Abstract
Colorectal cancer is one of the most prevalent cancer types. Although there have been breakthroughs in its treatments, a better understanding of the molecular mechanisms and genetic involvement in colorectal cancer will have a substantial role in producing novel and targeted treatments with better safety profiles. In this review, the main molecular pathways and driver genes that are responsible for initiating and propagating the cascade of signaling molecules reaching carcinoma and the aggressive metastatic stages of colorectal cancer were presented. Protein kinases involved in colorectal cancer, as much as other cancers, have seen much focus and committed efforts due to their crucial role in subsidizing, inhibiting, or changing the disease course. Moreover, notable improvements in colorectal cancer treatments with in silico studies and the enhanced selectivity on specific macromolecular targets were discussed. Besides, the selective multi-target agents have been made easier by employing in silico methods in molecular de novo synthesis or target identification and drug repurposing.
1. Introduction
Cancer does not develop from a single gene defect in a similar way to how it occurs in other diseases such as cystic fibrosis or muscular dystrophy. Instead, cancer becomes invasive in the event that there are multiple cancer gene mutations where the safeguarding mechanisms could not protect the normal and healthy mammalian cells from their lethal effects. As a result, it is better to think of cancer genes that have been altered as contributing to, rather than causing, cancer [1]. The development of colorectal cancer involves a multiple step process incited by a distinctive genomic instability which encourages the cancerous cells to multiply, as well as increases the chances of cell survival.
Colorectal cancer has three recognized primary molecular groupings in terms of molecular genetics. The most prevalent one is the “chromosomal instable” group, which is defined by an accumulation of mutations in certain oncogenes and tumor suppressor genes. Chromosomal instability is the most common type of genomic instability in CRC. It is characterized by various changes in chromosomal copy number and structure. The normal activities of certain tumor-suppressor genes, such as APC, P53, and SMAD4, can be altered via a mechanism triggered by chromosomal instability which is responsible for the physical loss of a wild-type copy of these tumor suppressor genes. The second group is the CpG Island Methylation phenotype (CIMP), which is defined by DNA hypermethylation [2], as additional genes were discovered to be influenced by the process, revealing that some groupings of genes had consistently elevated methylation in particular tumors. This was proved statistically by demonstrating that the methylation of two distinct genes in a specific tumor type was associated in cases such as colorectal cancer [3].
The third group is the “microsatellite instable” (MSI) colorectal cancer thatis caused by DNA mismatch repair gene failure, resulting in genetic hypermutability. High MSI was found in 75% of this group, which is often linked with hypermethylation and MLH1 gene silence, whereas the remaining 25% had mutations in the mismatch-repair and polymerase (POLE) genes [4]. Generally, genomic instability can cause aggregation of mutations in genes that are responsible for normal cell regulation and growth, such as proto-oncogenes and tumor suppressor genes [5]. It can also derange the normal cell repair system, induce epigenetic changes in DNA, and produce non-functional proteins that could threaten the healthy cells. Notably, the significant types of genomic instability involved in the development of colorectal cancer are chromosomal instability but microsatellite stable and microsatellite instability (MSI) [6]. Markedly, MSI is often associated with the CpG island methylator phenotype and hypermutation, which is essentially found in the right colon [7]. Furthermore, parallel investigations revealed that the mismatch repair gene MLH1 was hypermethylated and silenced in these MSI-positive tumors. The fact that inhibiting methylation repaired the mismatch repair deficit in colon cancer cell lines supported the hypothesis that hypermethylation causes MSI through MLH1 silencing [3]. MSI affects the size of the mononucleotide or dinucleotide repeats, which are also known as microsatellites, existing all over the genome. It occurs when the strand slippage within the repetitive DNA sequence element failed to be repaired. Such instability resulting from the loss of mismatch-repair function of proteins in DNA can further contribute to the inactivation of the tumor suppression pathway [6].
A cancerous tumor can be characterized by low frequency of somatic mutations such as single nucleotide variants (SNVs), copy number aberrations (CNAs), structural variations, and indels. As indicated by the name, SNVs are aroused by a single nucleotide variant that occurred in one particular genetic position, while CNAs are the amplifications or deletions of copies of a DNA region at a larger scale. However, structural variation is used to describe an area of DNA that is 1 kb or bigger in size and can include inversions, balanced translocations, and genomic imbalances, which are also known as copy number variations. Insertions and deletions, called indels, are changes to the DNA sequence that result in the addition or deletion of one or more nucleotides [8]. Only a small percentage of all somatic changes, known as driver mutations, offer a selective advantage to cancer cells, whereas the vast majority of somatic mutations are passenger mutations that do not contribute to the illness [9]. Inter-tumor heterogeneity, where cancer genomes do not share a similar set of somatic mutations and most of the different metastatic tumors bear a different kind of mutation in the same patient, is the most remarkable trait of the cancer mutational landscape [10]. Besides, in less than 5% of all patients with a specific cancer type, a small number of gene mutations are found in a large portion of tumors and mostly are affected by SNVs or CNAs [11]. Inter-tumor heterogeneity impedes efforts to discover driver genes with driver mutations by recognizing commonly mutated genes that are mutated in a statistically high proportion of patients [12]. The nature of the driver mutations in targeting normal functional genes, groups of interacting proteins, as well as signaling and molecular pathways, is one of the causes of inter-tumor heterogeneity [13].
In silico techniques have long been considered crucial in the efforts of predicting inhibitors, new targets, and diagnostic tools for CRC treatment plans. Exploring binding pockets, residue interactions, and different virtual screening methods are approaches, among others, that were utilized to target CRC [14]. Gene-mutated CRC was targeted by topological in-silico simulations to predict the best treatment combinations that can be successful in clinically advanced conditions [15]. Furthermore, other tactics, such as the simulations that predict the interplay between tumor microenvironment components, could enhance or reduce immunotherapy success or failure [16], and the gut-on-chip model that delineates the molecular mechanism of symbiotic effects on CRC genes’ expression [17] are examples of significant accomplishments in this field. The use of computational methods has also proved a distinguished efficacy by analyzing cell surface proteins overexpression in predicting disease progression, diagnosis, and drug resistance in CRC [18]. MicroRNA was employed as a biomarker for CRC through its attachment to the predicted target gene. The molecular pathways and functional analysis of this non-coding RNA with its target macromolecules can predict CRC pathogenesis [19]. In this review, we summarized the molecular pathways involved in colorectal cancer and the main driver genes that have the greatest triggering impacts. We also discussed the main tumor suppressor genes that can be inactivated, such as APC, TP53, and TGF-β, mainly the growth factor pathways VEGFR and EGFR, and the microsatellite instability mechanism involving genes. In each pathway, an overview of some landmark virtual screening studies that involves finding hits and/or optimizing lead compounds for each individual protein target were provided.
2. Driver Genes in CRC
Multistep tumorigenesis develops through the gradual collection and alterations of driver genes in colorectal cancer. Less than 1% of human genes can potentially turn into cancerous driver genes which are actively capable of controlling cell survival and fate, as well as affecting normal genome stability [10,20]. For a mature cell to become cancerous, it has to undergo phases of breakthrough, expansion, and invasion within 20 to 30 years, involving at least 2 to 3 driver gene mutations. It begins with the first driver mutation which minimally benefits the cell to survive and turns into a proliferating hyperplastic lesion. This could increase the risk of acquiring the second driver gene mutation and further leads to the third driver gene mutation as the cell gained autonomy and immortality, as well as the ability to self-renew. In the case when a third driver gene is involved, the tumor cell is upgraded to become invasive and metastatic. At this point, the malignant cells disseminate without the assistance of other driver mutations [21]. The International Cancer Genome Consortium (ICGC) platform shows the top 20 mutated genes in CRC such as APC, TP53, LRP1B, KRAS, and BRAF, which are significantly impacted by single somatic mutations that also have high functional impact as shown in Figure 1a. ICGC is a global platform that has compiled data on 670,946 unique somatic mutations and molecular profiles from 866 donors for CRC patients. These collected data are grouped into three CRC-related projects, namely, colon adenocarcinoma—TGCA, USA (COAD-US), non-Western colorectal cancer—China (COCA-CN), and rectum adenocarcinoma—USA (READ-US). In the same context, the Cancer Genome Atlas project profiled genomic changes in three cancer types; glioblastoma and ovarian carcinoma, in addition to colon and rectal cancer, among 20 different cancer types with a comprehensive molecular characterization for each one of them [7]. In this project, 276 samples were analyzed for a genome-scale investigation of promoter methylation, exome sequence, DNA copy number, and messenger and microRNA expression. Frequent mutations were revealed in ARID1A, SOX9, and FAM123B, in addition to the expected APC, TP53, SMAD4, PIK3CA, and KRAS mutations as shown in Figure 1b. Furthermore, amplifications in ERBB2 and the “newly-discovered” IGF2 that might be drug-targeted were also identified in the same project, are two examples of recurrent copy-number alterations.
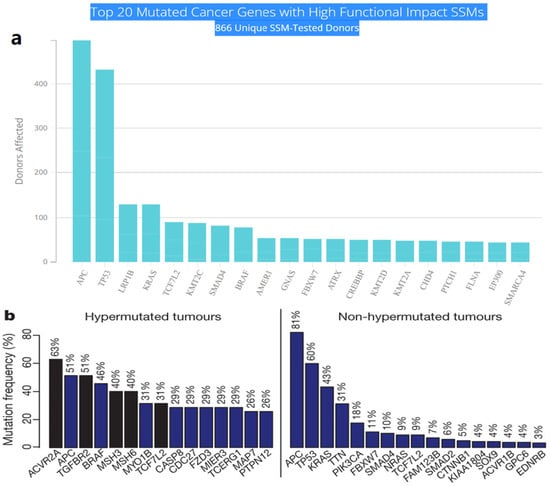
Figure 1.
(a) The top 20 mutated genes with high functional impact involved in colorectal cancer extracted from the ICGC Data Portal in three projects: Colon Adenocarcinoma—TCGA, US, Adenocarcinoma, non-Western (China), Rectum Adenocarcinoma—TCGA, US. https://dcc.icgc.org/ (accessed on 15 December 2021) (b) Significantly mutated genes in hypermutated and non-hypermutated tumors adopted from The Cancer Genome Atlas Network [7].
The genome-wide investigations strongly confirm the links between commonly altered driver genes and human colorectal cancer (Figure 2). Tumorigenesis is generated in the presence of mutant driver genes such as APC, KRAS, SMAD4, TP53, PIK3A, ARID1A, and SOX9, in intestinal epithelial cells using organoid culture systems [7,22]. In addition to the previously stated genes, other changed genes identified to be implicated in colorectal cancer carcinogenesis include FBXW7, BRAF, TCF7L2, PIK3CA, GNAS, CBX4, ADAMTS18, TAF1L, CSMD3, ITGB4, LRP1B, and SYNE1 [23]. APC, KRAS, BRAF, PIK3CA, SMAD4, and TP53 are the six CRC driver genes, with APC, KRAS, PIK3CA, and p53 being the most often altered. Mutations in APC, KRAS, and BRAF occur early in the transition phase from normal epithelium to adenoma, whereas PIK3CA mutation and loss of SMAD4 and P53 (due to mutations or epigenetic silencing) occur late, allowing tumor cells to invade surrounding tissues and metastasize, transforming the adenoma into a carcinoma. Mutations in APC, TP53, and KRAS, as well as, to a lesser extent, SMAD4, are related to metastatic conditions while being highly associated with MSI [24]. The APC (adenomatous polyposis coli) gene is thought to be the gatekeeper gene for CRC, with mutations reported in 83% of all cases [25]. KRAS contributes significantly to carcinogenesis by activating the RAF–MAPK and PI3K pathways. TGF-β signaling, on the other hand, promotes epithelial cell differentiation, acting as a tumor suppressor in colorectal cancer. Furthermore, FBXW7 is a component of the ubiquitin ligase complex, which eliminates proto-oncogene products by degradation, acting as a tumor suppressor, and Fbxw7 disruption promotes intestinal carcinogenesis. According to recent findings, mutant p53 affects gene expression globally via a gain-of-function mechanism, which promotes cancer [22]. APC mutations frequently occur concomitantly with KRAS or TP53 mutations, or both. This triad predicts poor prognosis, whereas BRAF, ITGB4, CBX4, CSMD3, SYNE1, FBXW7, and TAF1L are substantially linked to MSI but not to metastatic illness [20].
