
Cerebral Glutamate Regulation and Receptor Changes in Perioperative Neuroinflammation and Cognitive Dysfunction


{kind=link}
{kind=link}
Abstract
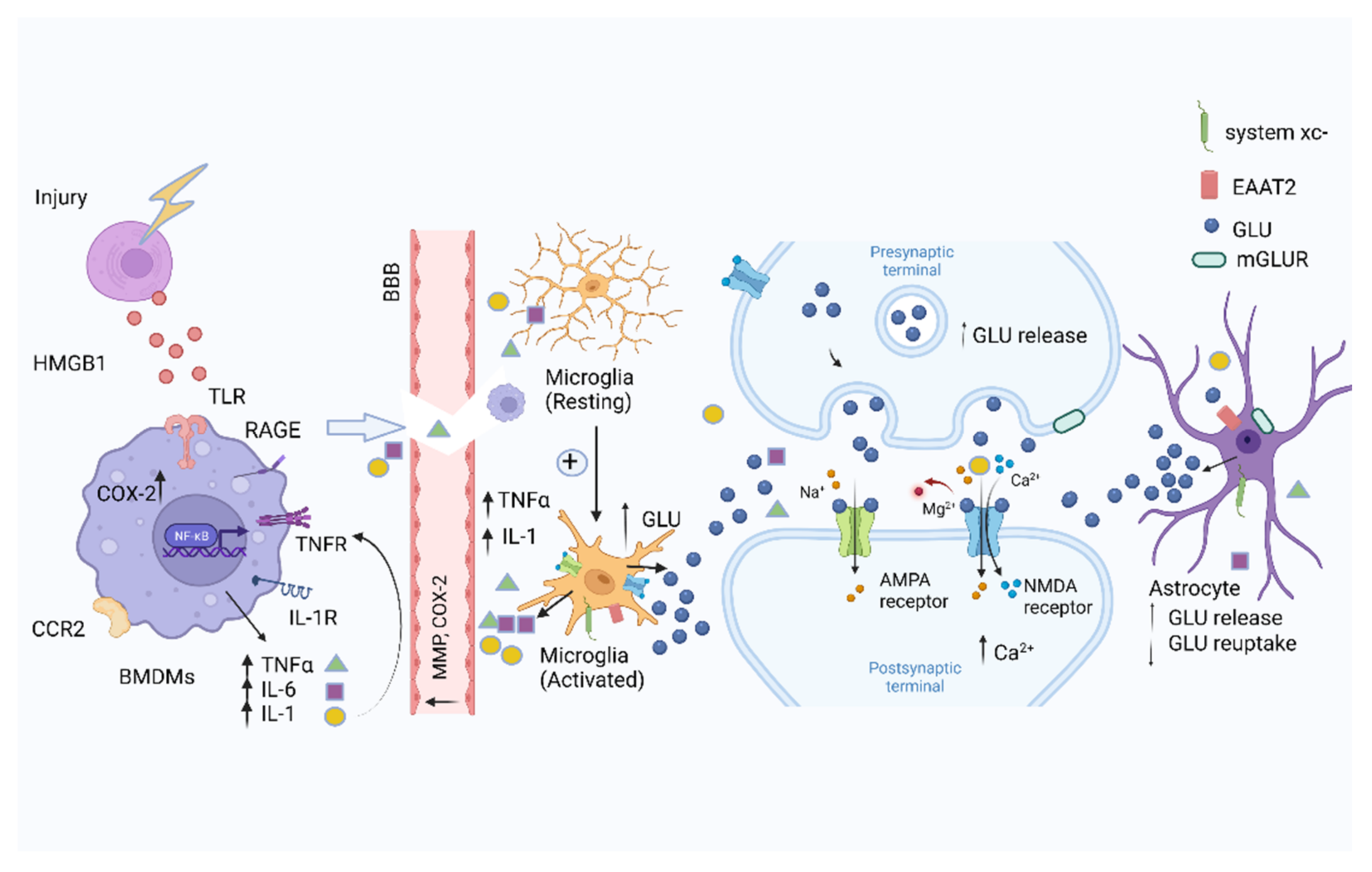
:1. Introduction
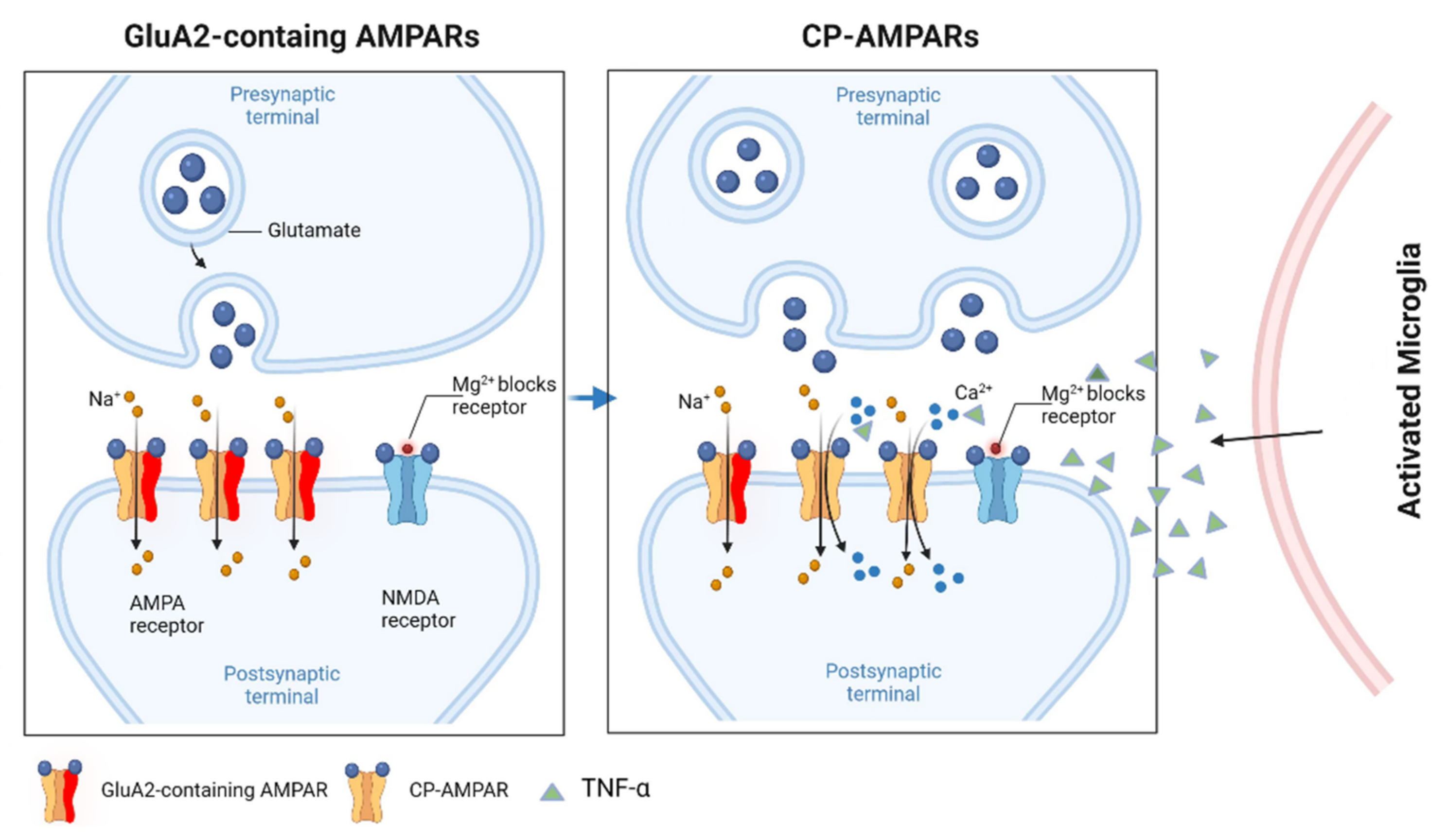
2. Glutamate Receptors
3. Glutamate Release and Handling
4. Regulation of Glutamatergic Neurotransmission by Glial Cells
4.1. Microglia
4.2. Astrocytes
4.3. Oligodendrocytes
5. Regulation of Glutamate Neurotransmission by Peripheral Immune Cells
5.1. Macrophages
5.2. T cells
6. The Effects of Pro-Inflammatory Cytokines on Glutamatergic Function
6.1. Interleukin IL-1β
6.2. IFN-γ
6.3. Interleukin 6 (IL-6)
6.4. TNFα
7. Anesthesia, Surgical Trauma, Glutamatergic Transmission, and Cognitive Dysfunction
7.1. Isoflurane
7.2. Sevoflurane
7.3. Surgical Trauma
8. Potential Therapeutics
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pluvinage, J.V.; Wyss-Coray, T. Author Correction: Systemic factors as mediators of brain homeostasis, ageing and neurodegeneration. Nat. Rev. Neurosci. 2020, 21, 298. [Google Scholar] [CrossRef] [Green Version]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Britschgi, M.; Wyss-Coray, T. Systemic and acquired immune responses in Alzheimer’s disease. Int. Rev. Neurobiol. 2007, 82, 205–233. [Google Scholar]
- Paouri, E.; Georgopoulos, S. Systemic and CNS inflammation crosstalk: Implications for Alzheimer’s disease. Curr. Alzheimer Res. 2019, 16, 559–574. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet. Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Collins, L.M.; Toulouse, A.; Connor, T.J.; Nolan, Y.M. Contributions of central and systemic inflammation to the pathophysiology of Parkinson’s disease. Neuropharmacology 2012, 62, 2154–2168. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, C.C.; Tarelli, R. Parkinson’s disease and systemic inflammation. Parkinson’s Dis. 2011, 2011, 436813. [Google Scholar] [CrossRef] [Green Version]
- Grigoriadis, N.; Grigoriadis, S.; Polyzoidou, E.; Milonas, I.; Karussis, D. Neuroinflammation in multiple sclerosis: Evidence for autoimmune dysregulation, not simple autoimmune reaction. Clin. Neurol. Neurosurg. 2006, 108, 241–244. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Matrix metalloproteinases and neuroinflammation in multiple sclerosis. Neuroscientist 2002, 8, 586–595. [Google Scholar] [CrossRef]
- Christensen, J.R.; Börnsen, L.; Ratzer, R.; Piehl, F.; Khademi, M.; Olsson, T.; Sørensen, P.S.; Sellebjerg, F. Systemic inflammation in progressive multiple sclerosis involves follicular T-helper, Th17-and activated B-cells and correlates with progression. PLoS ONE 2013, 8, e57820. [Google Scholar] [CrossRef]
- Tilleux, S.; Hermans, E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J. Neurosci. Res. 2007, 85, 2059–2070. [Google Scholar] [CrossRef]
- Michaelis, E.K. Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Prog. Neurobiol. 1998, 54, 369–415. [Google Scholar] [CrossRef]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [Green Version]
- Safavynia, S.A.; Goldstein, P.A. The Role of Neuroinflammation in Postoperative Cognitive Dysfunction: Moving From Hypothesis to Treatment. Front. Psychiatry 2018, 9, 752. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.; Selvakumar, G.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.; Zaheer, A. Neuroinflammation induces neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Shi, Y.; Jackson, A.C.; Bjorgan, K.; During, M.J.; Sprengel, R.; Seeburg, P.H.; Nicoll, R.A. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 2009, 62, 254–268. [Google Scholar] [CrossRef] [Green Version]
- Luchkina, N.V.; Huupponen, J.; Clarke, V.R.; Coleman, S.K.; Keinänen, K.; Taira, T.; Lauri, S.E. Developmental switch in the kinase dependency of long-term potentiation depends on expression of GluA4 subunit-containing AMPA receptors. Proc. Natl. Acad. Sci. USA 2014, 111, 4321–4326. [Google Scholar] [CrossRef] [Green Version]
- Rajasekaran, K.; Todorovic, M.; Kapur, J. Calcium-permeable AMPA receptors are expressed in a rodent model of status epilepticus. Ann. Neurol. 2012, 72, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Konen, L.M.; Wright, A.L.; Royle, G.A.; Morris, G.P.; Lau, B.K.; Seow, P.W.; Zinn, R.; Milham, L.T.; Vaughan, C.W.; Vissel, B. A new mouse line with reduced GluA2 Q/R site RNA editing exhibits loss of dendritic spines, hippocampal CA1-neuron loss, learning and memory impairments and NMDA receptor-independent seizure vulnerability. Mol. Brain 2020, 13, 27. [Google Scholar] [CrossRef] [Green Version]
