Res. Ther. 2011, 13, R125. [Google Scholar] [CrossRef] [PubMed]
- Loef, M.; Kroon, F.P.B.; Bergstra, S.A.; van der Pol, J.A.; Lems, W.F.; Kerstens, P.; Allaart, C.F.; Kloppenburg, M. TNF inhibitor treatment is associated with a lower risk of hand osteoarthritis progression in rheumatoid arthritis patients after 10 years. Rheumatology 2018, 57, 1917–1924. [Google Scholar] [CrossRef]
- Madry, H.; Cucchiarini, M. Tissue-Engineering Strategies to Repair Joint Tissue in Osteoarthritis: Nonviral Gene-Transfer Approaches. Curr. Rheumatol. Rep. 2014, 16, 450. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, R.; Chen, C.; Yang, F.; Xiao, H.; Geng, B.; Xia, Y. Tissue engineering strategies hold promise for the repair of articular cartilage injury. Biomed. Eng. Online 2024, 23, 92. [Google Scholar] [CrossRef]
- Hargreaves, K.; Dubner, R.; Brown, F.; Flores, C.; Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. PAIN 1988, 32, 77–88. [Google Scholar] [CrossRef]
- Bozkurt, A.; Deumens, R.; Scheffel, J.; O’Dey, D.M.; Weis, J.; Joosten, E.A.; Führmann, T.; Brook, G.A.; Pallua, N. CatWalk gait analysis in assessment of functional recovery after sciatic nerve injury. J. Neurosci. Methods 2008, 173, 91–98. [Google Scholar] [CrossRef]
- Deuis, J.R.; Dvorakova, L.S.; Vetter, I. Methods Used to Evaluate Pain Behaviors in Rodents. Front. Mol. Neurosci. 2017, 10, 284. [Google Scholar] [CrossRef]
- Guo, T.Z.; Wei, T.; Li, W.W.; Li, X.Q.; Clark, J.D.; Kingery, W.S. Immobilization contributes to exaggerated neuropeptide signaling, inflammatory changes, and nociceptive sensitization after fracture in rats. J. Pain 2014, 15, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Tate, C.C.; Tate, M.C.; LaPlaca, M.C. Fibronectin and laminin increase in the mouse brain after controlled cortical impact injury. J. Neurotrauma 2007, 24, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; Malfait, A.M. Osteoarthritis pain: What are we learning from animal models? Best Pract. Res. Clin. Rheumatol. 2017, 31, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Tatem, K.S.; Quinn, J.L.; Phadke, A.; Yu, Q.; Gordish-Dressman, H.; Nagaraju, K. Behavioral and locomotor measurements using an open field activity monitoring system for skeletal muscle diseases. J. Vis. Exp. 2014, 91, e51785. [Google Scholar] [CrossRef]
- Ma, L.; Liu, S.; Yi, M.; Wan, Y. Spontaneous pain as a challenge of research and management in chronic pain. Med. Rev. 2022, 2, 308–319. [Google Scholar] [CrossRef]