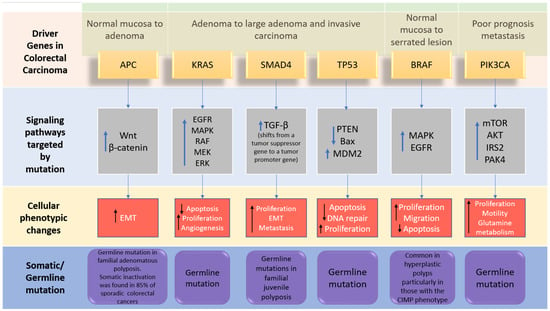
Figure 2.
The driver genes and signaling pathways involved across the CRC adenoma–carcinoma sequence from the transition of normal epithelium through to the metastasis stage in colorectal cancer (adopted from [6]). IRS2; insulin receptor substrate 2, MDM2; Mouse double minute 2 homolog, mTOR; Mammalian target of rapamycin. PAK4; p21 (RAC1) activated kinase 4, EMT; epithelial–mesenchymal transition.
3. Inactivation of Tumor-Suppressor Genes
3.1. Adenomatous Polyposis Coli (APC)
Apart from generating familial adenomatous polyposis (FAP), mutations in both alleles of the APC gene have a rate-limiting role in most sporadic CRC. The cascade of molecular events induced by the loss of APC function can subsequently contribute to the malignancy of the large bowel [26]. One of the crucial intracellular components, β-catenin, which is also the binding partner of APC, is found to be involved in the Wingless/Wnt signal transduction pathway. Wnt signaling pathway, which is promoted by the mutation of gene encoding the APC protein, initiates genomic colorectal carcinogenesis. Normally, the unoccupied, phosphorylated β-catenin is attached to the destruction complex in healthy cells without being stimulated by the extracellular Wnt signal. The destruction complex consists of the scaffolding protein axin, as well as other components such as APC, conductin, and glycogen synthase kinase 3-β (GSK3β). If not attached to that complex, the nuclear localization of β-catenin proteins will create a transcription factor favoring the cellular activation of oncogenic activities. Therefore, as the APC protein complex loses its function due to its encoding gene mutation, Wnt signaling pathway is activated with increasing oncogenic β-catenin protein nuclear localization. Somatic mutations and deletion of APC encoding gene are discovered in most sporadic colorectal adenomas and carcinomas, while germ-line mutations were found in familial adenomatous polyposis [6,27]. Figure 3 illustrates the detailed pathway.
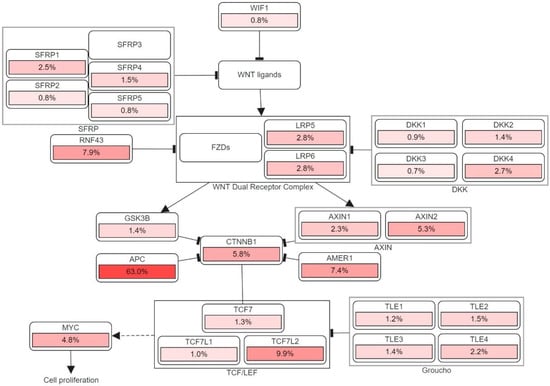
Figure 3.
The genetic pathways and frequencies of mutations collected from 13 studies and 4535 samples in the cBioportal platform that results in deregulation in Wnt signaling pathway, leading to the cell phenotypic modification. The dotted arrow illustrates induction. CTNNB1: Catenin Beta 1, TCF7: Transcription Factor 7, DKK: Dickkopf WNT Signaling Pathway Inhibitor, LRP: LDL Receptor Related Protein, SFRP: Secreted Frizzled Related Protein. The percentage under each gene represents the percent of mutated/altered samples related to the profiled ones in those studies [30,31,32,33,34,35,36,37,38].
CyclinD1 and MYC are the first two discovered downstream targets in Wnt signaling pathway responsible for tumor formation due to their capabilities in cell apoptosis, proliferation, and controlling or disrupting cell-cycle progression. Direct and indirect Myc activation via the Wnt/β-catenin pathway have distinct carcinogenic effects in the intestinal epithelium [28]. On the other hand, β-catenin overexpression in the cytoplasm, may accelerate malignant transformation in colorectal tumors by stimulating cyclin D1 expression [29]. Other Wnt target genes, including matrilysin, CD44, and the urokinase-type plasminogen activator receptor, appear to be more involved in tumor promotion than in tumor initiation [26].
3.2. TP53 Inactivation Pathway
Generally, the most frequent type of gene alterations that occur in human cancers are the p53 gene mutations. The transcriptional activity of the p53 protein is inactivated in most colorectal cancers by a missense mutation of the first allele and a 17p chromosomal deletion that extinguishes the second allele. The functional domains of TP53 are: transactivation domain (TAD), core domain that identifies specific DNA sequences, tetramerization domain, and the C-terminal domain that is responsible for the regulation of p53 activity [39]. As both p53 alleles are eliminated, tumor suppression activities in its pathway were shut down and the existing large adenomas become more invasive. The activity of p53 pathway can also be suppressed by the mutation in gene encoding BAX, which normally induces cell apoptosis, in colorectal cancers with mismatch-repair defects [40]. P53 protein is a stress-inducible transcription factor, acting as a functional regulator in a variety of downstream genes in multiple cell-signaling processes. In order to control the level of p53 from being excessive in normal cells, the negative regulator of p53 i.e., MDM2 will be upregulated to degrade p53 by regulating the ubiquination of p53. An abnormal amount of p53 can lead to cell apoptosis, cell cycle arrest or senescence triggered by DNA damage, hypoxia, and oncogene activation, as well as other cellular stresses [41].
Two pathways are triggered simultaneously upon the activation of p53, namely, the intrinsic mitochondrial and the extrinsic death-receptor-induced apoptotic pathways. Down along the intrinsic pathway, the pro-apoptotic B-cell lymphoma-2 (Ccl-2) family proteins (i.e., BAX, Noxa and PUMA) are induced while the pro-survival Bcl-2 are downregulated instead. As the result of the permeabilization of its outer membrane, the substance cytochrome c, which is released from the mitochondria, binds to Apaf-1 and forms a complex. The complex then activates initiator caspase-9, followed by executioner capase−3, −6, and −7 [42]. In the extrinsic pathway, the expressions of death receptors (DFs) Fas (CD95/APO-1), DR5 (TRAIL-R2), and PIDD (p53-induced protein with death domain) are upregulated as p53 is activated [43]. Additionally, a co-transcription factor named AFT3 assists p53 in maximizing the expression of DR5, which is a trans-membrane tumor necrosis factor (TNF), in CRC induced by DNA damage. DR5 consists of a death domain which binds to the tumor necrosis factor-related apoptosis-inducing ligand (TRIAL) and activates the extrinsic apoptotic pathway that triggers cell death [44].
A variety of small compounds have been designed to target and stabilize certain mutant versions of p53, restoring wild-type (WT)-like transcriptional activity and causing mutant tumor cells to undergo cell cycle arrest or apoptosis. The nine most common mutations of p53 protein (R175H, R248Q, R273H, R248W, R273C, R282W, G245S, R249S, Y220C) account for around 30% of all its cancer-driving mutations [45]. PRIMA-1 and its methyl analog APR-246 are potential small molecules that interact with the DNA binding domain of mutant p53, encouraging correct folding/function and, as a result, increase the production of pro-apoptotic genes Puma, Noxa, and Bax in p53 mutant cells [46]. The Y220C mutation is the ninth most common p53 missense mutation, that is linked to more than 100,000 new cancer cases each year. The Y220C pocket’s hydrophobic and “druggable” characteristics make it a good candidate to be targeted by small-molecule stabilizers. The mutation-induced crevice is far away from the p53 surfaces involved in DNA recognition or protein–protein interactions, allowing for creation of tailored chemical agents that stabilize the DNA binding domain without interfering with its natural substrate binding [45]. Several powerful lead compound families that bind Y220C pockets have been identified in recent years using fragment-based and in silico screening approaches. PK9328 is a carbazole derivative that was identified by computational screening techniques fit in the p53-Y220C binding pocket with a low micromolar affinity and has a significantly decreased cell viability in various Y220C cancer cell lines [47]. Moreover, the pyrazole derivative PK7088 restored p53-Y220C transactivation and downstream upregulation of p21 and Noxa expression, correlated with cell cycle arrest and apoptosis [48].
3.3. TGF-β Tumor Suppressor Pathway
Because it affects cell proliferation, differentiation, apoptosis, and homeostasis, TGF-β signaling is critical in the context of inflammation and cancer. TGF signaling suppresses epithelial growth in normal tissues but promotes tumor cell proliferation in malignant tissues. This phenomenon is called the TGF-β paradox, and instead of its typical nature of inhibiting the epithelial growth in normal tissues, the activated signaling pathway stimulates tumor progression in cancerous cells [49]. Tumor cells’ release of TGF-β also reduces the immune response to the tumor, allowing it to develop further [50]. Two serine/threonine protein kinases (Type I and Type II receptors) and a series of downstream substrates (SMADs) are involved in TGF-β signaling. Type 2 receptors work as activators to phosphorylate type I receptors, and type 1 operate as propagators to carry the signal downstream to cytoplasmic proteins [51]. Bone morphogenetic protein (BMP) type 1 receptors phosphorylate SMAD1/5/8 after ligand binding, whereas TGF- type I and activin type 1 receptors phosphorylate SMAD2/3. These sets of SMAD proteins are known as receptor-regulated SMAD (R-SMAD). Trimerization with a common-mediator SMAD4 and two R-SMAD molecules, which is facilitated by the phosphorylation of two C-terminal serine R-SMAD residues, leads to its translocation into the nucleus to bind to the DNA binding site [52]. The other non-canonical, SMAD-independent pathways that can be transduced by the TGF-β superfamily ligands include phosphoinositide 3-kinase (PI3K)/Akt, Rho/Rho-associated protein kinase (ROCK) pathways, as well as multiple types of mitogen-activated protein kinase (MAPK) [53].
TGFBR2 mutations are frequently found in MSI-H CRC (colorectal cancer with microsatellite instability-high frequency). Mismatch repair genes are silently expressed in MSI-H CRC cells due to germline mutations in genes such as MutL homolog 1 (MLH1), MutS homolog 2 (MSH2), MSH6, and Postmeiotic segregation increased 2 (PMS2), or MLH1 promoter hypermethylation. The genes that are affected by the germline mutations are usually MutL homolog 1 (MLH1), MutS homolog 2 (MSH2), MutS homolog 6 (MSH6), Postmeiotic segregation increased 2 (PMS2) or MLH1 promoter hypermethylation. TGFBR2 mutations, which are often discovered in MSI-H CRC, have the ability to convert normal epithelial cells into malignant ones in the colon [54]. Therefore, the malignant phenotype of the affected CRC cells will arise via Hippo, MAPK, and Wnt-β-catenin signaling pathways [55]. The second type of TGF-β Signaling in CRC is the mutation and deletion of the suppressor gene SMAD4 as a key transcription factor in this pathway. Many genes in the 18q21 chromosomal region are frequently affected by the loss of heterozygosity including SMAD2 and SMAD4 may contribute to forming microsatellite-stable CRC. Because it is a transcription factor for TGF-β signaling, the loss of tumor suppressor gene SMAD4 impairs canonical TGF-β signaling [7]. The non-canonical TGF-signaling route is the third signaling pathway. Although SMAD4 deletion inhibits canonical TGF-β signaling, it modifies BMP signaling via a non-canonical route to enhance CRC metastasis via activation of the Rho/ROCK pathway, resulting in EMT, migration, and invasion. SMAD4 deficiency also activates alternate MEK/ERK pathways, promoting cell death, migration, and invasion [56].
