1. Introduction
With the intense struggle between COVID-19 and mankind, there has been a growing number of investments and attention towards drug repositioning. Drug discovery is a time-consuming, expensive, and laborious process full of ups and downs [
1]. It generally takes more than 10 years to develop a new drug, with a success rate of only 2.01% [
2,
3]. Traditional drug development mainly consists of five stages [
2]: preclinical research, safety review, clinical trials, FDA review, and post-market safety monitoring. By comparison, drug repositioning provides more effective ways and alleviates the bottlenecks of time and cost for many countries.
Figure 1 displays the whole comparison process [
2]. In drug repositioning, the identification of potential drug–target interactions (DTIs) plays an extraordinary role in the early stage of drug development [
4,
5]. Luckily, with the accumulation of more targets, drugs, and their interaction data, various computational approaches have been developed and become accessible in recent years [
6]. It is rather promising and valuable to extract and refine their merits to realize unique contributions of knowledge and accelerate the process of drug repositioning. Therefore, we propose Multi-TransDTI for more effective predictions of potential drug–target interactions.
The computational methods for DTIs can be roughly categorized into the following four relatively superior and advanced methods, on the basis of theoretical and principal differences [
7,
8]: Firstly, matrix factorization has been implemented in DTIs tasks for a long time. Gonen [
9] implemented Bayesian matrix factorization with twin kernels and Ezzat [
10] used regularized matrix factorization methods to approximately multiply matrices representing the drug and target to obtain an interaction matrix and similarity score matrix; this method demonstrated the highest performance in comparison with previous methods at the time. Secondly, studies have focused on several docking methods which simulated the binding site between a molecule and a protein in a 3D structure. With the emergence of a series of open-source 3D docking programs such as AutoDock [
11] and Smina [
12], F. Wan [
13] conducted molecular docking studies for DTIs using AutoDock Vina [
11] on DUD-E, which is a benchmark dataset widely used for evaluating molecular docking. H. Li [
14] also proposed a docking method based on a random forest to improve predictive performance. Thirdly, machine learning-based prediction methods have also flourished in multiple aspects [
15], maintaining relatively superior merits. Optimized models based on machine learning, such as support vector machines (SVM) [
16], Gaussian interaction profiles (GIP) [
17], random forest (RM) [
18], random walk with restart (RWR) [
19,
20], and so on, have been implemented to effectively identify potential compound–protein interactions (CPIs). Despite the promising advances and prospects proposed in above methods, they still face some major obstacles. Most of the docking and machine learning-based methods rely on the assumption of structural similarity between different biological entities [
21,
22]; requiring a vast amount of domain knowledge makes it difficult to obtain the 3D structure of entities [
23], particularly when it comes to large-scale datasets. In addition, researchers have found that the similarity of protein sequences sharing an identical interacting drug is not strongly correlated [
21,
24]. Moreover, similarity-based methods work well for DTIs within specific target classes but not others. In addition, the matrix factorization method has been shown to be not generalizable to different target classes [
5,
22]. Luckily, another final category is the deep learning-based approach, which has risen rapidly in recent years and revolutionized DTIs.
Just as investment, consumption, and export are the major forces driving the economy, the deep learning-based approaches driving DTI performance have their characteristics as well. Differences among various deep learning methods mainly lie in data preprocessing, model architecture, and learning strategy. Lee [
21] simply used a deep network for drug fingerprints and CNN for proteins to obtain prediction outcomes. J. Peng [
25] implemented a restart random walk to extract features from the heterogeneous network and convolutional neural network to complete DTI predictions. F. Wan [
13] constructed corpora for generating both protein and drug feature vectors and then flowed them to multimodal neural networks to obtain binding scores. Both B. Y. Ji [
26] and [
27] proposed a novel network embedding for data preprocessing. K. Abbasi [
28] combined CNN [
29,
30] and LSTM to encode compounds and proteins for predicting DTIs. In recent years, with the breakthrough of transformer models in the field of natural language processing, transformer-based methods such as those described by L. Chen [
31] and K. Huang [
5] have been developed to further improve representation learning for DTIs. Although progress has been made in potential innovations, the following limitations remain: Current representation learning or feature generation approaches of both proteins and drugs are rather complicated. Moreover, current embedding methods such as that described by I. Lee [
21] do not take into account the relationship between each character in the sequence, which often results in a high-dimensional embedding matrix and produces partial data noise. In addition, it is still challenging to effectively obtain important local residues while focusing on global structure. As a result, we propose an end-to-end learning model with a multi-view strategy based on Simple Universal protein and drug dictionaries (SUPD and SUDD) for better embedding [
21,
32,
33], namely Multi-TransDTI, which fully takes into consideration the concerns discussed above.