- Turner, P.V.; Pang, D.S.; Lofgren, J.L. A Review of Pain Assessment Methods in Laboratory Rodents. Comp. Med. 2019, 69, 451–467. [Google Scholar] [CrossRef]
- Lakes, E.H.; Allen, K.D. Gait analysis methods for rodent models of arthritic disorders: Reviews and recommendations. Osteoarthr. Cartil. 2016, 24, 1837–1849. [Google Scholar] [CrossRef]
- Christiansen, C.L.; Stevens-Lapsley, J.E. Weight-bearing asymmetry in relation to measures of impairment and functional mobility for people with knee osteoarthritis. Arch. Phys. Med. Rehabil. 2010, 91, 1524–1528. [Google Scholar] [CrossRef]
- Malfait, A.M.; Little, C.B.; McDougall, J.J. A commentary on modelling osteoarthritis pain in small animals. Osteoarthr. Cartil. 2013, 21, 1316–1326. [Google Scholar] [CrossRef]
- Zheng, Y.; Lunn, A.; Gao, J.; Chen, H.; Yao, Y. Quantitative evaluation of hindlimb grip strength in mice as a measure of neuromuscular function. MethodsX 2025, 14, 103118. [Google Scholar] [CrossRef] [PubMed]
- Nugent, S.M.; Lovejoy, T.I.; Shull, S.; Dobscha, S.K.; Morasco, B.J. Associations of Pain Numeric Rating Scale Scores Collected during Usual Care with Research Administered Patient Reported Pain Outcomes. Pain Med. 2021, 22, 2235–2241. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Valente, M.A.; Pais-Ribeiro, J.L.; Jensen, M.P. Validity of four pain intensity rating scales. PAIN 2011, 152, 2399–2404. [Google Scholar] [CrossRef] [PubMed]
- Åström, M.; Thet Lwin, Z.M.; Teni, F.S.; Burström, K.; Berg, J. Use of the visual analogue scale for health state valuation: A scoping review. Qual. Life Res. 2023, 32, 2719–2729. [Google Scholar] [CrossRef]
- Tsai, P.F.; Beck, C.; Richards, K.C.; Phillips, L.; Roberson, P.K.; Evans, J. The Pain Behaviors for Osteoarthritis Instrument for Cognitively Impaired Elders (PBOICIE). Res. Gerontol. Nurs. 2008, 1, 116–122. [Google Scholar] [CrossRef]
- Stewart, C. Practical Management of Pain; Mosby: Maryland Heights, MO, USA, 2014; pp. 467–473. [Google Scholar]
- Melzack, R. The McGill Pain Questionnaire: Major properties and scoring methods. PAIN 1975, 1, 277–299. [Google Scholar] [CrossRef]
- Im, D.D.; Jambaulikar, G.D.; Kikut, A.; Gale, J.; Weiner, S.G. Brief Pain Inventory—Short Form: A New Method for Assessing Pain in the Emergency Department. Pain Med. 2020, 21, 3263–3269. [Google Scholar] [CrossRef]
- Harato, K.; Iwama, Y.; Kaneda, K.; Kobayashi, S.; Niki, Y.; Nagura, T. Pain detect questionnaire and pain catastrophizing scale affect gait pattern in patients with knee osteoarthritis. J. Exp. Orthop. 2022, 9, 52. [Google Scholar] [CrossRef]