The three above-mentioned inactivation of tumor suppressor genes pathways have witnessed many attempts to develop inhibitors against a certain molecular signaling that was inhibited by the APC, TGF-β, and other genes. In Table 1, we collected a number of representing in-silico studies by computer aided drug discovery and high throughput virtual screening to show the targets that were used and the results of these studies. Due to fundamental roles played by TGF-β suppressor gene, its downstream pathways, and the diverse mutations on its main pathway components, many computational approaches were considered to identify potential small molecules to restore is original function. Nicklas et al. [57] established a computer modeling-based technique capable of statistically analyzing the signaling cascade in order to identify possible treatment targets. They investigated a model that incorporated the exact dynamics of the system, mutations that impact system parameters, and a collection of potentially targetable pathway components, such as the suppression of protein association or production. Interestingly, they also found a collection of mutations that significantly change the signaling dynamics for each cell line, as well as a number of molecular interventions that may be employed to effectively target the effects of these mutations, based on the findings of the molecular intervention optimization method. In a different manner, other in silico studies were established to study the negative regulation on the TGF-β/Smad signaling system on different time scales [58]. This also includes a set of computer models that illustrate the individual and combined impacts of R-Smad negative regulation. Comparisons of models and data indicated that negative regulation occurs at several temporal scales. It has been revealed that a model would need to include at least one fast-mode and one slow-mode effect in order to describe the phospho-R-Smad dynamics in both short- and long-exposure studies. A second important discovery in the aforementioned study was a unique negative feedback effect, which has been verified experimentally, in which the phosphatase PPM1A is increased following TGF- β stimulation. Another addition provided by the same study is an explanation for an earlier debate over proteasomal degradation of phospho-R-Smad. Nevertheless, studies that inhibited proteasomal degradation reported either substantial or no impact on phospho-R-Smad levels. Both of these seemingly contradicting tendencies were mathematically compatible with the mentioned model, and the gap may be explained by varied TGF- β exposure durations.

Table 1.
In silico screening studies that tackle tumor suppressor genes with a library of compounds used and the summaries of those findings.
4. Growth Factor Pathways
The main growth factor pathways include vascular endothelial growth factor receptor-2 (VEGFR-2) and epidermal growth factor receptor (EGFR), as well as other protein kinases.
4.1. Vascular Endothelial Growth Factor Receptor-2 (VEGFR-2)
A majority of central cellular activities are carried out by a total of 518 protein kinases present in the human genome which account for about 2% of all human genes [80]. The protein data bank (PDB) has collected 185 unique structures of human protein kinase domain as well as 197 kinases of other species [81]. VEGF protein kinases are greatly involved in many vascular physiologies, such as the development of blood vessels, formation of lymphatic vessels, and homeostasis. Among the VEGF family, VEGF-A, which is also known as vascular permeability factor, is significant for angiogenesis synchronization and vasculogenesis during embryonic development. In addition, VEGF-A plays a substantial role in repairing the function of damaged tissues [82]. However, it could aggravate cancer in the event of an “angiogenic switch”, which occurs due to the imbalance in anti- and pro-angiogenic activities induced by the recruitment of inflammatory cells into the tumor cells [83]. VEGF-A mediates its biological response through VEGFR2, therefore, it is believed that the protein tyrosine kinase VEGFR2 is a potential target for anti-cancer therapy, as it acts as a medium for VEGF-A to exert its biological activities [84]. A large number of α helical C-terminal lobes, together with smaller portions of β strands-filled N-terminal lobe, construct the catalytic protein kinase domains. In the cleft between the two lobes, an active site which is surrounded by a flexible activation loop on its circumference exists. The activation loop is made of a polypeptide which usually consists of serine, threonine, or tyrosine residues that are ready to be phosphorylated. As phosphorylation occurs, the catalytic activity in the protein kinases will increase dramatically (Figure 4) [85].
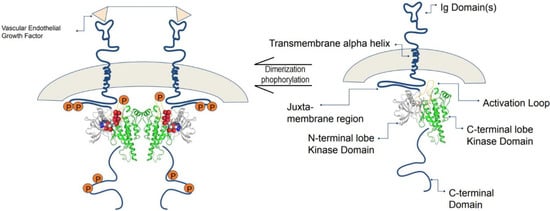
Figure 4.
The composition of VEGFR consists of seven immunoglobin-like motifs. VEGF binds to the extracellular domain, and VEGFRs dimerize, leading to a conformational change that is transmitted across the membrane, which leads to activation. Adapted from Schrodinger tutorials [86].
According to the pattern of conformations, the protein tyrosine kinase inhibitors are classified into 4 types: Type I, Type II, Type III, and Type IV [87]. The competitive Type I and II enzyme inhibitors, which interact with ATP-binding pocket and Mg2+ ion in the active site of the domain between N-terminal and C-terminal lobes, work in the presence of ATP. Type II inhibitors, specifically, extend to new pockets generated by flipped DFG motif next to ATP-binding pocket, and this pocket is formed by DFG motif rearrangement in the inactive conformation [88]. Type II inhibitors have an advantage over type I inhibitors in that they are selective inhibitors with greater chemical space to be exploited compared to type I inhibitors [87]. Despite the high sequence conservation throughout this huge protein family, the breakthrough drug imatinib demonstrated some years ago that the flexibility of kinase structure can permit the generation of specific kinase inhibitors. Imatinib is classified as a “type II” kinase inhibitor because it binds to both the ATP cofactor binding site and an adjacent “allosteric” site that is only available when the kinase adopts a catalytically inactive conformation in which the “Asp-Phe-Gly (DFG)” motif at the N terminus of the activation loop is flipped “out” [89]. Type I inhibitors, such as dasatinib, bind at the ATP site but not the allosteric pocket, hence they are not dependent on certain kinase conformations for binding. Hari et al. [85] address this matter, arguing that underlying disparities in kinase capacity to adopt the DFG-out conformation might contribute to type II inhibitor selectivity.
Magnesium ion-ATP is positioned in a deep cleft between the N- and C-terminal lobes of the highly conserved kinase domain. The bulk of small-molecule kinase inhibitors produced to date target the ATP binding site, with the kinase assuming a conformation that is almost comparable to that of the ATP binding site (the active conformation). The discovery of a second family of kinase inhibitors, whose members preferentially bind to an inactive conformation of the kinase, blocking activation, has been made possible through medicinal chemistry [90]. Type II inhibitors exploit the ATP binding cleft and a nearby hydrophobic pocket generated by the activation loop’s “out” conformation (which contains the conserved DFG motif). Type I inhibitors attach to the ATP binding site by forming hydrogen bonds with the kinase “hinge” residues and by hydrophobic contacts in and around the adenine ring of ATP [91]. Type II inhibitors primarily target the ATP binding site, but they also take advantage of unique hydrogen bonding and hydrophobic interactions enabled by the activation loop’s DFG residues being folded away from the ATP phosphate transfer conformation, as shown in Figure 5 [87].
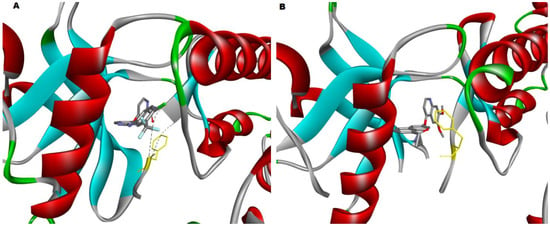
Figure 5.
(A) The crystal structure of the VEGFR2 kinase domain in complex with a benzimidazole inhibitor (2QU5) has the phenylalanine (highlighted in yellow) of the DFG motif facing much closer to the surface of the active site; therefore, it is in the inactive DFG-out state, and (B) The crystal structure of the VEGFR2 kinase domain in complex with a naphthamide inhibitor (3B8R), showing that the DFG motif has the phenylalanine (highlighted in yellow) facing in towards the center of the pocket between the N-lobe and C-lobe; therefore, it is in the active DFG-in state. The two PDB-derived structures were visualized by Discovery Studio v21.1.
Many virtual screening campaigns were established to design potent inhibitors for VEFGR2. Virtual screening uses computer models to assess a specific biological activity of compounds in order to filter existing databases or virtual libraries for the purpose of identifying molecules that have a specific activity against the target of interest. Pharmacophoric, docking, and shape similarity screening studies are carried out in a different setting in order to optimize leads suitable for VEGF receptor-2. Table 2 summarizes the known VEGFR2 inhibitors, their PDB ID, and the effect of those inhibitors against other receptor tyrosine kinases. Additionally, Table 3 shows some in silico campaigns to find hits that can be possible inhibitors against VEGFR2. Since natural products offer immense promise in drug development as the largest source of novel molecules with active biological activities, natural products no doubt continue to be a key part of drug discovery, as they are generally perceived as less toxic. On the contrary, synthetic small molecules and monoclonal antibodies have exhibited a more severe adverse drug reaction profile. In the treatment of cancers by targeting VEGFR-2, bevacizumab, for example, is likely to produce significant ophthalmic inflammation [92], whereas sunitinib can cause multiple adverse drug reactions, including thrombopenia and hypertension [93]. Based on this, many virtual screening and computer aided drug discovery campaigns were initiated to find VEGFR-2 inhibitors based on natural products and natural products library of compounds. Sharma et al. [94] established ligand-based pharmacophore models from the most potent VEGFR-2 inhibitors, then screened a library of 62,082 natural compounds from InterBioscreen natural compound database. The yielded results were passed through many filters to guarantee good binding affinities, biological activity prediction, drug-likeness study, ADMET prediction, and molecular dynamic simulations. Others studied the FDA-approved anti-malarial artemisinin derivatives to be repurposed against VEGFR-2 and other cancer targets [95]. Artemisinin, artenimol, artemether, artemotil, and artesunate were found to interact more potently with CDK-6 and VEGFR-2 than other receptors, in addition to other density functional theory calculations that provided good insight on the electronic and structural properties, as well as various reactivity measures. Furthermore, designing inhibitors that may interact with several cancer targets at the same time, is a promising technique; hence, blocking these three receptor tyrosine kinases (RTKs) with a single chemical component may provide an effective and safe chemotherapeutic option. The polypharmacology of the flavonol “cediodarin” against three RTKs was performed by structure-based pharmacophore mapping and virtual screening of natural products library of compounds. Good affinity results were found for cediodarin against c-MET, EGFR, and VEGFR-2 [96].
Table 2.
An overview for some Vascular Endothelial Growth Factor Receptor-2 inhibitors, their PDB-ID, resolution, and their effects on other receptor kinase targets.
Table 3.
Summaries of high throughput virtual screening that aim at finding hits against vascular endothelial growth factor receptor-2.