- Vargas, J.L.C.; Osswald, I.K.; Unsain, N.; Aurousseau, M.R.; Barker, P.A.; Bowie, D.; Di Polo, A. Soluble tumor necrosis factor alpha promotes retinal ganglion cell death in glaucoma via calcium-permeable AMPA receptor activation. J. Neurosci. 2015, 35, 12088–12102. [Google Scholar] [CrossRef] [Green Version]
- Conrad, K.L.; Tseng, K.Y.; Uejima, J.L.; Reimers, J.M.; Heng, L.-J.; Shaham, Y.; Marinelli, M.; Wolf, M.E. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 2008, 454, 118–121. [Google Scholar] [CrossRef]
- Paoletti, P.; Neyton, J. NMDA receptor subunits: Function and pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Fazio, F.; Ulivieri, M.; Volpi, C.; Gargaro, M.; Fallarino, F. Targeting metabotropic glutamate receptors for the treatment of neuroinflammation. Curr. Opin. Pharmacol. 2018, 38, 16–23. [Google Scholar] [CrossRef]
- Julio-Pieper, M.; Flor, P.J.; Dinan, T.G.; Cryan, J.F. Exciting times beyond the brain: Metabotropic glutamate receptors in peripheral and non-neural tissues. Pharmacol. Rev. 2011, 63, 35–58. [Google Scholar] [CrossRef] [Green Version]
- Conn, P.J.; Pin, J.-P. Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 205–237. [Google Scholar] [CrossRef] [Green Version]
- Swanson, C.J.; Bures, M.; Johnson, M.P.; Linden, A.-M.; Monn, J.A.; Schoepp, D.D. Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat. Rev. Drug Discov. 2005, 4, 131–144. [Google Scholar] [CrossRef]
- Conn, P.J. Physiological roles and therapeutic potential of metabotropic glutamate receptors. Ann. N. Y. Acad. Sci. 2003, 1003, 12–21. [Google Scholar] [CrossRef]
- Shigemoto, R.; Kinoshita, A.; Wada, E.; Nomura, S.; Ohishi, H.; Takada, M.; Flor, P.J.; Neki, A.; Abe, T.; Nakanishi, S.; et al. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J. Neurosci. 1997, 17, 7503–7522. [Google Scholar] [CrossRef] [Green Version]
- Skeberdis, V.A.; Lan, J.; Opitz, T.; Zheng, X.; Bennett, M.V.; Zukin, R.S. mGluR1-mediated potentiation of NMDA receptors involves a rise in intracellular calcium and activation of protein kinase C. Neuropharmacology 2001, 40, 856–865. [Google Scholar] [CrossRef]
- Matta, J.A.; Ashby, M.C.; Sanz-Clemente, A.; Roche, K.W.; Isaac, J.T. mGluR5 and NMDA receptors drive the experience- and activity-dependent NMDA receptor NR2B to NR2A subunit switch. Neuron 2011, 70, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Südhof, T.C. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron 2013, 80, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215. [Google Scholar] [CrossRef]
- Seki, Y.; Feustel, P.J.; Keller, R.W., Jr.; Tranmer, B.I.; Kimelberg, H.K. Inhibition of ischemia-induced glutamate release in rat striatum by dihydrokinate and an anion channel blocker. Stroke 1999, 30, 433–440. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Kashi-Malina, K.; Cooper, I.; Teichberg, V.I. Mechanisms of glutamate efflux at the blood-brain barrier: Involvement of glial cells. J. Cereb. Blood Flow Metab. 2012, 32, 177–189. [Google Scholar] [CrossRef]
- Zhang, Z.; Tao, Z.; Gameiro, A.; Barcelona, S.; Braams, S.; Rauen, T.; Grewer, C. Transport direction determines the kinetics of substrate transport by the glutamate transporter EAAC1. Proc. Natl. Acad. Sci. USA 2007, 104, 18025–18030. [Google Scholar] [CrossRef] [Green Version]
- Grewer, C.; Gameiro, A.; Zhang, Z.; Tao, Z.; Braams, S.; Rauen, T. Glutamate forward and reverse transport: From molecular mechanism to transporter-mediated release after ischemia. IUBMB Life 2008, 60, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Haydon, P.G.; Carmignoto, G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 2006, 86, 1009–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjørn-Yoshimoto, W.E.; Underhill, S.M. The importance of the excitatory amino acid transporter 3 (EAAT3). Neurochem. Int. 2016, 98, 4–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šerý, O.; Sultana, N.; Kashem, M.A.; Pow, D.V.; Balcar, V.J. GLAST but not least—Distribution, function, genetics and epigenetics of L-glutamate transport in brain—focus on GLAST/EAAT1. Neurochem. Res. 2015, 40, 2461–2472. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hassel, B.; Eid, T.; Danbolt, N.C. Axon-terminals expressing EAAT2 (GLT-1; Slc1a2) are common in the forebrain and not limited to the hippocampus. Neurochem. Int. 2019, 123, 101–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Peled, O.; Ben-Hur, H.; Biegon, A.; Groner, Y.; Dewhurst, S.; Furuta, A.; Rothstein, J.D. Distribution of glutamate transporter subtypes during human brain development. J. Neurochem. 1997, 69, 2571–2580. [Google Scholar] [CrossRef] [Green Version]