- Freynhagen, R.; Baron, R.; Gockel, U.; Tölle, T.R. painDETECT: A new screening questionnaire to identify neuropathic components in patients with back pain. Curr. Med. Res. Opin. 2006, 22, 1911–1920. [Google Scholar] [CrossRef]
- Bouhassira, D.; Attal, N.; Alchaar, H.; Boureau, F.; Brochet, B.; Bruxelle, J.; Cunin, G.; Fermanian, J.; Ginies, P.; Grun-Overdyking, A.; et al. Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). PAIN 2005, 114, 29–36. [Google Scholar] [CrossRef]
- Braaksma, C.; Wolterbeek, N.; Veen, M.R.; Prinsen, C.A.C.; Ostelo, R. Systematic review and meta-analysis of measurement properties of the Hip disability and Osteoarthritis Outcome Score—Physical Function Shortform (HOOS-PS) and the Knee Injury and Osteoarthritis Outcome Score—Physical Function Shortform (KOOS-PS). Osteoarthr. Cartil. 2020, 28, 1525–1538. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.F.; Schalet, B.D.; Kallen, M.A.; Rutsohn, J.P.; Cella, D. Establishing a common metric for self-reported pain: Linking BPI Pain Interference and SF-36 Bodily Pain Subscale scores to the PROMIS Pain Interference metric. Qual. Life Res. 2015, 24, 2305–2318. [Google Scholar] [CrossRef] [PubMed]
- Riddle, D.L.; Perera, R.A. The WOMAC Pain Scale and Crosstalk From Co-occurring Pain Sites in People With Knee Pain: A Causal Modeling Study. Phys. Ther. 2020, 100, 1872–1881. [Google Scholar] [CrossRef]
- Anderson, A.F.; Irrgang, J.J.; Kocher, M.S.; Mann, B.J.; Harrast, J.J. The International Knee Documentation Committee Subjective Knee Evaluation Form: Normative data. Am. J. Sports Med. 2006, 34, 128–135. [Google Scholar] [CrossRef]
- Barber-Westin, S.D.; Noyes, F.R.; McCloskey, J.W. Rigorous statistical reliability, validity, and responsiveness testing of the Cincinnati knee rating system in 350 subjects with uninjured, injured, or anterior cruciate ligament-reconstructed knees. Am. J. Sports Med. 1999, 27, 402–416. [Google Scholar] [CrossRef]
- Briggs, K.K.; Lysholm, J.; Tegner, Y.; Rodkey, W.G.; Kocher, M.S.; Steadman, J.R. The reliability, validity, and responsiveness of the Lysholm score and Tegner activity scale for anterior cruciate ligament injuries of the knee: 25 years later. Am. J. Sports Med. 2009, 37, 890–897. [Google Scholar] [CrossRef]