4.2. Epidermal Growth Factor Receptor (EGFR)
Epidermal Growth Factor Receptor (EGFR), which is also known as human EGF receptor (HER), is a 170-kilodalton transmembrane cell-surface receptor with c-erb-B proto-oncogene-encoded tyrosine kinase activity [104]. EGFR acts as a catalyst in the transfer of phosphate molecules from ATP to the active site of tyrosine kinase. The resulting signals trigger cellular activities such as anti-apoptotic, tumor cells invasion, and angiogenesis promoting reaction. Subsequently, the intracellular EGFR signaling pathway is initiated together with the activation of AKT and STAT proteins as well as MAP and SRC family kinases. Thus, it further amplifies the transcription of genes that promote cell division and increase survival rate [105]. The overexpression of EGFR protein is discovered in 25 to 75 percent of colorectal cancers with poor prognosis and a high risk of developing metastasis. [106]. Furthermore, EGFR and its ligands, epidermal growth factor (EGF), and transforming growth factor-α (TGF-α) are usually co-expressed at a high level in malignant tissue compared to those in the surrounding mucosa [107]. Generally, such a phenomenon is connected with severe disease or aggressive conditions such as advanced tumor stage cancer with major mesenteric lymph-node involvement [108].
All of the EGFR family members are designated with a greatly glycosylated extracellular region containing 11 sites for glycosylation across 620 amino acids approximately. Each transmembrane domain consists of 23 residues with a juxtamembrane regulatory domain on each side, linking down to a TK domain and C-terminal regulatory region of 232 amino acids [109]. ErbB extracellular region is made up of 4 protein domains: domains I, II, III, and IV [110]. Domains II (CR1) and IV (CR2) are rich in cysteine. Furthermore, leucine-rich domains I and III are favored as binding sites for their competent growth factor ligands. On top of that, numerous studies have shown a variety of mutated EGFR coupled with domains I and III provide a high-affinity binding site for EGF [109]. The activation of EGFR results in a downstream signaling cascade of several pathways such as the RAS-RAF-MAP kinase, phosphatidyl inositol-3-kinase (PI3K), and AKT pathway as well as the activation of other malicious oncogenes such as KRAS, BRAF, MEK, and MAPK [53]. The phosphorylation of phosphatidylinositol-2-phosphate (PIP2) to phosphatidylinositol-3-phosphate (PIP3) leads to the activation of AKT and initiation of carcinoma [111,112].
The strategy of targeting the allosteric site with potent small molecule EGFR degrader has obtained more selective cancer cell killing, disrupting aberrant signaling in mutant tumors and reducing drug resistance. EAI045 is a fourth-generation allosteric EGFR inhibitor that binds away from the ATP-binding site rather than relying on Cys 797 binding. Patel et al. [113] described compound ZINC20531199 as an allosteric inhibitor to overcome the EGFR T790M/C797S Tyrosine Kinase mutation problem using virtual based screening methods. The docked compound was also shown to be stable in the allosteric pocket of the C797S EGFR tyrosine kinase after a 10-ns molecular dynamics simulation. Another attempt was carried out to target the allosteric binding site of C797S mutant EGFR enzyme [114]. Subsequently, the discovery of a Y-shaped structure has paved the way for the development of allosteric fourth-generation EGFR inhibitors. Various enumeration libraries, such as scaffold hopping and R-group enumeration, assisted in the construction of as many novel structural compounds as is feasible. The screening of chemicals from the enumerated library yielded promising allosteric inhibitor hits. Different filters, such as Lipinski’s Rule of Five, ADMET filters, and Jargan’s Rule of Three, were used to further screen the top docking score compounds. The top potential hit was put through a molecular dynamic simulation, which validated the compound’s binding ability and potency. Top-ranked virtual hit compounds binding to the allosteric site of the EGFR enzyme can function as strong EGFR inhibitors in the treatment of non-small cell lung cancer mutations. Moreover, the binding of glucokinase activator to EGFR C797S was investigated using structure-based virtual screening, which revealed that mutant-selective allosteric inhibition might overcome EGFR resistance. EAI045 was shown to be an allosteric, non-ATP competitive inhibitor of mutant C797S EGFR with a Y-shaped structure. Glucokinase activators meet all pharmacophoric requirements, similar to EAI045, and they also occur in a Y-shaped structure, similar to the allosteric inhibitor EAI045, according to a 3D pharmacophoric search. A library of 143 glucokinase activators was tested against all forms of mutant EGFR (C797S, T790M, L858R, TMLR) and WT EGFR, yielding seventeen compounds found to be potential inhibitors for all mutant EGFR in addition to wild type EGFR [115].
4.3. Other Receptor and Protein Kinases in CRC
The Ras-Raf-MAPK/ERK kinase and extracellular signal-regulated kinase 1 and 2 (ERK1/2) are two of the most dysregulated signaling cascades in human cancer, which are included by the MAPK pathway. In addition to the growth factors and cytokines which act via receptor tyrosine kinase signals, RAS and RAF genes mutation can also activate the RAS-RAF-MEK-ERK pathway [116]. Ras and its isotopes NRas, HRas, and Kras, in particular, bind to GDP and are inactive (‘off’ state) in normal quiescent cells, while it binds to GTP (“on” state) in response to external stimuli, which possesses an additional phosphate group. Ras binds GTP to Raf and mobilizes the inactive protein from the cytoplasm, where it recruits the Raf kinases (ARAF, BRAF, and CRAF) to the plasma membrane [117]. Ras also stimulates the serine/threonine kinase action of Raf isoforms after the Ras—Raf complex is translocated to the cell membrane. On the other hand, Raf functions as a MAPK kinase kinase (MAPKKK) when Ras is recruited, activating MEK1 and MEK2, which then catalyze the activation of the effector ERK1 and ERK2 kinases, as well as their translocation into the nucleus. Upon activation, ERK1/ERK2 phosphorylates a number of nuclear and cytoplasmic effector genes involved in a variety of physiological responses, including cell proliferation, survival, differentiation, motility, and angiogenesis [118]. Other downstream signaling pathways that Ras can activate include PI3K, p38 MAPK, and the JNK stress-activated protein kinase pathway. Furthermore, the phosphoinositide 3-kinase (PI3K) enzyme is involved in cancer cell proliferation, survival, and motility/metastasis. Phosphoinositide-dependent protein kinase-1 (PDK1), Akt, the mammalian target of rapamycin (mTOR), and the ribosomal protein S6 kinase (S6K) are all involved in PI3K signaling, which governs cell growth, proliferation, and survival. The fact that mutations in the tumor suppressor gene PTEN are common in human cancers emphasizes the relevance of PI3K/Akt/mTOR signaling in cancer [119,120], as depicted in Figure 6.
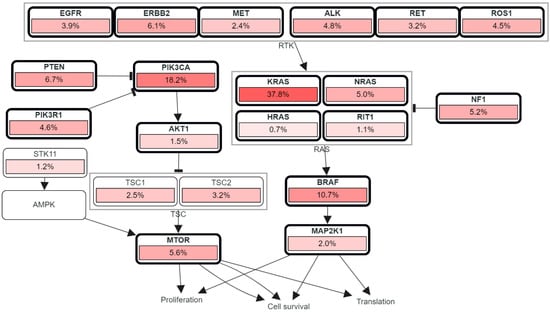
Figure 6.
RTK, RAS, and PI3K signaling in colorectal cancer showing the genetic pathways and frequencies of mutations in 13 studies and 4535 samples in cBioportal platform that led to deregulation in this pathway reaching the cell phenotypic modification. The percentage under each gene represents the percent of mutated/altered samples relative to profiled ones in those studies [30,31,32,33,34,35,36,37,38].
On top of that, IGF-2 has been proposed to act as an auto-/paracrine growth factor in human CRC via binding to IGF-1R. IGF-1 promotes the production of vascular endothelial growth factor (VEGF) in human colon cancer cells by inducing VEGF gene transcription. IGFs are also anti-apoptotic compounds that play a role in cell proliferation and the renewal of epithelial cell populations [121]. Among 22 known ligands of the fibroblast growth factors (FGFs) family, there are 5 highly conserved transmembrane tyrosine kinase receptors (FGFR1-5) that were identified. FGFs interact with the cell surface and its cellular matrix via heparan sulphate proteoglycans (HSPGs) stabilization [122]. A cascade of downstream signaling pathways, such as DAG-PKC and IP3-Ca2+ signaling branches via PLCγ activation, mitogen-activated protein kinase (MAPK), phosphoinositide-3-kinase (PI3K)/Akt pathways, and signal transducer and activator of transcription (STAT), are triggered upon ligand binding and dimerization of FGFRs [123]. Similar to most of the signaling pathways mentioned, FGFR pathway activation contributes to carcinogenesis with somatic abnormalities [124]. The causes of FGFR overexpression include gene alterations (i.e., point mutations and translocations) in the process of post-transcription which results in constitutive activation of receptors or diminished sensitivity in ligand binding as well as production of fusion proteins with uncontrolled cellular activities. Other than that, isoform switching and alternative splicing, which reduces FGFs specificities, can also lead to FGFR overexpression [125]. In Table 4, we summarized some of the receptor tyrosine kinases with examples for virtual screening studies for discovering new lead compounds to the respective receptor/protein.
Table 4.
The characteristics of virtual screening, protein kinases, and the resulting compounds of the screening.
5. Microsatellite Instability Pathways
5.1. Epigenetic Silencing of Gene Expression
In the process of DNA methylation, the enzyme DNA methylase introduces a methylated form of cytosine to the 5′-position as the fifth DNA base by modifying the cytosines within the CpG dinucleotides. In adult cells, the majority of the remaining CpG sites are methylated. A CpG island is found in the promoter region of around half of all genes, and this gene arrangement has received the most attention recently [3]. In colorectal cancer, a remarkable level of abnormal methylation occurs within the CpG-rich region even though there is a global depletion of cytosine methylation in the genome. As a result, it leads to epigenetic silencing of gene expressions and subsequently, the inactivation of the relevant gene (i.e., MLH1) followed by mutation of tumor suppression genes encoding tumor-suppression proteins (i.e., TGFBR2 and BAX) [6]. For instance, the Hereditary nonpolyposis colon cancer (HNPCC) or Lynch syndrome is characterized by germ-line defects in mismatch repair MHL1 and MSH2 genes due to the methylation-induced silencing phenomenon [155]. Somatic inactivation of the wild-type parental allele or more specifically, methylation-inactivated MHL1 gene is also the cause for loss of mismatch-repair function in HNPCC [156]. Therefore, the genomic pattern of HNPCC could be characterized by the combination of somatic and germ-line defects. A specific subgroup resulting from an aberrant methylation mechanism known as CpG island methylator phenotype (CIMP) is discovered in 15% of colorectal cancer cases where it is presented with MHL1 gene expressions silencing. This phenotype is categorized into 2 different subtypes: CIMP-low and CIMP-high in which the magnitude of the methylation is parallel with the clinical manifestations as moderate or aggressive respectively [157].
Cytoskeletal proteins are believed to be a potential therapeutic target as malignant cell transformation commonly displayed interactions among the mismatch-repair system, especially MLH1 protein, due to cytoskeletal reorganization. The other cytoskeletal scaffolding proteins that are involved in such interaction include Actin gamma, Annexin A2, Cathepsin B, Desmin, and Thymosin beta 4 [158]. In CRC with MLH1-deficient cell lines, low levels of cytoskeletal SPTAN1 scaffolding proteins are associated with decreased cell migration whereas high levels of SPTAN1 could promote tumor progression and invasion [159,160]. Furthermore, sporadic tumors with microsatellite instability (MSI) were shown to have higher rates of promoter methylation in numerous genes, including CDKN2A, which encodes the protein INK4A, and THBS1 (thrombosponsin 1) [161]. Other investigations have included HPP1 (hyperplastic polyposis gene 1, also known as TMEFF2) and CDKN2A, which encodes ARF and other proteins, to the list of genes that are preferentially hypermethylated in sporadic MSI positive cases. [162].