- Bridges, R.J.; Esslinger, C.S. The excitatory amino acid transporters: Pharmacological insights on substrate and inhibitor specificity of the EAAT subtypes. Pharmacol. Ther. 2005, 107, 271–285. [Google Scholar] [CrossRef]
- Holmseth, S.; Scott, H.A.; Real, K.; Lehre, K.P.; Leergaard, T.B.; Bjaalie, J.G.; Danbolt, N.C. The concentrations and distributions of three C-terminal variants of the GLT1 (EAAT2; slc1a2) glutamate transporter protein in rat brain tissue suggest differential regulation. Neuroscience 2009, 162, 1055–1071. [Google Scholar] [CrossRef]
- Chen, W.; Mahadomrongkul, V.; Berger, U.V.; Bassan, M.; DeSilva, T.; Tanaka, K.; Irwin, N.; Aoki, C.; Rosenberg, P.A. The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J. Neurosci. 2004, 24, 1136–1148. [Google Scholar] [CrossRef]
- Colton, C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Dissing-Olesen, L.; MacVicar, B.A.; Stevens, B. Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol. 2015, 36, 605–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Dissing-Olesen, L.; Stevens, B. New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 2016, 36, 128–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelucci, A.; Heurtaux, T.; Grandbarbe, L.; Morga, E.; Heuschling, P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J. Neuroimmunol. 2009, 210, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef]
- Park, J.S.; Gamboni-Robertson, F.; He, Q.; Svetkauskaite, D.; Kim, J.Y.; Strassheim, D.; Sohn, J.W.; Yamada, S.; Maruyama, I.; Banerjee, A.; et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell Physiol. 2006, 290, C917–C924. [Google Scholar] [CrossRef]
- Skvarc, D.R.; Berk, M.; Byrne, L.K.; Dean, O.M.; Dodd, S.; Lewis, M.; Marriott, A.; Moore, E.M.; Morris, G.; Page, R.S.; et al. Post-Operative Cognitive Dysfunction: An exploration of the inflammatory hypothesis and novel therapies. Neurosci. Biobehav. Rev. 2018, 84, 116–133. [Google Scholar] [CrossRef]
- Saxena, S.; Maze, M. Impact on the brain of the inflammatory response to surgery. Presse Med. 2018, 47, e73–e81. [Google Scholar] [CrossRef]
- Amantea, D.; Nappi, G.; Bernardi, G.; Bagetta, G.; Corasaniti, M.T. Post-ischemic brain damage: Pathophysiology and role of inflammatory mediators. FEBS J. 2009, 276, 13–26. [Google Scholar] [CrossRef]
- Ye, L.; Huang, Y.; Zhao, L.; Li, Y.; Sun, L.; Zhou, Y.; Qian, G.; Zheng, J.C. IL-1β and TNF-α induce neurotoxicity through glutamate production: A potential role for neuronal glutaminase. J. Neurochem. 2013, 125, 897–908. [Google Scholar] [CrossRef]
- Mascarucci, P.; Perego, C.; Terrazzino, S.; De Simoni, M. Glutamate release in the nucleus tractus solitarius induced by peripheral lipopolysaccharide and interleukin-1β. Neuroscience 1998, 86, 1285–1290. [Google Scholar] [CrossRef]
- Noda, M.; Nakanishi, H.; Nabekura, J.; Akaike, N. AMPA-kainate subtypes of glutamate receptor in rat cerebral microglia. J. Neurosci. 2000, 20, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Joh, T.H. Microglia, major player in the brain inflammation: Their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 2006, 38, 333–347. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Gonçalves, F.Q.; Cristóvão, G.; Rodrigues, L.; Meyer Fernandes, J.R.; Gonçalves, T.; Cunha, R.A.; Gomes, C.A. Different danger signals differently impact on microglial proliferation through alterations of ATP release and extracellular metabolism. Glia 2015, 63, 1636–1645. [Google Scholar] [CrossRef]
- Taylor, D.L.; Jones, F.; Kubota, E.S.; Pocock, J.M. Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor alpha-induced neurotoxicity in concert with microglial-derived Fas ligand. J. Neurosci. 2005, 25, 2952–2964. [Google Scholar] [CrossRef] [Green Version]
- Pinteaux-Jones, F.; Sevastou, I.G.; Fry, V.A.; Heales, S.; Baker, D.; Pocock, J.M. Myelin-induced microglial neurotoxicity can be controlled by microglial metabotropic glutamate receptors. J. Neurochem. 2008, 106, 442–454. [Google Scholar] [CrossRef]
- Piers, T.M.; Heales, S.J.; Pocock, J.M. Positive allosteric modulation of metabotropic glutamate receptor 5 down-regulates fibrinogen-activated microglia providing neuronal protection. Neurosci. Lett. 2011, 505, 140–145. [Google Scholar] [CrossRef]
- Marina, N.; Turovsky, E.; Christie, I.N.; Hosford, P.S.; Hadjihambi, A.; Korsak, A.; Ang, R.; Mastitskaya, S.; Sheikhbahaei, S.; Theparambil, S.M.; et al. Brain metabolic sensing and metabolic signaling at the level of an astrocyte. Glia 2018, 66, 1185–1199. [Google Scholar] [CrossRef] [Green Version]