- Malviya, S.; Voepel-Lewis, T.; Burke, C.; Merkel, S.; Tait, A.R. The revised FLACC observational pain tool: Improved reliability and validity for pain assessment in children with cognitive impairment. Pediatr. Anesth. 2006, 16, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Mogil, J.S.; Pang, D.S.J.; Silva Dutra, G.G.; Chambers, C.T. The development and use of facial grimace scales for pain measurement in animals. Neurosci. Biobehav. Rev. 2020, 116, 480–493. [Google Scholar] [CrossRef]
- Kennedy, D.L.; Kemp, H.I.; Ridout, D.; Yarnitsky, D.; Rice, A.S.C. Reliability of conditioned pain modulation: A systematic review. PAIN 2016, 157, 2410–2419. [Google Scholar] [CrossRef]
- Knazovicky, D.; Helgeson, E.S.; Case, B.; Gruen, M.E.; Maixner, W.; Lascelles, B.D.X. Widespread somatosensory sensitivity in naturally occurring canine model of osteoarthritis. PAIN 2016, 157, 1325–1332. [Google Scholar] [CrossRef]
- Ji, J.; Huh, Y.; Ji, R.-R. Inflammatory Mediators, Nociceptors, and Their Interactions in Pain. In Neuroimmune Interactions in Pain: Mechanisms and Therapeutics; Ji, R.-R., Cheng, J., Ji, J., Eds.; Springer International Publishing: Cham, Switzerland, 2023; pp. 87–119. [Google Scholar]
- Pinho-Ribeiro, F.A.; Verri, W.A.; Chiu, I.M. Nociceptor Sensory Neuron–Immune Interactions in Pain and Inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Perrot, S.; Anne-Priscille, T. Pain in osteoarthritis from a symptom to a disease. Best Pract. Res. Clin. Rheumatol. 2023, 37, 101825. [Google Scholar] [CrossRef]
- Panchal, Y.; Pardeshi, A.; Singhal, A.; Soni, H.; Singh, G.; Gomathy, G.; Shhroye, N.; Thakur, N.; Thorat, P. Effective Strategies for Managing Chronic Pain in Patients: A Review of the Current Evidence. Universe Int. J. Interdiscip. Res. 2024, 5, 85–96. [Google Scholar]
- Liao, J.; Gu, Q.; Liu, Z.; Wang, H.; Yang, X.; Yan, R.; Zhang, X.; Song, S.; Wen, L.; Wang, Y. Edge advances in nanodrug therapies for osteoarthritis treatment. Front. Pharmacol. 2024, 15, 1402825. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, T. Multimodal approach to intraarticular drug delivery in knee osteoarthritis. Rheumatol. Int. 2020, 40, 1763–1769. [Google Scholar] [CrossRef]
- Veronese, N.; Cooper, C.; Bruyère, O.; Al-Daghri, N.M.; Branco, J.; Cavalier, E.; Cheleschi, S.; da Silva Rosa, M.C.; Conaghan, P.G.; Dennison, E.M.; et al. Multimodal Multidisciplinary Management of Patients with Moderate to Severe Pain in Knee Osteoarthritis: A Need to Meet Patient Expectations. Drugs 2022, 82, 1347–1355. [Google Scholar] [CrossRef]