5.2. Base Excision Repair Defects
From prokaryotic to eukaryotic cells, base excision repair has been employed to repair the high volume of endogenous DNA damage that occurs as part of the normal physiology process. It is also necessary for normal mammalian development, and its absence has been linked to neurological diseases and cancer. [163]. MutY homolog base excision repair gene (MUTYH) which encodes its MYH protein functions to excise the 8-oxoguanine product from the DNA. The product excised is due to the oxidative damage to Guanine base in the DNA strain [164]. The germ-line inactivation of MYH base-excision gene can result in the development of colorectal cancer. The risk of polyposis phenotype can reach as high as 100% in people by the age of 60 years old, who carry two inactive germline MHY alleles. Genetic testing has proven two common mutations, G382D and Y165C, that are account for 85% of cases of MYH-associated polyposis [6].
Virtual screening was used to identify cytotoxic compounds that would bind to MSH2/MSH6 while the protein is in the death-signaling conformation, causing apoptosis. A DNA-Escherichia coli MutS “as a MSH homolog” complex modified to incorporate the cisplatin adduct cross-linking DNA and performed molecular simulation for the complex [165]. The generated ensemble of conformations was docked with a small library of commercially available drugs to determine which compounds had the highest binding affinities. It was discovered that the E. coli MutS-DNA complex in vitro on MSH2/MSH6 may really employ a selectively binding ligand to choose the proteins’ death-signaling conformation. This study revealed the predictive capacity of in silico molecular dynamics and virtual screening for drug selection. Based on the previous work, the dynamics of MutSα-DNA complexes were studied in order to better understand the physiological response to DNA damage signaling by mismatch-repair proteins. Negureanu et al. [166] used 50 ns molecular dynamic simulations to study correlated movements in response to MutSα binding of mismatched and platinum cross-linked DNA fragments. Firstly, the protein dynamics in response to mismatched and damaged DNA recognition show that MutS signals their recognition via distinct pathways, giving support for the molecular basis of mismatch repair-dependent death. Secondly, the MSH2 subunit is implicated in signaling both mismatched and damaged DNA recognition; localized and collective movements within the protein enable identifying locations on the MSH2 surface that may be relevant in recruiting proteins responsible for downstream actions. This verifies MSH2’s involvement in signaling DNA damage-induced apoptosis and implies that deficiencies in mismatch repair alone are sufficient to cause carcinogenesis, lending credence to the experimental data that mismatch repair-damage response function might protect against tumor initiation. Identifying these specific communication locations might have significance for the treatment of malignancies that are not mismatch repair–deficient but are unable to function adequately for mismatch repair–dependent responses following DNA damage, such as cisplatin resistance.
6. Conclusions
The diverse yet intertwined CRC molecular pathways were reviewed, focusing mainly on the ligand–target based interactions. Furthermore, the importance of in silico studies for the genes that are having a pivotal role in changing the course of the disease was presented. After such studies, it has been found that some had an important impact on the de novo synthesis or repurposing of known commercial drugs to be used as anticancer agents. Moreover, computer-aided drug discovery facilitated the identification of lead compounds for targets that have only a partial or no crystal structure yet identified. When compared to the experimental results, in-silico techniques such as docking, pharmacophoric, shape similarity screening, and molecular dynamics were found to be significantly correlated with wet laboratory results, and this was illustrated in the examples cited in the tables above. Of note, the advances that are being made in virtual drug discovery models and algorithms are time, effort, and cost-saving in discovering new selective inhibitors for allosteric cancer targets and complicated pathways.
Author Contributions
Conceptualization, S.M. and L.C.M.; methodology, S.M., L.C.M. and C.A.; software, C.A. and S.M.; validation, K.W.G.; investigation, S.M., L.C.M. and N.K.; resources, K.W.G., L.C.M. and H.P.G.; data curation, C.A., K.W.G. and Q.J.; writing—original draft preparation, S.M.; writing—review and editing, S.M., L.C.M., C.A., K.W.G. and A.F.L.; visualization, S.M. and A.F.L.; supervision, N.K., H.P.G. and L.C.M.; project administration, L.C.M. All authors have read and agreed to the published version of the manuscript.
Funding
Universiti Brunei Darussalam FIC Research Grant (UBD/RSCH/1.6/, FICBF(b)/2020/007).
Acknowledgments
Figure 1 is, in whole or part, based upon data generated by the National Cancer Institute’s Genomic Data Commons (GDC) data portal: https://portal.gdc.cancer.gov/. Figure 4 is adopted and modified from Schrodinger training tutorial slides. We thank them for allowing us to reproduce these images in our manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, J.; Prenen, H. Molecular genetics of colorectal cancer. Ann. Gastroenterol. 2014, 27, 9. [Google Scholar] [PubMed]
- Issa, J.-P. CpG island methylator phenotype in cancer. Nat. Rev. Cancer 2004, 4, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Castelli, G.; Pelosi, E. Genetic alterations of metastatic colorectal cancer. Biomedicines 2020, 8, 414. [Google Scholar] [CrossRef]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Willett, C.G.; Chang, D.T.; Czito, B.G.; Meyer, J.; Wo, J. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012.(5). Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 1. [Google Scholar] [CrossRef]
- Lin, M.; Whitmire, S.; Chen, J.; Farrel, A.; Shi, X.; Guo, J.-T. Effects of short indels on protein structure and function in human genomes. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Garraway, L.A.; Lander, E.S. Lessons from the Cancer Genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.; Mardis, E.R. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012, 22, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, L.; Muthusamy, K.; Jayaraj, J.M.; Kajamaideen, A.; Balthasar, J.J. In silico insights on tankyrase protein: A potential target for colorectal cancer. J. Biomol. Struct. Dyn. 2018, 37, 3637–3648. [Google Scholar] [CrossRef]
- Baur, F.; Nietzer, S.L.; Kunz, M.; Saal, F.; Jeromin, J.; Matschos, S.; Linnebacher, M.; Walles, H.; Dandekar, T.; Dandekar, G. Connecting cancer pathways to tumor engines: A stratification tool for colorectal cancer combining human in vitro tissue models with boolean in silico models. Cancers 2019, 12, 28. [Google Scholar] [CrossRef]
- Kather, J.N.; Poleszczuk, J.; Suarez-Carmona, M.; Krisam, J.; Charoentong, P.; Valous, N.A.; Weis, C.-A.; Tavernar, L.; Leiss, F.; Herpel, E. In silico modeling of immunotherapy and stroma-targeting therapies in human colorectal cancer. Cancer Res. 2017, 77, 6442–6452. [Google Scholar] [CrossRef]
- Greenhalgh, K.; Ramiro-Garcia, J.; Heinken, A.; Ullmann, P.; Bintener, T.; Pacheco, M.P.; Baginska, J.; Shah, P.; Frachet, A.; Halder, R.; et al. Integrated In Vitro and In Silico Modeling Delineates the Molecular Effects of a Synbiotic Regimen on Colorectal-Cancer-Derived Cells. Cell Rep. 2019, 27, 1621–1632.e1629. [Google Scholar] [CrossRef]
- Nazempour, N.; Taleqani, M.H.; Taheri, N.; Najafabadi, A.H.H.A.A.; Shokrollahi, A.; Zamani, A.; Fattahi Dolatabadi, N.; Peymani, M.; Mahdevar, M. The role of cell surface proteins gene expression in diagnosis, prognosis, and drug resistance of colorectal cancer: In silico analysis and validation. Exp. Mol. Pathol. 2021, 123, 104688. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Klein, A.; Pretorius, A. In silico identification of microRNAs as candidate colorectal cancer biomarkers. Tumor Biol. 2019, 41, 1010428319883721. [Google Scholar] [CrossRef]
- Raskov, H.; Søby, J.H.; Troelsen, J.; Bojesen, R.D.; Gögenur, I. Driver gene mutations and epigenetics in colorectal cancer. Ann. Surg. 2020, 271, 75–85. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. The path to cancer—Three strikes and you’re out. N. Engl. J. Med. 2015, 373, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Sakai, E.; Nakayama, M.; Oshima, H.; Kouyama, Y.; Niida, A.; Fujii, S.; Ochiai, A.; Nakayama, K.I.; Mimori, K.; Suzuki, Y. Combined mutation of Apc, Kras, and Tgfbr2 effectively drives metastasis of intestinal cancer. Cancer Res. 2018, 78, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Schell, M.J.; Yang, M.; Teer, J.K.; Lo, F.Y.; Madan, A.; Coppola, D.; Monteiro, A.N.A.; Nebozhyn, M.V.; Yue, B.; Loboda, A.; et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat. Commun. 2016, 7, 11743. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.; Little, P.; Hayes, D.N.; Lee, M.S. Characterization of the Number and Site of APC Mutations in Sporadic Colorectal Cancer; American Society of Clinical Oncology: Alexandria, VA, USA, 2017. [Google Scholar]
- Fodde, R. The APC gene in colorectal cancer. Eur. J. Cancer 2002, 38, 867–871. [Google Scholar] [CrossRef]
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.-E. Molecular mechanisms of colon cancer progression and metastasis: Recent insights and advancements. Int. J. Mol. Sci. 2021, 22, 130. [Google Scholar] [CrossRef]
- Finch, A.J.; Soucek, L.; Junttila, M.R.; Swigart, L.B.; Evan, G.I. Acute overexpression of Myc in intestinal epithelium recapitulates some but not all the changes elicited by Wnt/β-catenin pathway activation. Mol. Cell. Biol. 2009, 29, 5306–5315. [Google Scholar] [CrossRef][Green Version]
- Utsunomiya, T.; Doki, Y.; Takemoto, H.; Shiozaki, H.; Yano, M.; Sekimoto, M.; Tamura, S.; Yasuda, T.; Fujiwara, Y.; Monden, M. Correlation of beta-catenin and cyclin D1 expression in colon cancers. Oncology 2001, 61, 226–233. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef]
- Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e296. [Google Scholar] [CrossRef] [PubMed]
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014, 15, 454. [Google Scholar] [CrossRef] [PubMed]
- Mondaca, S.; Walch, H.; Nandakumar, S.; Chatila, W.K.; Schultz, N.; Yaeger, R. Specific Mutations in APC, but Not Alterations in DNA Damage Response, Associate With Outcomes of Patients With Metastatic Colorectal Cancer. Gastroenterology 2020, 159, 1975–1978.e1974. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e123. [Google Scholar] [CrossRef]
- Guda, K.; Veigl, M.L.; Varadan, V.; Nosrati, A.; Ravi, L.; Lutterbaugh, J.; Beard, L.; Willson, J.K.; Sedwick, W.D.; Wang, Z.J.; et al. Novel recurrently mutated genes in African American colon cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 1149–1154. [Google Scholar] [CrossRef]