- Tasdemir-Yilmaz, O.E.; Freeman, M.R. Astrocytes engage unique molecular programs to engulf pruned neuronal debris from distinct subsets of neurons. Genes Dev. 2014, 28, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Matos, M.; Augusto, E.; Oliveira, C.R.; Agostinho, P. Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: Involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience 2008, 156, 898–910. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, X.; Qian, L.; O’Callaghan, J.P.; Hong, J.-S. Astrogliosis in CNS pathologies: Is there a role for microglia? Mol. Neurobiol. 2010, 41, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte crosstalk in CNS inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- O’Kane, R.L.; Martínez-López, I.; DeJoseph, M.R.; Viña, J.R.; Hawkins, R.A. Na(+)-dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) of the blood-brain barrier. A mechanism for glutamate removal. J. Biol. Chem. 1999, 274, 31891–31895. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.L.; Kong, Q.; Cuny, G.D.; Glicksman, M.A. Glutamate transporter EAAT2: A new target for the treatment of neurodegenerative diseases. Future Med. Chem. 2012, 4, 1689–1700. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Li, J.; Li, M.; Yang, X.; Qi, Z.; Xu, B.; Liu, W.; Xu, Z.; Deng, Y. Research progress on the role of type I vesicular glutamate transporter (VGLUT1) in nervous system diseases. Cell Biosci. 2020, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist 2014, 20, 160–172. [Google Scholar] [CrossRef]
- Lutgen, V.; Narasipura, S.D.; Sharma, A.; Min, S.; Al-Harthi, L. β-Catenin signaling positively regulates glutamate uptake and metabolism in astrocytes. J. Neuroinflamm. 2016, 13, 242. [Google Scholar] [CrossRef] [Green Version]
- Vesce, S.; Rossi, D.; Brambilla, L.; Volterra, A. Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. Int. Rev. Neurobiol. 2007, 82, 57–71. [Google Scholar] [CrossRef]
- Noch, E.; Khalili, K. Molecular mechanisms of necrosis in glioblastoma: The role of glutamate excitotoxicity. Cancer Biol. Ther. 2009, 8, 1791–1797. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Friese, M.A.; Craner, M.J.; Palace, J.; Newcombe, J.; Esiri, M.M.; Fugger, L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 2008, 172, 146–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentin-Torres, A.; Savarin, C.; Barnett, J.; Bergmann, C.C. Blockade of sustained tumor necrosis factor in a transgenic model of progressive autoimmune encephalomyelitis limits oligodendrocyte apoptosis and promotes oligodendrocyte maturation. J. Neuroinflamm. 2018, 15, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Maeda, Y.; Ming, X.; Cook, S.; Chapin, J.; Husar, W.; Dowling, P. Apoptotic death following Fas activation in human oligodendrocyte hybrid cultures. J. Neurosci. Res. 2002, 69, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J. CXCR4-activated astrocyte glutamate release via TNFα: Amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Evonuk, K.S.; Doyle, R.E.; Moseley, C.E.; Thornell, I.M.; Adler, K.; Bingaman, A.M.; Bevensee, M.O.; Weaver, C.T.; Min, B.; DeSilva, T.M. Reduction of AMPA receptor activity on mature oligodendrocytes attenuates loss of myelinated axons in autoimmune neuroinflammation. Sci. Adv. 2020, 6, eaax5936. [Google Scholar] [CrossRef] [Green Version]
- McDonald, J.W.; Althomsons, S.P.; Hyrc, K.L.; Choi, D.W.; Goldberg, M.P. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat. Med. 1998, 4, 291–297. [Google Scholar] [CrossRef]
- Domercq, M.; Sánchez-Gómez, M.V.; Sherwin, C.; Etxebarria, E.; Fern, R.; Matute, C. System xc- and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J. Immunol. 2007, 178, 6549–6556. [Google Scholar] [CrossRef] [Green Version]
- Nutma, E.; van Gent, D.; Amor, S.; Peferoen, L.A.N. Astrocyte and Oligodendrocyte Cross-Talk in the Central Nervous System. Cells 2020, 9, 600. [Google Scholar] [CrossRef] [Green Version]
- Semple, B.D.; Kossmann, T.; Morganti-Kossmann, M.C. Role of chemokines in CNS health and pathology: A focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J. Cereb. Blood Flow Metab. 2010, 30, 459–473. [Google Scholar] [CrossRef] [Green Version]
- Piani, D.; Frei, K.; Do, K.Q.; Cuénod, M.; Fontana, A. Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci. Lett. 1991, 133, 159–162. [Google Scholar] [CrossRef]
- Klegeris, A.; McGeer, P.L. beta-amyloid protein enhances macrophage production of oxygen free radicals and glutamate. J. Neurosci. Res. 1997, 49, 229–235. [Google Scholar] [CrossRef]