- Ho, M.J.; Kim, S.R.; Choi, Y.W.; Kang, M.J. Recent advances in intra-articular drug delivery systems to extend drug retention in joint. J. Pharm. Investig. 2019, 49, 9–15. [Google Scholar] [CrossRef]
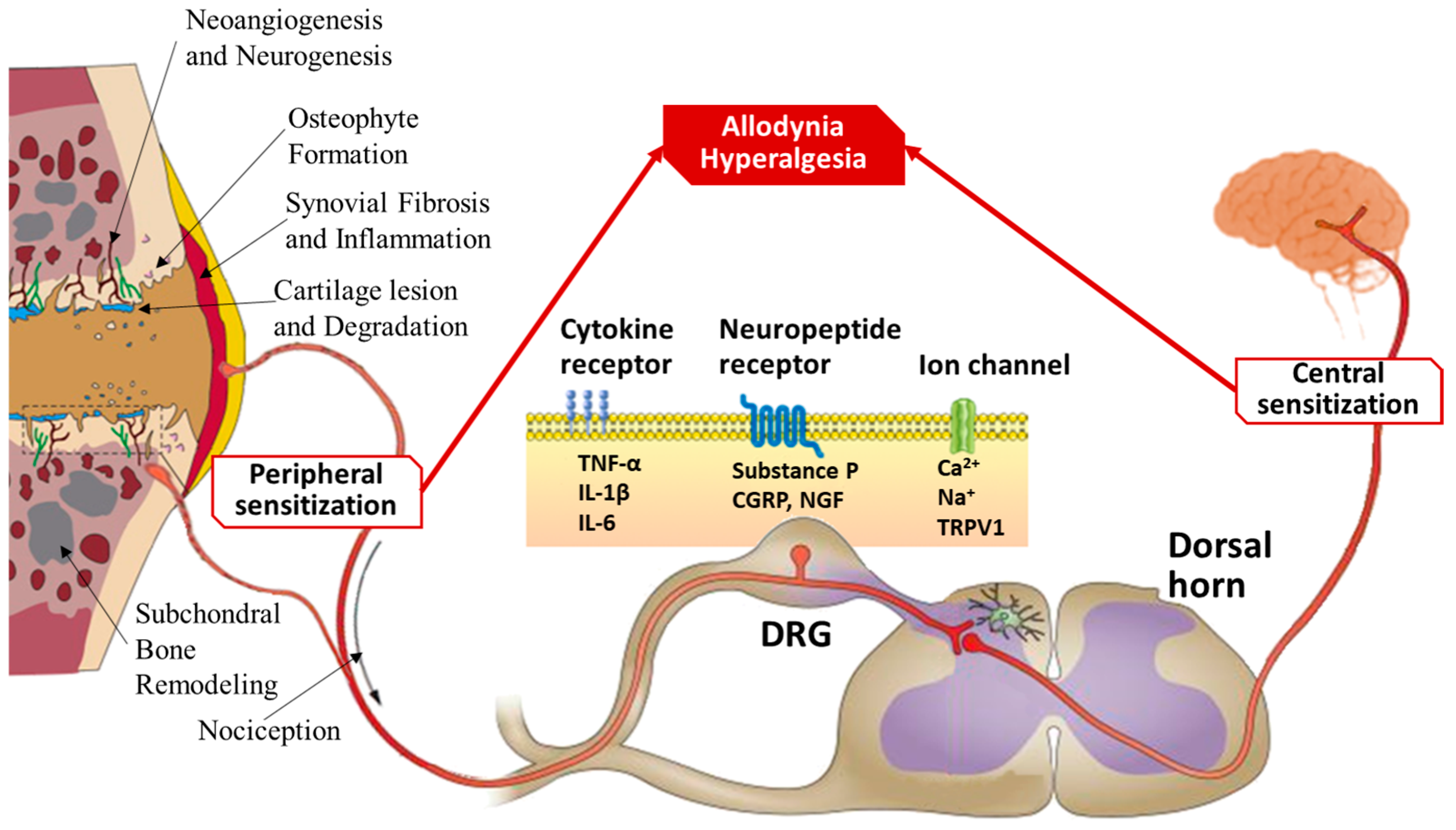
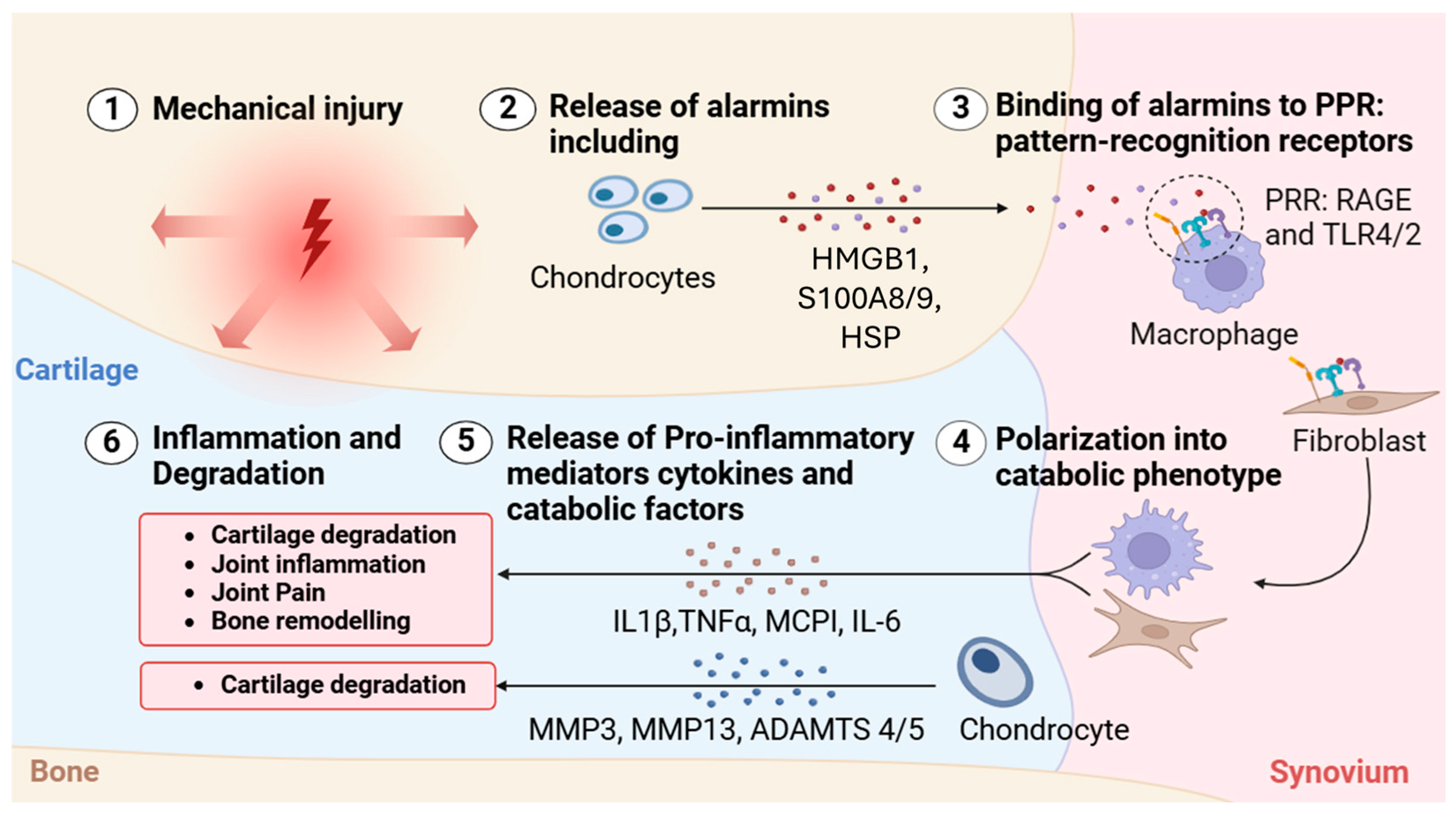
| Current OA Treatments | Mode of Action | Target | Potential Therapeutic Benefits |
|---|---|---|---|
| Corticosteroid Drugs (CSDs) | Reduction in inflammation and pain | Inflammatory mediators | Short-term relief of pain and inflammation |
| NSAIDs (Ibuprofen; Diclofenac) | Inhibit COX-1 and -2 enzymes | COX enzymes | Pain relief and reduced inflammation |
| Hyaluronic AcidIntraarticular Injections | Lubrication and pain reduction | Synovial fluid | Improved joint mobility and symptom relief |
| Tanezumab | NGF inhibition | Nerve growth factor | Reduction in pain and inflammation in OA |
| Cannabinoids | Activates CB1 and CB2 receptors | Cannabinoid receptors | Pain reduction and anti-inflammatory effects |
| Knee ReplacementSurgery | Surgical intervention for functional restoration | Knee joint | Long-term pain relief and functional improvement |
| Pain Assessment | Species Used | Measurement/ Observation | Relevance to Human OA Pain |
|---|---|---|---|
| Mechanical or Thermal Sensitivity | |||
| Von Frey Filaments [217,220] | rats; mice | Mechanical allodynia in hind paw using calibrated filaments | pain hypersensitivity seen in chronic OA |
| Pressure Application Measurement (PAM) [220] | rats | Knee hyperalgesia induced by applying controlled force to the joint until withdrawal or vocalization occurs | mechanical joint pain |
| Thermal Withdrawal Test [220] | rats; mice | Latency to withdraw from heat/cold stimuli | altered pain thresholds in OA |
| Behavioral Assessment | |||
| Open Field Test [221] | rats; mice | Spontaneous locomotor activity and rearing | reduced mobility and pain-related behavioral changes |
| Conditioned Place Preference (CPP) [222,223] | rats; mice | Relief of spontaneous pain by linking the environment with analgesia | affective dimension of pain in humans |
| Burrowing Behavior Test [223] | rats; mice | Changes in innate behavior using a custom-made burrowing device | spontaneous pain |
| Affective/Mimetic Assessment | |||
| Facial Grimace Scale [217] | mice, rats, sheep | Facial expressions (e.g., orbital tightening) to assess spontaneous pain | chronic human OA pain |
| Video Monitoring [224] | rats; mice | Continuous video tracking of spontaneous behaviors like licking, guarding, or rearing | spontaneous pain behaviors |