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177, 1035–1049.e1019. [Google Scholar] [CrossRef]
- Huszno, J.; Grzybowska, E. TP53 mutations and SNPs as prognostic and predictive factors in patients with breast cancer. Oncol. Lett. 2018, 16, 34–40. [Google Scholar] [CrossRef]
- He, X.; Liao, J.; Liu, F.; Yan, J.; Yan, J.; Shang, H.; Dou, Q.; Chang, Y.; Lin, J.; Song, Y. Functional repair of p53 mutation in colorectal cancer cells using trans-splicing. Oncotarget 2015, 6, 2034. [Google Scholar] [CrossRef]
- Li, X.-L.; Zhou, J.; Chen, Z.-R.; Chng, W.-J. P53 mutations in colorectal cancer-molecular pathogenesis and pharmacological reactivation. World J. Gastroenterol. 2015, 21, 84. [Google Scholar] [CrossRef]
- Shen, J.; Vakifahmetoglu, H.; Stridh, H.; Zhivotovsky, B.; Wiman, K. PRIMA-1 MET induces mitochondrial apoptosis through activation of caspase-2. Oncogene 2008, 27, 6571–6580. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M.; Phillips, A.C.; Vousden, K.H. Regulation and function of the p53 tumor suppressor protein. Curr. Opin. Cell Biol. 2001, 13, 332–337. [Google Scholar] [CrossRef]
- Taketani, K.; Kawauchi, J.; Tanaka-Okamoto, M.; Ishizaki, H.; Tanaka, Y.; Sakai, T.; Miyoshi, J.; Maehara, Y.; Kitajima, S. Key role of ATF3 in p53-dependent DR5 induction upon DNA damage of human colon cancer cells. Oncogene 2012, 31, 2210–2221. [Google Scholar] [CrossRef] [PubMed]
- Chasov, V.; Mirgayazova, R.; Zmievskaya, E.; Khadiullina, R.; Valiullina, A.; Stephenson Clarke, J.; Rizvanov, A.; Baud, M.G.; Bulatov, E. Key players in the mutant p53 team: Small molecules, gene editing, immunotherapy. Front. Oncol. 2020, 10, 1460. [Google Scholar] [CrossRef] [PubMed]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1Met (APR-246): From mutant/wild type p53 reactivation to unexpected mechanisms underlying their potent anti-tumor effect in combinatorial therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef]
- Bauer, M.R.; Jones, R.N.; Tareque, R.K.; Springett, B.; Dingler, F.A.; Verduci, L.; Patel, K.J.; Fersht, A.R.; Joerger, A.C.; Spencer, J. A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Future Med. Chem. 2019, 11, 2491–2504. [Google Scholar] [CrossRef]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Yingling, J.M.; Blanchard, K.L.; Sawyer, J.S. Development of TGF-β signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 2004, 3, 1011–1022. [Google Scholar] [CrossRef]
- Antony, M.L.; Nair, R.; Sebastian, P.; Karunagaran, D. Changes in expression, and/or mutations in TGF-β receptors (TGF-β RI and TGF-β RII) and Smad 4 in human ovarian tumors. J. Cancer Res. Clin. Oncol. 2010, 136, 351–361. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming growth factor-β signaling pathway in colorectal cancer and its tumor microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.H.; Enns, R.; Heidelbaugh, J.; Barkun, A.; Adams, M.A.; Dorn, S.D.; Dudley-Brown, S.L.; Flamm, S.L.; Gellad, Z.F.; Gruss, C.B. American Gastroenterological Association Institute guideline on the diagnosis and management of Lynch syndrome. Gastroenterology 2015, 149, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M.; Davison, J.; Carter, K.T.; O’Leary, R.M.; Trobridge, P.; Knoblaugh, S.E.; Myeroff, L.L.; Markowitz, S.D.; Brett, B.T.; Scheetz, T.E. Transposon mutagenesis identifies candidate genes that cooperate with loss of transforming growth factor-beta signaling in mouse intestinal neoplasms. Int. J. Cancer 2017, 140, 853–863. [Google Scholar] [CrossRef]
- Voorneveld, P.W.; Kodach, L.L.; Jacobs, R.J.; Liv, N.; Zonnevylle, A.C.; Hoogenboom, J.P.; Biemond, I.; Verspaget, H.W.; Hommes, D.W.; De Rooij, K. Loss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCK. Gastroenterology 2014, 147, 196–208.e113. [Google Scholar] [CrossRef]
- Nicklas, D.; Saiz, L. In silico identification of potential therapeutic targets in the TGF-β signal transduction pathway. Mol. BioSystems 2014, 10, 537–548. [Google Scholar] [CrossRef]
- Wang, J.; Tucker-Kellogg, L.; Ng, I.C.; Jia, R.; Thiagarajan, P.; White, J.K.; Yu, H. The self-limiting dynamics of TGF-β signaling in silico and in vitro, with negative feedback through PPM1A upregulation. PLoS Comput. Biol. 2014, 10, e1003573. [Google Scholar] [CrossRef]
- Jadav, S.S.; Macalino, S.J.Y.; Alluri, R. Structure-based discovery of small molecule APC-Asef interaction inhibitors: In silico approaches and molecular dynamics simulations. J. Mol. Modeling 2020, 26, 1–11. [Google Scholar] [CrossRef]
- Li, B.; Liang, J.; Lu, F.; Zeng, G.; Zhang, J.; Ma, Y.; Liu, P.; Wang, Q.; Zhou, Q.; Chen, L. Discovery of novel inhibitor for Wnt/β-catenin pathway by tankyrase 1/2 structure-based virtual screening. Molecules 2020, 25, 1680. [Google Scholar] [CrossRef]
- Zhang, W.; Lu, W.; Ananthan, S.; Suto, M.J.; Li, Y. Discovery of novel frizzled-7 inhibitors by targeting the receptor’s transmembrane domain. Oncotarget 2017, 8, 91459. [Google Scholar] [CrossRef]
- Lee, H.-M.; Chan, D.S.-H.; Yang, F.; Lam, H.-Y.; Yan, S.-C.; Che, C.-M.; Ma, D.-L.; Leung, C.-H. Identification of natural product Fonsecin B as a stabilizing ligand of c-myc G-quadruplex DNA by high-throughput virtual screening. Chem. Commun. 2010, 46, 4680–4682. [Google Scholar] [CrossRef] [PubMed]
- Carabet, L.A.; Lallous, N.; Leblanc, E.; Ban, F.; Morin, H.; Lawn, S.; Ghaidi, F.; Lee, J.; Mills, I.G.; Gleave, M.E.; et al. Computer-aided drug discovery of Myc-Max inhibitors as potential therapeutics for prostate cancer. Eur. J. Med. Chem. 2018, 160, 108–119. [Google Scholar] [CrossRef]
- Mokgautsi, N.; Wang, Y.-C.; Lawal, B.; Khedkar, H.; Sumitra, M.R.; Wu, A.T.; Huang, H.-S. Network pharmacological analysis through a bioinformatics approach of novel NSC765600 and NSC765691 compounds as potential inhibitors of CCND1/CDK4/PLK1/CD44 in cancer types. Cancers 2021, 13, 2523. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.-H.; Shih, J.-W.; Chen, J.-S.; Mokgautsi, N.; Wei, P.-L.; Huang, Y.-J. Preclinical Identification of Sulfasalazine’s Therapeutic Potential for Suppressing Colorectal Cancer Stemness and Metastasis through Targeting KRAS/MMP7/CD44 Signaling. Biomedicines 2022, 10, 377. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, P.; Mezei, M.; Zhou, M.-M.; Ossowski, L. Computer aided identification of small molecules disrupting uPAR/α5β1-integrin interaction: A new paradigm for metastasis prevention. PLoS ONE 2009, 4, e4617. [Google Scholar] [CrossRef]
- Dolezal, R.; Melikova, M.; Mesicek, J.; Kuca, K. Rational discovery of GSK3-beta modulators aided by protein pocket prediction and high-throughput molecular docking. In Proceedings of the International Conference on Computational Collective Intelligence, Wrocław, Poland, 5–7 October 2009; pp. 429–439. [Google Scholar]
- Nagaraj, A.; Wang, Q.; Joseph, P.; Zheng, C.; Chen, Y.; Kovalenko, O.; Singh, S.; Armstrong, A.; Resnick, K.; Zanotti, K. Using a novel computational drug-repositioning approach (DrugPredict) to rapidly identify potent drug candidates for cancer treatment. Oncogene 2018, 37, 403–414. [Google Scholar] [CrossRef]
- Tian, W.; Han, X.; Yan, M.; Xu, Y.; Duggineni, S.; Lin, N.; Luo, G.; Li, Y.M.; Han, X.; Huang, Z. Structure-based discovery of a novel inhibitor targeting the β-catenin/Tcf4 interaction. Biochemistry 2012, 51, 724–731. [Google Scholar] [CrossRef]
- Enayatkhani, M.; Salimi, M.; Azadmanesh, K.; Teimoori-Toolabi, L. In-silico identification of new inhibitors for Low-density lipoprotein receptor-related protein6 (LRP6). J. Biomol. Struct. Dyn. 2020, 40, 1–11. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.-X.; Lin, Y.-X.; Xu, X.-M.; Li, L.; Yang, J.-B. Virtual Screening Based on Ensemble Docking Targeting Wild-Type p53 for Anticancer Drug Discovery. Chem. Biodivers. 2019, 16, e1900170. [Google Scholar] [CrossRef]
- Park, I.-S.; Seo, H.R.; Kim, K.; Lee, H.; Shum, D.; Choi, I.; Kim, J. Identification of inhibitors of Bcl-2 family protein-protein interaction by combining the BRET screening platform with virtual screening. Biochem. Biophys. Res. Commun. 2020, 527, 709–715. [Google Scholar] [CrossRef]
- Atatreh, N.; Ghattas, M.A.; Bardaweel, S.K.; Al Rawashdeh, S.; Al Sorkhy, M. Identification of new inhibitors of Mdm2–p53 interaction via pharmacophore and structure-based virtual screening. Drug Des. Dev. Ther. 2018, 12, 3741. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, S.M.; Bagherzadeh, K.; Amanlou, M. A new attempt to introduce efficient inhibitors for Caspas-9 according to structure-based Pharmacophore Screening strategy and Molecular Dynamics Simulations. Medbiotech J. 2017, 1, 1–8. [Google Scholar]
- Lakshmi, P.J.; Kumar, B.S.; Nayana, R.S.; Mohan, M.S.; Bolligarla, R.; Das, S.K.; Bhanu, M.U.; Kondapi, A.K.; Ravikumar, M. Design, synthesis, and discovery of novel non-peptide inhibitor of Caspase-3 using ligand based and structure based virtual screening approach. Bioorganic Med. Chem. 2009, 17, 6040–6047. [Google Scholar] [CrossRef]
- Tahir, R.A.; Sehgal, S.A.; Khattak, N.A.; Khan Khattak, J.Z.; Mir, A. Tumor necrosis factor receptor superfamily 10B (TNFRSF10B): An insight from structure modeling to virtual screening for designing drug against head and neck cancer. Theor. Biol. Med. Model. 2013, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sessions, R.B.; Prime, S.S.; Shoemark, D.K.; Allen, S.J.; Hong, W.; Narayanan, S.; Paterson, I.C. Identification of novel small molecule TGF-β antagonists using structure-based drug design. J. Comput. -Aided Mol. Des. 2013, 27, 365–372. [Google Scholar] [CrossRef]
- Singh, J.; Chuaqui, C.E.; Boriack-Sjodin, P.A.; Lee, W.-C.; Pontz, T.; Corbley, M.J.; Cheung, H.K.; Arduini, R.M.; Mead, J.N.; Newman, M.N.; et al. Successful shape-Based virtual screening: The discovery of a potent inhibitor of the type I TGFβ receptor kinase (TβRI). Bioorganic Med. Chem. Lett. 2003, 13, 4355–4359. [Google Scholar] [CrossRef]
- Huang, S.; Mei, H.; Lu, L.; Qiu, M.; Liang, X.; Xu, L.; Kuang, Z.; Heng, Y.; Pan, X. De Novo Molecular Design of Caspase-6 Inhibitors by a GRU-Based Recurrent Neural Network Combined with a Transfer Learning Approach. Pharmaceuticals 2021, 14, 1249. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D. 1998, 54, 1078–1084. [Google Scholar] [CrossRef]
- Nagy, J.A.; Dvorak, A.M.; Dvorak, H.F. VEGF-A and the induction of pathological angiogenesis. Annu. Rev. Pathol. Mech. Dis. 2007, 2, 251–275. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular endothelial growth factor as a target for anticancer therapy. Oncol. 2004, 9, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Hari, S.B.; Merritt, E.A.; Maly, D.J. Sequence Determinants of a Specific Inactive Protein Kinase Conformation. Chem. Biol. 2013, 20, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2022-1. Maestro; Schrödinger LLC: New York, NY, USA. Available online: https://www.schrodinger.com/learn/training/schrodinger-online-learning (accessed on 10 November 2021).