- Hendriks, J.J.; Teunissen, C.E.; de Vries, H.E.; Dijkstra, C.D. Macrophages and neurodegeneration. Brain Res. Brain Res. Rev. 2005, 48, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Kipnis, J.; Gadani, S.; Derecki, N.C. Pro-cognitive properties of T cells. Nat. Rev. Immunol. 2012, 12, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Ganor, Y.; Grinberg, I.; Reis, A.; Cooper, I.; Goldstein, R.S.; Levite, M. Human T-leukemia and T-lymphoma express glutamate receptor AMPA GluR3, and the neurotransmitter glutamate elevates the cancer-related matrix-metalloproteinases inducer CD147/EMMPRIN, MMP-9 secretion and engraftment of T-leukemia in vivo. Leuk. Lymphoma 2009, 50, 985–997. [Google Scholar] [CrossRef]
- Ganor, Y.; Levite, M. The neurotransmitter glutamate and human T cells: Glutamate receptors and glutamate-induced direct and potent effects on normal human T cells, cancerous human leukemia and lymphoma T cells, and autoimmune human T cells. J. Neural Transm. 2014, 121, 983–1006. [Google Scholar] [CrossRef] [PubMed]
- Viviani, B.; Bartesaghi, S.; Gardoni, F.; Vezzani, A.; Behrens, M.M.; Bartfai, T.; Binaglia, M.; Corsini, E.; Di Luca, M.; Galli, C.L.; et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 2003, 23, 8692–8700. [Google Scholar] [CrossRef]
- Curran, B.P.; Murray, H.J.; O’Connor, J.J. A role for c-Jun N-terminal kinase in the inhibition of long-term potentiation by interleukin-1beta and long-term depression in the rat dentate gyrus in vitro. Neuroscience 2003, 118, 347–357. [Google Scholar] [CrossRef]
- Hu, S.; Sheng, W.S.; Ehrlich, L.C.; Peterson, P.K.; Chao, C.C. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 2000, 7, 153–159. [Google Scholar] [CrossRef]
- Pemberton, L.A.; Kerr, S.J.; Smythe, G.; Brew, B.J. Quinolinic acid production by macrophages stimulated with IFN-gamma, TNF-alpha, and IFN-alpha. J. Interferon Cytokine Res. 1997, 17, 589–595. [Google Scholar] [CrossRef]
- Mizuno, T.; Zhang, G.; Takeuchi, H.; Kawanokuchi, J.; Wang, J.; Sonobe, Y.; Jin, S.; Takada, N.; Komatsu, Y.; Suzumura, A. Interferon-gamma directly induces neurotoxicity through a neuron specific, calcium-permeable complex of IFN-gamma receptor and AMPA GluR1 receptor. FASEB J. 2008, 22, 1797–1806. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Castell, J.V.; Andus, T. Interleukin-6 and the acute phase response. Biochem. J. 1990, 265, 621–636. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Hatanaka, H. Interleukin-6 protects cultured rat hippocampal neurons against glutamate-induced cell death. Brain Res. 1994, 643, 173–180. [Google Scholar] [CrossRef]
- Merrill, J.E. Tumor necrosis factor alpha, interleukin 1 and related cytokines in brain development: Normal and pathological. Dev. Neurosci. 1992, 14, 1–10. [Google Scholar] [CrossRef]
- D’Arcangelo, G.; Tancredi, V.; Onofri, F.; D’Antuono, M.; Giovedì, S.; Benfenati, F. Interleukin-6 inhibits neurotransmitter release and the spread of excitation in the rat cerebral cortex. Eur. J. Neurosci. 2000, 12, 1241–1252. [Google Scholar] [CrossRef]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNFalpha. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef]
- Derkach, V.A.; Oh, M.C.; Guire, E.S.; Soderling, T.R. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci. 2007, 8, 101–113. [Google Scholar] [CrossRef]
- Ferguson, A.R.; Christensen, R.N.; Gensel, J.C.; Miller, B.A.; Sun, F.; Beattie, E.C.; Bresnahan, J.C.; Beattie, M.S. Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J. Neurosci. 2008, 28, 11391–11400. [Google Scholar] [CrossRef]
- Bliss, R.M.; Finckbone, V.L.; Trice, J.; Strahlendorf, H.; Strahlendorf, J. Tumor necrosis factor-α (TNF-α) augments AMPA-induced Purkinje neuron toxicity. Brain Res. 2011, 1386, 1–14. [Google Scholar] [CrossRef]
- Zou, J.Y.; Crews, F.T. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: Neuroprotection by NF kappa B inhibition. Brain Res. 2005, 1034, 11–24. [Google Scholar] [CrossRef]
- Le, Y.; Liu, S.; Peng, M.; Tan, C.; Liao, Q.; Duan, K.; Ouyang, W.; Tong, J. Aging differentially affects the loss of neuronal dendritic spine, neuroinflammation and memory impairment at rats after surgery. PLoS ONE 2014, 9, e106837. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, X.; Zhang, S.; Duan, S.; Qing, W.; Chen, G.; Ye, F.; Le, Y.; Ouyang, W. Pre-existing weakness is critical for the occurrence of postoperative cognitive dysfunction in mice of the same age. PLoS ONE 2017, 12, e0182471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rundshagen, I. Postoperative cognitive dysfunction. Deutsch. Ärztebl. Int. 2014, 111, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, J.T.; Cluitmans, P.; Rasmussen, L.S.; Houx, P.; Rasmussen, H.; Canet, J.; Rabbitt, P.; Jolles, J.; Larsen, K.; Hanning, C. Long-term postoperative cognitive dysfunction in the elderly: ISPOCD1 study. Lancet 1998, 351, 857–861. [Google Scholar] [CrossRef]