| Functional Assessment | |||
| Static Weight-Bearing Test [225] | rats; mice | Unequal weight distribution using a limb loading capacitance meter | indicates joint discomfort and mechanical pain |
| Dynamic Weight-Bearing Test [226] | rats; mice | Voluntary limb loading during movement | mimics activity-related OA pain |
| Gait Analysis (CatWalk and TreadScan) [224] | rats, mice, Guinea pigs | Stride length, swing speed, and stance time using automated gait systems | reflects mobility impairments similar to human OA |
| Hind Limb Grip Strength [227] | rats | Limb muscle strength and coordination through grip meter | indirectly reflects joint discomfort or weakness |
| Tools | Common Use Case | Measurement/ Observation |
|---|---|---|
| Self-Report Tools | ||
| Numeric Rating Scale (NRS) [228,229] | General clinical use (acute/chronic pain) | Rates pain from 0 (no pain) to 10 (worst pain) |
| Visual Analog Scale (VAS) [229,230] | 10 cm line for marking perceived pain level | Postoperative pain; research trials |
| Verbal Descriptor Scale (VDS) [229,231] | Elderly, cognitively impaired patients | Uses words (e.g., mild, moderate, severe) to describe pain |
| McGill Pain Questionnaire (MPQ) [232,233] | Neuropathic pain; chronic conditions | Evaluates sensory, affective, and evaluative qualities of pain |
| Brief Pain Inventory (BPI) [234] | Chronic pain; cancer pain | Assesses pain severity and functional interference |
| Pain DETECT Questionnaire [235,236] | Neuropathic pain screening | Screens for neuropathic pain characteristics |
| DN4 Questionnaire [237] | Neuropathic pain diagnosis | Checklist combining sensory symptoms and clinical signs |
| Knee Injury and Osteoarthritis Outcome Score (KOOS) [238] | Common in knee OA trials, especially post-surgery | Includes a pain subscale with items related to specific joint-loading activities |
| Hip Disability and Osteoarthritis Outcome Score (HOOS) [238] | Used in clinical studies and outcome assessments for hip OA | Adapted from KOOS; includes hip-specific pain and function items |
| SF-36 Bodily Pain Subscale [239] | Applied in OA studies to assess overall well-being | Part of a general health survey measuring pain and overall health-related QoL |
| WOMAC Pain Subscale [240] | Gold standard in OA clinical trials and interventions | Part of the Western Ontario and McMaster Universities Osteoarthritis Index; evaluates pain during five activities (e.g., walking and using stairs) |