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Rathi, E.; Kumar, A.; Kini, S.G. Molecular dynamics guided insight, binding free energy calculations and pharmacophore-based virtual screening for the identification of potential VEGFR2 inhibitors. J. Recept. Signal Transduct. 2019, 39, 415–433. [Google Scholar] [CrossRef]
- Treiber, D.K.; Shah, N.P. Ins and outs of kinase DFG motifs. Chem. Biol. 2013, 20, 745–746. [Google Scholar] [CrossRef]
- Mol, C.D.; Fabbro, D.; Hosfield, D.J. Structural insights into the conformational selectivity of STI-571 and related kinase inhibitors. Curr. Opin. Drug Discov. Dev. 2004, 7, 639–648. [Google Scholar]
- Traxler, P.; Furet, P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol. Ther. 1999, 82, 195–206. [Google Scholar] [CrossRef]
- Sharma, S.; Johnson, D.; Abouammoh, M.; Hollands, S.; Brissette, A. Rate of serious adverse effects in a series of bevacizumab and ranibizumab injections. Can. J. Ophthalmol. 2012, 47, 275–279. [Google Scholar] [CrossRef]
- Li, X.-S.; Wu, X.; Zhao, P.-J.; Huang, L.-H.; Song, Y.; Gong, K.; Shen, C.Y.W.; Song, G.; Zhao, Z.; Zhang, Z. Efficacy and safety of sunitinib in the treatment of metastatic renal cell carcinoma. Chin. Med. J. 2011, 124, 2920–2924. [Google Scholar]
- Sharma, N.; Sharma, M.; Rahman, Q.I.; Akhtar, S.; Muddassir, M. Quantitative structure activity relationship and molecular simulations for the exploration of natural potent VEGFR-2 inhibitors: An in silico anti-angiogenic study. J. Biomol. Struct. Dyn. 2021, 39, 2806–2823. [Google Scholar] [CrossRef]
- Parveen, S. In silico drug repurposing of fda-approved artemisinins as potent chemotherapeutics targeting BCL-2, CDK-6 & VEGFR-2: Density functional exploration and molecular docking study. Biointerface Res. Appl. Chem. 2021, 11, 9604–9618. [Google Scholar]
- Varma, D.A.; Singh, M.; Wakode, S.; Dinesh, N.; Vinaik, S.; Asthana, S.; Tiwari, M. Structure-based pharmacophore mapping and virtual screening of natural products to identify polypharmacological inhibitor against c-MET/EGFR/VEGFR-2. J. Biomol. Struct. Dyn. 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Ban, H.S.; Kawada, J.; Hirokawa, T.; Nakamura, H. Discovery of indenopyrazoles as EGFR and VEGFR-2 tyrosine kinase inhibitors by in silico high-throughput screening. Bioorganic Med. Chem. Lett. 2008, 18, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, S.; Jiao, Y.; Liu, H.; Yuan, H.; Lu, S.; Ran, T.; Yao, S.; Ke, Z.; Xu, J. An integrated virtual screening approach for VEGFR-2 inhibitors. J. Chem. Inf. Modeling 2013, 53, 3163–3177. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Patidar, K.; Ali, M.A.; Patil, P.; Goud, H.; Hussain, T.; Nayarisseri, A.; Singh, S.K. Structure-based virtual screening for the identification of high affinity compounds as potent VEGFR2 inhibitors for the treatment of renal cell carcinoma. Curr. Top. Med. Chem. 2018, 18, 2174–2185. [Google Scholar] [CrossRef]
- Li, J.; Zhou, N.; Luo, K.; Zhang, W.; Li, X.; Wu, C.; Bao, J. In silico discovery of potential VEGFR-2 inhibitors from natural derivatives for anti-angiogenesis therapy. Int. J. Mol. Sci. 2014, 15, 15994–16011. [Google Scholar] [CrossRef]
- Harris, P.A.; Cheung, M.; Hunter, R.N.; Brown, M.L.; Veal, J.M.; Nolte, R.T.; Wang, L.; Liu, W.; Crosby, R.M.; Johnson, J.H. Discovery and evaluation of 2-anilino-5-aryloxazoles as a novel class of VEGFR2 kinase inhibitors. J. Med. Chem. 2005, 48, 1610–1619. [Google Scholar] [CrossRef]
- Lee, K.; Jeong, K.-W.; Lee, Y.; Song, J.Y.; Kim, M.S.; Lee, G.S.; Kim, Y. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur. J. Med. Chem. 2010, 45, 5420–5427. [Google Scholar] [CrossRef]
- Kankanala, J.; Latham, A.; Johnson, A.; Homer-Vanniasinkam, S.; Fishwick, C.; Ponnambalam, S. A combinatorial in silico and cellular approach to identify a new class of compounds that target VEGFR2 receptor tyrosine kinase activity and angiogenesis. Br. J. Pharmacol. 2012, 166, 737–748. [Google Scholar] [CrossRef]
- Goldstein, N.S.; Armin, M. Epidermal growth factor receptor immunohistochemical reactivity in patients with American Joint Committee on Cancer Stage IV colon adenocarcinoma: Implications for a standardized scoring system. Cancer 2001, 92, 1331–1346. [Google Scholar] [CrossRef]
- Spano, J.P.; Fagard, R.; Soria, J.C.; Rixe, O.; Khayat, D.; Milano, G. Epidermal growth factor receptor signaling in colorectal cancer: Preclinical data and therapeutic perspectives. Ann. Oncol. 2005, 16, 189–194. [Google Scholar] [CrossRef]
- Cohen, R.B. Epidermal growth factor receptor as a therapeutic target in colorectal cancer. Clin. Colorectal Cancer 2003, 2, 246–251. [Google Scholar] [CrossRef]
- Messa, C.; Russo, F.; Gabriella Caruso, M.; Di Leo, A. EGF, TGF-a, and EGF-R in human colorectal adenocarcinoma. Acta Oncol. 1998, 37, 285–289. [Google Scholar] [CrossRef]
- Markman, B.; Javier Ramos, F.; Capdevila, J.; Tabernero, J. EGFR and KRAS in colorectal cancer. In Advances in Clinical Chemistry; Academic Press: New York, NY, USA, 2010; Volume 51, p. 72. [Google Scholar]
- Snyder, L.C.; Astsaturov, I.; Weiner, L.M. Overview of monoclonal antibodies and small molecules targeting the epidermal growth factor receptor pathway in colorectal cancer. Clin. Colorectal Cancer 2005, 5, S71–S80. [Google Scholar] [CrossRef]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.-S.; Leahy, D.J.; Lemmon, M.A. EGF Activates Its Receptor by Removing Interactions that Autoinhibit Ectodomain Dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef]
- Yarom, N.; Jonker, D.J. The role of the epidermal growth factor receptor in the mechanism and treatment of colorectal cancer. Discov. Med. 2011, 11, 95–105. [Google Scholar]
- Berg, M.; Soreide, K. EGFR and downstream genetic alterations in KRAS/BRAF and PI3K/AKT pathways in colorectal cancer—Implications for targeted therapy. Discov. Med. 2012, 14, 207–214. [Google Scholar]
- Patel, H.M.; Ahmad, I.; Pawara, R.; Shaikh, M.; Surana, S. In silico search of triple mutant T790M/C797S allosteric inhibitors to conquer acquired resistance problem in non-small cell lung cancer (NSCLC): A combined approach of structure-based virtual screening and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2021, 39, 1491–1505. [Google Scholar] [CrossRef]
- Karnik, K.S.; Sarkate, A.P.; Lokwani, D.K.; Narula, I.S.; Burra, P.V.; Wakte, P.S. Development of triple mutant T790M/C797S allosteric EGFR inhibitors: A computational approach. J. Biomol. Struct. Dyn. 2021, 39, 5376–5398. [Google Scholar] [CrossRef]
- Patel, H.; Pawara, R.; Surana, S. In-silico evidences for binding of Glucokinase activators to EGFR C797S to overcome EGFR resistance obstacle with mutant-selective allosteric inhibition. Comput. Biol. Chem. 2018, 74, 167–189. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Et. Biophys. Acta (BBA)-Mol. Cell Res. 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Vikis, H.G.; Guan, K.-L. Mechanisms of regulating the Raf kinase family. Cell. Signal. 2003, 15, 463–469. [Google Scholar] [CrossRef]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef]
- Arcaro, A.; Guerreiro, A.S. The phosphoinositide 3-kinase pathway in human cancer: Genetic alterations and therapeutic implications. Curr. Genom. 2007, 8, 271–306. [Google Scholar] [CrossRef] [PubMed]
- Wojtalla, A.; Salm, F.; Christiansen, D.G.; Cremona, T.; Cwiek, P.; Shalaby, T.; Gross, N.; Grotzer, M.A.; Arcaro, A. Novel agents targeting the IGF-1R/PI3K pathway impair cell proliferation and survival in subsets of medulloblastoma and neuroblastoma. PLoS ONE 2012, 7, e47109. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.; Gongoll, S.; Langner, C.; Mengel, M.; Piso, P.; Klempnauer, J.; Rüschoff, J.; Kreipe, H.; von Wasielewski, R. IGF-1R, IGF-1 and IGF-2 expression as potential prognostic and predictive markers in colorectal-cancer. Virchows Arch. 2003, 443, 139–145. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, 1–12. [Google Scholar] [CrossRef]
- Katoh, M.; Nakagama, H. FGF receptors: Cancer biology and therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef]
- Chen, Y.; Li, X.; Eswarakumar, V.P.; Seger, R.; Lonai, P. Fibroblast growth factor (FGF) signaling through PI 3-kinase and Akt/PKB is required for embryoid body differentiation. Oncogene 2000, 19, 3750–3756. [Google Scholar] [CrossRef]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052. [Google Scholar] [CrossRef] [PubMed]
- Mahajanakatti, A.B.; Murthy, G.; Sharma, N.; Skariyachan, S. Exploring inhibitory potential of Curcumin against various cancer targets by in silico virtual screening. Interdiscip. Sci. 2014, 6, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Sathishkumar, N.; Karpagam, V.; Sathiyamoorthy, S.; Woo, M.J.; Kim, Y.-J.; Yang, D.-C. Computer-aided identification of EGFR tyrosine kinase inhibitors using ginsenosides from Panax ginseng. Comput. Biol. Med. 2013, 43, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Rasyid, H.; Purwono, B.; Pranowo, H.D. Design of New Quinazoline Derivative as EGFR (Epidermal Growth Factor Receptor) Inhibitor through Molecular Docking and Dynamics Simulation. Indones. J. Chem. 2021, 21, 201–211. [Google Scholar] [CrossRef]
- Gómez-Ganau, S.; Castillo, J.; Cervantes, A.; de Julián-Ortiz, J.V.; Gozalbes, R. Computational Evaluation and In Vitro Validation of New Epidermal Growth Factor Receptor Inhibitors. Curr. Top. Med. Chem. 2020, 20, 1628–1639. [Google Scholar] [CrossRef]