- Eckenhoff, R.G. The Perioperative Neurocognitive Disorders; Cambridge University Press: Cambridge, UK, 2019. [Google Scholar]
- Subramaniyan, S.; Terrando, N. Narrative Review Article: Neuroinflammation and Perioperative Neurocognitive Disorders. Anesth. Analg. 2019, 128, 781–788. [Google Scholar] [CrossRef]
- Shen, X.; Dong, Y.; Xu, Z.; Wang, H.; Miao, C.; Soriano, S.G.; Sun, D.; Baxter, M.G.; Zhang, Y.; Xie, Z. Selective anesthesia-induced neuroinflammation in developing mouse brain and cognitive impairment. J. Am. Soc. Anesthesiol. 2013, 118, 502–515. [Google Scholar] [CrossRef] [Green Version]
- Lord, J.M.; Midwinter, M.J.; Chen, Y.F.; Belli, A.; Brohi, K.; Kovacs, E.J.; Koenderman, L.; Kubes, P.; Lilford, R.J. The systemic immune response to trauma: An overview of pathophysiology and treatment. Lancet 2014, 384, 1455–1465. [Google Scholar] [CrossRef] [Green Version]
- Toft, P.; Tønnesen, E. The systemic inflammatory response to anaesthesia and surgery. Curr. Anaesth. Crit. Care 2008, 19, 349–353. [Google Scholar] [CrossRef]
- Wu, L.-J.; Toyoda, H.; Zhao, M.-G.; Lee, Y.-S.; Tang, J.; Ko, S.W.; Jia, Y.H.; Shum, F.W.; Zerbinatti, C.V.; Bu, G. Upregulation of forebrain NMDA NR2B receptors contributes to behavioral sensitization after inflammation. J. Neurosci. 2005, 25, 11107–11116. [Google Scholar] [CrossRef]
- Zeron, M.M.; Fernandes, H.B.; Krebs, C.; Shehadeh, J.; Wellington, C.L.; Leavitt, B.R.; Baimbridge, K.G.; Hayden, M.R.; Raymond, L.A. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington’s disease. Mol. Cell. Neurosci. 2004, 25, 469–479. [Google Scholar] [CrossRef]
- Cheung, N.S.; Beart, P.M.; Pascoe, C.J.; John, C.A.; Bernard, O. Human Bcl-2 protects against AMPA receptor-mediated apoptosis. J. Neurochem. 2000, 74, 1613–1620. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Lu, Y.; Dong, Y.; Zhang, G.; Zhang, Y.; Xu, Z.; Culley, D.J.; Crosby, G.; Marcantonio, E.R.; Tanzi, R.E.; et al. The inhalation anesthetic isoflurane increases levels of proinflammatory TNF-α, IL-6, and IL-1β. Neurobiol. Aging 2012, 33, 1364–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, M.; Hegstad, E.; Berg-Johnsen, J.; Langmoen, I.A. Isoflurane increases the uptake of glutamate in synaptosomes from rat cerebral cortex. Br. J. Anaesth. 1997, 78, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.M.; Drummond, J.C.; Cole, D.J.; Goskowicz, R.L. Isoflurane reduces ischemia-induced glutamate release in rats subjected to forebrain ischemia. Anesthesiology 1995, 82, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zuo, Z. Isoflurane induces a protein kinase C alpha-dependent increase in cell-surface protein level and activity of glutamate transporter type 3. Mol. Pharmacol. 2005, 67, 1522–1533. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Wang, Z.; Mi, W.; Zuo, Z. Isoflurane unveils a critical role of glutamate transporter type 3 in regulating hippocampal GluR1 trafficking and context-related learning and memory in mice. Neuroscience 2014, 272, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.A.; Choi, J.G.; Zuo, Z. Volatile anesthetics attenuate oxidative stress-reduced activity of glutamate transporter type 3. Anesth. Analg. 2009, 109, 1506–1510. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Park, S.H.; Zuo, Z. Effects of isoflurane on learning and memory functions of wild-type and glutamate transporter type 3 knockout mice. J. Pharm. Pharmacol. 2012, 64, 302–307. [Google Scholar] [CrossRef]
- Wang, X.; Shan, Y.; Tang, Z.; Gao, L.; Liu, H. Neuroprotective effects of dexmedetomidine against isoflurane-induced neuronal injury via glutamate regulation in neonatal rats. Drug Des. Dev. Ther. 2019, 13, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Huang, Y.; Zuo, Z. The different responses of rat glutamate transporter type 2 and its mutant (tyrosine 403 to histidine) activity to volatile anesthetics and activation of protein kinase C. Brain Res. 2002, 953, 255–264. [Google Scholar] [CrossRef]
- Vutskits, L.; Xie, Z. Lasting impact of general anaesthesia on the brain: Mechanisms and relevance. Nat. Rev. Neurosci. 2016, 17, 705–717. [Google Scholar] [CrossRef]