3. Results
In our work, we introduce Multi-TransDTI, an end-to-end learning model based on the transformer and newly encoded dictionaries. Not only do we avoid the complex feature generation process, but also hugely reduce the dimensionality of embedding methods for both drugs and proteins without losing information. In addition, we use a multi-view strategy and transformer module to further extract local important residues for proteins while focusing on the global structure, demonstrating promising prospects. Finally, we conducted comprehensive comparison experiments on the BindingDB dataset and evaluated the performance of different state-of-the-art models. Results show that Multi-TransDTI is very competitive in predicting potential DTIs, while maintaining the leading prediction capability on small sample datasets.
3.1. Evaluation Indicators
Selection of threshold: We take AUC as the most important indicator to comprehensively measure the advantages and disadvantages of different models. AUC curve is a monotonic increasing function which represents the dynamic classification capability of the model under a series of thresholds. The optimal threshold comes from the point where the distance between the ordinate and abscissa value on the AUC curve is the maximum. Consequently, we define its calculation formula as follows:
where L is the threshold list and
are values at the i-th threshold.
AUC: Area Under Curve. The curve refers to the receiver operating characteristic curve. A series of threshold points drawn by the abscissa value of False Positive Rates (FPR) and ordinate value of True Positive Rates (TPR) are connected together to form the AUC curve. The area under the curve represents its value. The calculation formulas of FPR and TPR are as follows:
AUPR: Area Under Precision-Recall. The drawing process is rather similar to the AUC curve where the only differences lie in the meaning of abscissa and ordinate. The final value of the AUPR curve is also obtained by calculating the area. The calculation formulas of Recall and Precision are as follows:
ACC: Accuracy. The calculation formula is as follows:
F1 score: A trade-off indicator between precision and recall values of the model which represents model stability. The calculation formula is as follows:
3.2. Baseline Methods
DNN: we combined Morgan fingerprints with 100-dimensional encoded tokens based on SUDD as drug feature input, and 800-dimensional encoded tokens based on SUPD as protein features. Then, we flowed them to four layers of the deep neural network, with hidden sizes of 128 and 32 to complete the drug–target interaction prediction task.
Model-CPI: the paper [
41] proposes an end-to-end representation learning for compounds and proteins, developing a new CPI prediction by combing a graph neural network (GNN) for compounds and a convolutional neural network (CNN) for proteins. We used the same parameters in the source code, without making any changes.
Moltrans: the paper [
5] applies an augmented transformer to extract and capture the semantic relationships among substructures generated from massive unlabeled biomedical data. It conducts extensive experiments on different datasets and achieves promising performance. We employed the same parameter settings from the paper.
DeepConv-DTI: as one of the state-of-the-art models in DTI binary prediction tasks, the paper [
21] implements CNN, global max pooling and batch normalization layers to extract local patterns in protein sequences, using dense layers on Morgan fingerprints of the drugs. We used all the same optimal hyperparameters described in that paper.
3.3. Comparisons of Different Models
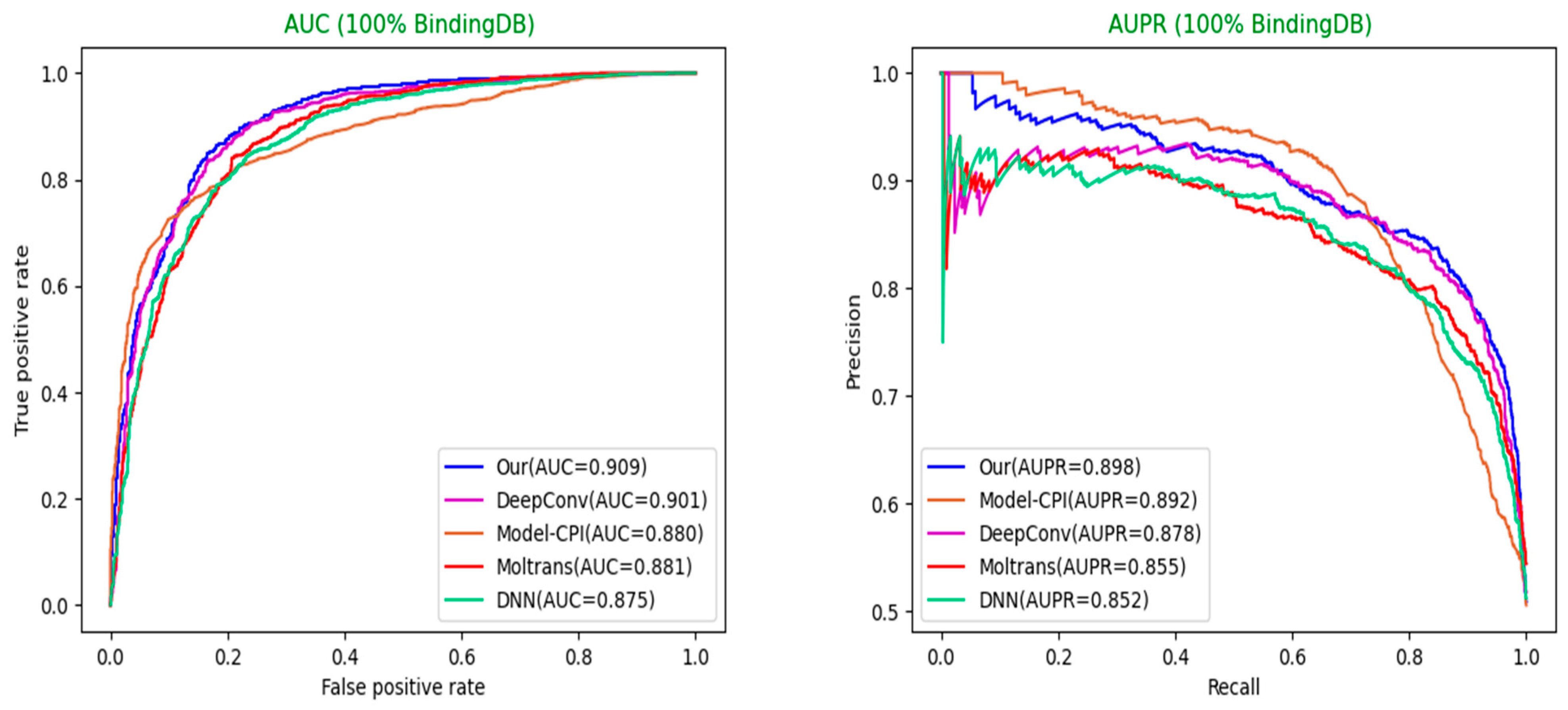