- Sharda, S.; Khandelwal, R.; Adhikary, R.; Sharma, D.; Majhi, M.; Hussain, T.; Nayarisseri, A.; Singh, S.K. A Computer-Aided Drug Designing for Pharmacological Inhibition of Mutant ALK for the Treatment of Non-small Cell Lung Cancer. Curr. Top. Med. Chem. 2019, 19, 1129–1144. [Google Scholar] [CrossRef] [PubMed]
- Arulanandam, C.D.; Prathiviraj, R.; Kaveriyappan, G.R. Repurposing of an Antifungal Drug against Gastrointestinal Stromal Tumors. Biorxiv 2021. [Google Scholar] [CrossRef]
- Rocca, R.; Moraca, F.; Costa, G.; Talarico, C.; Ortuso, F.; Da Ros, S.; Nicoletto, G.; Sissi, C.; Alcaro, S.; Artese, A. In silico identification of piperidinyl-amine derivatives as novel dual binders of oncogene c-myc/c-Kit G-quadruplexes. ACS Med. Chem. Lett. 2018, 9, 848–853. [Google Scholar] [CrossRef]
- Zhu, J.; Li, K.; Xu, L.; Cai, Y.; Chen, Y.; Zhao, X.; Li, H.; Huang, G.; Jin, J. Discovery of novel selective PI3Kγ inhibitors through combining machine learning-based virtual screening with multiple protein structures and bio-evaluation. J. Adv. Res. 2022, 36, 1–13. [Google Scholar] [CrossRef]
- Liu, X.; Ma, X.H.; Tan, C.; Jiang, Y.; Go, M.; Low, B.C.; Chen, Y.Z. Virtual screening of Abl inhibitors from large compound libraries by support vector machines. J. Chem. Inf. Modeling 2009, 49, 2101–2110. [Google Scholar] [CrossRef]
- Singh, V.K.; Chang, H.-H.; Kuo, C.-C.; Shiao, H.-Y.; Hsieh, H.-P.; Coumar, M.S. Drug repurposing for chronic myeloid leukemia: In silico and in vitro investigation of DrugBank database for allosteric Bcr-Abl inhibitors. J. Biomol. Struct. Dyn. 2017, 35, 1833–1848. [Google Scholar] [CrossRef]
- Kumar, H.; Raj, U.; Gupta, S.; Varadwaj, P.K. In-silico identification of inhibitors against mutated BCR-ABL protein of chronic myeloid leukemia: A virtual screening and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2016, 34, 2171–2183. [Google Scholar] [CrossRef]
- Corradi, V.; Mancini, M.; Manetti, F.; Petta, S.; Santucci, M.A.; Botta, M. Identification of the first non-peptidic small molecule inhibitor of the c-Abl/14-3-3 protein–protein interactions able to drive sensitive and Imatinib-resistant leukemia cells to apoptosis. Bioorganic Med. Chem. Lett. 2010, 20, 6133–6137. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Xie, P.; Marmorstein, R. Identification of BRAF inhibitors through in silico screening. J. Med. Chem. 2008, 51, 6121–6127. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Umar, A.B.; Uzairu, A.; Shallangwa, G.A.; Uba, S. In silico evaluation of some 4-(quinolin-2-yl) pyrimidin-2-amine derivatives as potent V600E-BRAF inhibitors with pharmacokinetics ADMET and drug-likeness predictions. Future J. Pharm. Sci. 2020, 6, 1–10. [Google Scholar] [CrossRef]
- Kulkarni, A.M.; Kumar, V.; Parate, S.; Lee, G.; Yoon, S.; Lee, K.W. Identification of New KRAS G12D Inhibitors through Computer-Aided Drug Discovery Methods. Int. J. Mol. Sci. 2022, 23, 1309. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhuang, C.; Lu, J.; Jiang, Y.; Sheng, C. Discovery of novel KRAS-PDEδ inhibitors by fragment-based drug design. J. Med. Chem. 2018, 61, 2604–2610. [Google Scholar] [CrossRef]
- Ishola, A.A.; Adewole, K.E. In Silico Screening Reveals Histone Deacetylase 7 and ERK1/2 as Potential Targets for Artemisinin Dimer and Artemisinin Dimer Hemisuccinate. Curr. Drug Discov. Technol. 2019, 17, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Pathania, S.; Singh, P.K.; Narang, R.K.; Rawal, R.K. Identifying novel putative ERK1/2 inhibitors via hybrid scaffold hopping–FBDD approach. J. Biomol. Struct. Dyn. 2021, 39, 1–16. [Google Scholar] [CrossRef]
- Xi, D.; Niu, Y.; Li, H.; Noha, S.M.; Temml, V.; Schuster, D.; Wang, C.; Xu, F.; Xu, P. Discovery of carbazole derivatives as novel allosteric MEK inhibitors by pharmacophore modeling and virtual screening. Eur. J. Med. Chem. 2019, 178, 802–817. [Google Scholar] [CrossRef]
- Ashtekar, S.S.; Bhatia, N.M.; Bhatia, M.S. Exploration of Leads from Natural Domain Targeting HER2 in Breast Cancer: An In-Silico Approach. Int. J. Pept. Res. Ther. 2019, 25, 659–667. [Google Scholar] [CrossRef]
- Pasha, M.K.; Jabeen, I.; Samarasinghe, S. 3D QSAR and pharmacophore studies on inhibitors of insuline like growth factor 1 receptor (IGF-1R) and insulin receptor (IR) as potential anti-cancer agents. Curr. Res. Chem. Biol. 2022, 2, 100019. [Google Scholar] [CrossRef]
- Muthumanickam, S.; Indhumathi, T.; Boomi, P.; Balajee, R.; Jeyakanthan, J.; Anand, K.; Ravikumar, S.; Kumar, P.; Sudha, A.; Jiang, Z. In silico approach of naringin as potent phosphatase and tensin homolog (PTEN) protein agonist against prostate cancer. J. Biomol. Struct. Dyn. 2020, 40, 1629–1638. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.-H.; Cheng, T.-C.; Leu, Y.-L.; Chuang, K.-H.; Tzou, S.-C.; Chen, C.-S. Discovery of Akt kinase inhibitors through structure-based virtual screening and their evaluation as potential anticancer agents. Int. J. Mol. Sci. 2015, 16, 3202–3212. [Google Scholar] [CrossRef] [PubMed]
- Saidel, M.É.; dos Santos, K.C.; Nagano, L.F.P.; Montanari, C.A.; Leitão, A. Novel anti-prostate cancer scaffold identified by the combination of in silico and cell-based assays targeting the PI3K-AKT-mTOR pathway. Bioorganic Med. Chem. Lett. 2017, 27, 4001–4006. [Google Scholar] [CrossRef]
- Peddi, S.R.; Sivan, S.K.; Manga, V. Discovery and design of new PI3K inhibitors through pharmacophore-based virtual screening, molecular docking, and binding free energy analysis. Struct. Chem. 2018, 29, 1753–1766. [Google Scholar] [CrossRef]
- Zahler, S.; Tietze, S.; Totzke, F.; Kubbutat, M.; Meijer, L.; Vollmar, A.M.; Apostolakis, J. Inverse In Silico Screening for Identification of Kinase Inhibitor Targets. Chem. Biol. 2007, 14, 1207–1214. [Google Scholar] [CrossRef]
- Yang, W.; AbdulHameed, M.D.M.; Hamza, A.; Zhan, C.-G. New inhibitor of 3-phosphoinositide dependent protein kinase-1 identified from virtual screening. Bioorganic Med. Chem. Lett. 2012, 22, 1629–1632. [Google Scholar] [CrossRef][Green Version]
- Xiao, Z.; Riccardi, D.; Velazquez, H.A.; Chin, A.L.; Yates, C.R.; Carrick, J.D.; Smith, J.C.; Baudry, J.; Quarles, L.D. A computationally identified compound antagonizes excess FGF-23 signaling in renal tubules and a mouse model of hypophosphatemia. Sci. Signal. 2016, 9, ra113. [Google Scholar] [CrossRef]
- Velazquez, H.A.; Riccardi, D.; Xiao, Z.; Quarles, L.D.; Yates, C.R.; Baudry, J.; Smith, J.C. Ensemble docking to difficult targets in early-stage drug discovery: Methodology and application to fibroblast growth factor 23. Chem. Biol. Drug Des. 2018, 91, 491–504. [Google Scholar] [CrossRef]
- Wahlberg, S.S.; Schmeits, J.; Thomas, G.; Loda, M.; Garber, J.; Syngal, S.; Kolodner, R.D.; Fox, E. Evaluation of microsatellite instability and immunohistochemistry for the prediction of germ-line MSH2 and MLH1 mutations in hereditary nonpolyposis colon cancer families. Cancer Res. 2002, 62, 3485–3492. [Google Scholar] [PubMed]
- Evans, D.G.; Walsh, S.; Hill, J.; McMahon, R.T. Strategies for identifying hereditary nonpolyposis colon cancer. Semin. Oncol. 2007, 34, 411–417. [Google Scholar]
- Kawasaki, T.; Ohnishi, M.; Nosho, K.; Suemoto, Y.; Kirkner, G.J.; Meyerhardt, J.A.; Fuchs, C.S.; Ogino, S. CpG island methylator phenotype-low (CIMP-low) colorectal cancer shows not only few methylated CIMP-high-specific CpG islands, but also low-level methylation at individual loci. Mod. Pathol. 2008, 21, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Brieger, A.; Adryan, B.; Wolpert, F.; Passmann, S.; Zeuzem, S.; Trojan, J. Cytoskeletal scaffolding proteins interact with Lynch-Syndrome associated mismatch repair protein MLH1. Proteomics 2010, 10, 3343–3355. [Google Scholar] [CrossRef] [PubMed]
- Hinrichsen, I.; Ernst, B.P.; Nuber, F.; Passmann, S.; Schäfer, D.; Steinke, V.; Friedrichs, N.; Plotz, G.; Zeuzem, S.; Brieger, A. Reduced migration of MLH1 deficient colon cancer cells depends on SPTAN1. Mol. Cancer 2014, 13, 1–12. [Google Scholar] [CrossRef]
- Ackermann, A.; Schrecker, C.; Bon, D.; Friedrichs, N.; Bankov, K.; Wild, P.; Plotz, G.; Zeuzem, S.; Herrmann, E.; Hansmann, M.-L. Downregulation of SPTAN1 is related to MLH1 deficiency and metastasis in colorectal cancer. PLoS ONE 2019, 14, e0213411. [Google Scholar]
- Ahuja, N.; Mohan, A.L.; Li, Q.; Stolker, J.M.; Herman, J.G.; Hamilton, S.R.; Baylin, S.B.; Issa, J.-P.J. Association between CpG island methylation and microsatellite instability in colorectal cancer. Cancer Res. 1997, 57, 3370–3374. [Google Scholar]
- Shibata, D.M.; Sato, F.; Mori, Y.; Perry, K.; Yin, J.; Wang, S.; Xu, Y.; Olaru, A.; Selaru, F.; Spring, K. Hypermethylation of HPP1 is associated with hMLH1 hypermethylation in gastric adenocarcinomas. Cancer Res. 2002, 62, 5637–5640. [Google Scholar]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef]
- Hazra, T.K.; Hill, J.W.; Izumi, T.; Mitra, S. Multiple DNA glycosylases for repair of 8-oxoguanine and their potential in vivo functions. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 193–205. [Google Scholar]
- Godwin, R.C.; Melvin, R.; Salsbury, F.R. Molecular dynamics simulations and computer-aided drug discovery. In Computer-aided Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–30. [Google Scholar]
- Negureanu, L.; Salsbury, F.R., Jr. The molecular origin of the MMR-dependent apoptosis pathway from dynamics analysis of MutSα-DNA complexes. J. Biomol. Struct. Dyn. 2012, 30, 347–361. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).