- Saatman, K.E.; Bozyczko-Coyne, D.; Marcy, V.; Siman, R.; McIntosh, T.K. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J. Neuropathol. Exp. Neurol. 1996, 55, 850–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Zhang, X.; Li, S.; Chi, X.; Luo, A.; Zhao, Y. NR2B receptor- and calpain-mediated KCC2 cleavage resulted in cognitive deficiency exposure to isoflurane. Neurotoxicology 2020, 76, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Rammes, G.; Starker, L.K.; Haseneder, R.; Berkmann, J.; Plack, A.; Zieglgänsberger, W.; Ohl, F.; Kochs, E.F.; Blobner, M. Isoflurane anaesthesia reversibly improves cognitive function and long-term potentiation (LTP) via an up-regulation in NMDA receptor 2B subunit expression. Neuropharmacology 2009, 56, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Li, L.; Lin, D.; Zuo, Z. Isoflurane induces learning impairment that is mediated by interleukin 1β in rodents. PLoS ONE 2012, 7, e51431. [Google Scholar] [CrossRef]
- Lin, D.; Zuo, Z. Isoflurane induces hippocampal cell injury and cognitive impairments in adult rats. Neuropharmacology 2011, 61, 1354–1359. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, P.; Zhang, X.; Zhang, W.; Gu, G. Effects of different concentration and duration time of isoflurane on acute and long-term neurocognitive function of young adult C57BL/6 mouse. Int. J. Clin. Exp. Pathol. 2014, 7, 5828–5836. [Google Scholar]
- Qu, X.; Xu, C.; Wang, H.; Xu, J.; Liu, W.; Wang, Y.; Jia, X.; Xie, Z.; Xu, Z.; Ji, C.; et al. Hippocampal glutamate level and glutamate aspartate transporter (GLAST) are up-regulated in senior rat associated with isoflurane-induced spatial learning/memory impairment. Neurochem. Res. 2013, 38, 59–73. [Google Scholar] [CrossRef]
- Wang, N.; Wang, M. Dexmedetomidine suppresses sevoflurane anesthesia-induced neuroinflammation through activation of the PI3K/Akt/mTOR pathway. BMC Anesthesiol. 2019, 19, 134. [Google Scholar] [CrossRef] [Green Version]
- Moe, M.C.; Berg-Johnsen, J.; Larsen, G.A.; Røste, G.K.; Vinje, M.L. Sevoflurane reduces synaptic glutamate release in human synaptosomes. J. Neurosurg. Anesthesiol. 2002, 14, 180–186. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, F.; Shi, J. Neonatal exposure to sevoflurane caused cognitive deficits by dysregulating SK2 channels and GluA2-lacking AMPA receptors in juvenile rat hippocampus. Neuropharmacology 2018, 141, 66–75. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, T.; Chen, Y.; Sun, Z.; Lu, J.; Shi, Z.; Song, X. Prenatal sevoflurane exposure causes abnormal development of the entorhinal cortex in rat offspring. J. Integr. Neurosci. 2021, 20, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cheng, Y.; Liu, G.; Tian, X.; Tu, X.; Wang, J. Chronic exposure of gestation rat to sevoflurane impairs offspring brain development. Neurol. Sci. 2012, 33, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Satomoto, M.; Sun, Z.; Adachi, Y.U.; Makita, K. Neonatal Sevoflurane Exposure Induces Adulthood Fear-induced Learning Disability and Decreases Glutamatergic Neurons in the Basolateral Amygdala. J. Neurosurg. Anesthesiol. 2018, 30, 59–64. [Google Scholar] [CrossRef]
- Wang, W.; Lu, R.; Feng, D.Y.; Zhang, H. Sevoflurane Inhibits Glutamate-Aspartate Transporter and Glial Fibrillary Acidic Protein Expression in Hippocampal Astrocytes of Neonatal Rats Through the Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) Pathway. Anesth. Analg. 2016, 123, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Haseneder, R.; Starker, L.; Berkmann, J.; Kellermann, K.; Jungwirth, B.; Blobner, M.; Eder, M.; Kochs, E.; Rammes, G. Sevoflurane anesthesia improves cognitive performance in mice, but does not influence in vitro long-term potentation in hippocampus CA1 stratum radiatum. PLoS ONE 2013, 8, e64732. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Li, P.; Liu, P.; Yan, H.; Wang, J.; Lu, W.; Liu, C.; Zhou, Y. Cistanches alleviates sevoflurane-induced cognitive dysfunction by regulating PPAR-γ-dependent antioxidant and anti-inflammatory in rats. J. Cell. Mol. Med. 2020, 24, 1345–1359. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, F.; Shi, J. Sevoflurane anesthesia impairs metabotropic glutamate receptor-dependent long-term depression and cognitive functions in senile mice. Geriatr. Gerontol. Int. 2019, 19, 357–362. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Hadavi, M.; Hassanshahi, G.; Rezaeian, M.; Vazirinejad, R. General anesthetics on immune system cytokines: A narrative review article. Anesth. Pain Med. 2020, 10, e103033. [Google Scholar] [CrossRef]