| International Knee Documentation Committee (IKDC) Subjective Knee Evaluation Form [241] | Knee ligament and cartilage injuries | Pain, stiffness, swelling, and function during daily and sports activities |
| Cincinnati Knee Rating [242] | Knee injuries in athletes | Physician and patient ratings of pain, swelling, and functional activity |
| Lysholm Knee Scoring [243] | Post-ACL or meniscal | Knee function, including pain, instability, and stair climbing |
| Tegner Activity Scale [243] | Physical activity level | Activity levels from sedentary (level 0) to elite (level 10) |
| Observational Tools | ||
| FLACC Scale [244] | Infants, ICU, non-verbal adults | Rates pain based on face, legs, activity, cry, consolability (0–10) |
| Facial Grimace Scale [245] | Neonates, unconscious, dementia patients | Evaluates facial expressions for spontaneous pain cues |
| Experimental/Physiological Tools | ||
| Conditioned Pain Modulation (CPM) [246] | Central sensitization research | Measures descending inhibitory control through dual-pain paradigm |
| Quantitative Sensory Testing (QST) [247] | Neuropathic pain; sensory profiling | Measures mechanical and thermal thresholds to stimuli |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alad, M.; Yousef, F.; Epure, L.M.; Lui, A.; Grant, M.P.; Merle, G.; Eliopoulos, N.; Barralet, J.; Antoniou, J.; Mwale, F. Unraveling Osteoarthritis: Mechanistic Insights and Emerging Therapies Targeting Pain and Inflammation. Biomolecules 2025, 15, 874. https://doi.org/10.3390/biom15060874
Alad M, Yousef F, Epure LM, Lui A, Grant MP, Merle G, Eliopoulos N, Barralet J, Antoniou J, Mwale F. Unraveling Osteoarthritis: Mechanistic Insights and Emerging Therapies Targeting Pain and Inflammation. Biomolecules. 2025; 15(6):874. https://doi.org/10.3390/biom15060874
Chicago/Turabian StyleAlad, Muskan, Fajer Yousef, Laura M. Epure, Angelina Lui, Michael P. Grant, Geraldine Merle, Nicoletta Eliopoulos, Jake Barralet, John Antoniou, and Fackson Mwale. 2025. "Unraveling Osteoarthritis: Mechanistic Insights and Emerging Therapies Targeting Pain and Inflammation" Biomolecules 15, no. 6: 874. https://doi.org/10.3390/biom15060874
APA StyleAlad, M., Yousef, F., Epure, L. M., Lui, A., Grant, M. P., Merle, G., Eliopoulos, N., Barralet, J., Antoniou, J., & Mwale, F. (2025). Unraveling Osteoarthritis: Mechanistic Insights and Emerging Therapies Targeting Pain and Inflammation. Biomolecules, 15(6), 874. https://doi.org/10.3390/biom15060874