To evaluate the competitiveness of our model, we conducted comparative experiments with state-of-art models proposed previously for drug–target interaction prediction. Our model generally outperforms state-of-the-art models in datasets of different proportions.
Selection of different optimal models: For each model, we used the optimal hyperparameters either given in the source codes or described in the paper. Moreover, each model sets sufficient epochs until the loss value converges completely, so that the weights of the model reach optimality. After that, we repeated the training, validation, and test process on each model three times and adopted the best model among them. The optimal model weights of different models are saved based on the AUC values in the validation set. Finally, we used weights saved to perform the predictions on the test set for each model and calculated a series of indicator values. The comparison results of different models in various indicators are shown in
Table 3,
Table 4 and
Table 5 below. The comparison diagrams for AUC and AUPR are shown in
Figure 5 and
Figure 6.
After all the models are set to the optimal threshold, the bar charts of ACC and F1-score of different models are as follows in
Figure 7 and
Figure 8. As the results prove once again, our method is highly competitive.
3.4. Ablation Experiments
In the end, we also compared different channels of our model to observe the contribution of different channels and the stability of model performance. For drugs and proteins, we separately measured the operation importance of Morgan fingerprints, drug_CNN, protein_CNN, and protein_transformer. Among them, protein_CNN represents proteins that are processed merely by the CNN channel while keeping two drug channels constant. The same is true for the other three channels. The ALL channel denotes the integrated model without removing any parts or channels. We carried out each experiment three times and selected an optimal value based on the AUC value. The specific results are shown in
Table 6. Among them, AUC and AUPR show a tiny difference, which means higher AUC or AUPR channels have relatively more than one optimal threshold. On the other hand, all channels display very competitive performance in ACC and F1-score under the best threshold. Another merit observed in
Table 6 is that the use of a multi-view strategy and multiple information features could make the model more stable and improve the performance in every aspect.
4. Discussion
The biological process of targets (proteins) in our body uniquely influences the fundamental way of life every day, in which their inhibition and activation are highly related to drug interactions. In consequence, predicting potential drug–target interactions has great value in drug repositioning, discovery, etc. Given that biological assays for identifying drug candidates are time consuming and labor intensive, computational prediction approaches have been introduced. However, existing methods generally extract important local residues of protein sequences through CNN-based models, ignoring their global structure and generating an embedding matrix of high dimension. At the same time, the feature generation of drugs (SMILES, InchI, and so on) relies heavily on complex libraries, such as RDKit for fingerprints, resulting in the unavailability of some drug features. Furthermore, the weak generalization ability of different models has become a common problem due to the unicity of features and insufficient learning in the case of small sample data. In view of all the above concerns, we implement a transformer, simple universal dictionaries, and a multi-view strategy, respectively, achieving highly competitive performance in experiments.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