- Hietbrink, F.; Koenderman, L.; Rijkers, G.; Leenen, L. Trauma: The role of the innate immune system. World J. Emerg. Surg. 2006, 1, 15. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Helmy, S.A.; Wahby, M.A.; El-Nawaway, M. The effect of anaesthesia and surgery on plasma cytokine production. Anaesthesia 1999, 54, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Gu, C.; Mandeville, E.T.; Dong, Y.; Esposito, E.; Zhang, Y.; Yang, G.; Shen, Y.; Fu, X.; Lo, E.H. Anesthesia and surgery impair blood–brain barrier and cognitive function in mice. Front. Immunol. 2017, 8, 902. [Google Scholar] [CrossRef] [PubMed]
- Stover, J.; Kempski, O. Anesthesia increases circulating glutamate in neurosurgical patients. Acta Neurochir. 2005, 147, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Kawano, T.; Tamura, T.; Iwata, H.; Takahashi, Y.; Eguchi, S.; Yamazaki, F.; Kumagai, N.; Yokoyama, M. Postoperative pain impairs subsequent performance on a spatial memory task via effects on N-methyl-D-aspartate receptor in aged rats. Life Sci. 2013, 93, 986–993. [Google Scholar] [CrossRef]
- Zhang, X.; Xin, X.; Dong, Y.; Zhang, Y.; Yu, B.; Mao, J.; Xie, Z. Surgical incision-induced nociception causes cognitive impairment and reduction in synaptic NMDA receptor 2B in mice. J. Neurosci. 2013, 33, 17737–17748. [Google Scholar] [CrossRef] [Green Version]
- Riazi, K.; Galic, M.A.; Kentner, A.C.; Reid, A.Y.; Sharkey, K.A.; Pittman, Q.J. Microglia-dependent alteration of glutamatergic synaptic transmission and plasticity in the hippocampus during peripheral inflammation. J. Neurosci. 2015, 35, 4942–4952. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xiao, B.; Li, C.X.; Wang, Y. Fingolimod (FTY720) improves postoperative cognitive dysfunction in mice subjected to D-galactose-induced aging. Neural Regen. Res. 2020, 15, 1308–1315. [Google Scholar] [CrossRef]
- Hu, N.; Wang, M.; Xie, K.; Wang, H.; Wang, C.; Wang, C.; Wang, C.; Li, Y.; Yu, Y.; Wang, G. Internalization of GluA2 and the underlying mechanisms of cognitive decline in aged rats following surgery and prolonged exposure to sevoflurane. Neurotoxicology 2015, 49, 94–103. [Google Scholar] [CrossRef]
- Wang, T.; Zhu, H.; Hou, Y.; Gu, W.; Wu, H.; Luan, Y.; Xiao, C.; Zhou, C. Galantamine reversed early postoperative cognitive deficit via alleviating inflammation and enhancing synaptic transmission in mouse hippocampus. Eur. J. Pharmacol. 2019, 846, 63–72. [Google Scholar] [CrossRef]
- Xie, W.; Yang, Y.; Gu, X.; Zheng, Y.; Sun, Y.E.; Liang, Y.; Bo, J.; Ma, Z. Senegenin attenuates hepatic ischemia-reperfusion induced cognitive dysfunction by increasing hippocampal NR2B expression in rats. PLoS ONE 2012, 7, e45575. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Seubert, S.; Huhn, K.; Brecht, L.; Rötger, C.; Waschbisch, A.; Schlachetzki, J.; Klausmeyer, A.; Melms, A.; Wiese, S.; et al. Fingolimod effects in neuroinflammation: Regulation of astroglial glutamate transporters? PLoS ONE 2017, 12, e0171552. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, B.; Shan, S.; Zhao, X. Neuroprotective effects of vitexin against isoflurane-induced neurotoxicity by targeting the TRPV1 and NR2B signaling pathways. Mol. Med. Rep. 2016, 14, 5607–5613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.W.; Su, H.C.; Nyam, T.E.; Chio, C.C.; Kuo, J.R.; Wang, C.C. Ceftriaxone therapy attenuates brain trauma in rats by affecting glutamate transporters and neuroinflammation and not by its antibacterial effects. BMC Neurosci. 2021, 22, 54. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Chu, J.-M.-T.; Wong, G.-T.-C. Cerebral Glutamate Regulation and Receptor Changes in Perioperative Neuroinflammation and Cognitive Dysfunction. Biomolecules 2022, 12, 597. https://doi.org/10.3390/biom12040597
Zhang Y, Chu J-M-T, Wong G-T-C. Cerebral Glutamate Regulation and Receptor Changes in Perioperative Neuroinflammation and Cognitive Dysfunction. Biomolecules. 2022; 12(4):597. https://doi.org/10.3390/biom12040597
Chicago/Turabian StyleZhang, Yan, John-Man-Tak Chu, and Gordon-Tin-Chun Wong. 2022. "Cerebral Glutamate Regulation and Receptor Changes in Perioperative Neuroinflammation and Cognitive Dysfunction" Biomolecules 12, no. 4: 597. https://doi.org/10.3390/biom12040597
APA StyleZhang, Y., Chu, J.-M.-T., & Wong, G.-T.-C. (2022). Cerebral Glutamate Regulation and Receptor Changes in Perioperative Neuroinflammation and Cognitive Dysfunction. Biomolecules, 12(4), 597. https://doi.org/10.3390/biom12040597