Abstract
The diffusion of next-generation sequencing (NGS)-based approaches allows for the identification of pathogenic mutations of cardiomyopathies and channelopathies in more than 200 different genes. Since genes considered uncommon for a clinical phenotype are also now included in molecular testing, the detection rate of disease-causing variants has increased. Here, we report the prevalence of genetic variants detected by using a NGS custom panel in a cohort of 133 patients with inherited cardiomyopathies (n = 77) or channelopathies (n = 56). We identified 82 variants, of which 50 (61%) were identified in genes without a strong or definitive evidence of disease association according to the NIH-funded Clinical Genome Resource (ClinGen; “uncommon genes”). Among these, 35 (70%) were variants of unknown significance (VUSs), 13 (26%) were pathogenic (P) or likely pathogenic (LP) mutations, and 2 (4%) benign (B) or likely benign (LB) variants according to American College of Medical Genetics (ACMG) classifications. These data reinforce the need for the screening of uncommon genes in order to increase the diagnostic sensitivity of the genetic testing of inherited cardiomyopathies and channelopathies by allowing for the identification of mutations in genes that are not usually explored due to a currently poor association with the clinical phenotype.
1. Introduction
Inheritable cardiomyopathies and channelopathies are disorders with phenotypic and genetic heterogeneous features caused by the presence of structural or electrical heart abnormalities [1]. Variable penetrance and incomplete expression are also common and may be due to the interaction of the causal mutation with modifier genes, epigenetic changes, environmental factors, or individual factors such as age, gender, ethnicity, or physical activity [2]. According to their functional and morphological features, cardiomyopathies are commonly classified as hypertrophic cardiomyopathy (HCM), arrhythmogenic cardiomyopathy (ACM), dilated cardiomyopathy (DCM), or restrictive cardiomyopathy (RCM) [3]. Channelopathies are arrhythmic disorders in patients without structural heart abnormalities that are usually due to genetic alterations in genes coding for cardiac ion channels or their associated proteins. The main channelopathies are: Brugada syndrome (BrS), long QT syndrome (LQTS), short QT syndrome (SQTS), and catecholaminergic polymorphic ventricular tachycardia (CPVT) [4].
Both cardiomyopathies and channelopathies can cause sudden cardiac death (SCD), and this tragic event may represent the onset of an inherited heart disease in asymptomatic individuals, including subjects who practice intense physical activity, such as elite athletes [5,6,7,8,9].
Over the last two decades, the knowledge of the molecular bases of cardiomyopathies and channelopathies has gradually increased, and putatively associated variants have been identified in more than 200 genes [10,11,12,13,14,15,16,17,18,19,20,21,22]. Indeed, while some genes are strongly related to a clinical phenotype and are highly penetrant causative genes, others have only rarely been identified in affected subjects, and the association with the disease seems to be poor. Currently, mutations in about 100 different genes are associated to HCM [5,23]. Nevertheless, eight main genes, MYBPC3, MYH7, TNNT2, TNNI3, TPM1, ACTC1, MYL2, and MYL3, account for up to 65–70% of all HCM cases [24,25,26], while other uncommon genes seem to be globally involved in about 10% of HCM cases [12,27,28,29,30]. Similarly, 51 curated genes were associated with idiopathic DCM [31], explaining up to 40–50% of DCM cases [24]. Among these, mutations in TTN gene account for up to 20–25% of DCM cases, while mutations occurring in other genes have rarely been identified [32,33,34,35,36,37,38,39,40,41,42]. With regard to ACM, pathogenic variants in each of the main cardiac desmosome genes were identified in more than 50% of the affected subjects [24,43,44], a smaller fraction carrying pathogenic mutations in nondesmosomal genes [43,45,46]. Lastly, a combination of sarcomeric and cytoskeletal genes were detected in half of RCM patients [47].
Similar considerations also apply to channelopathies. About 20–25% of BrS patients carry a mutation in the SCN5A gene, while other disease-related genes explain an additional 5% of all BrS cases [5,48,49,50]. With regard to LQTS, mutations in the KCNQ1, KCNH2, and SCN5A genes are found in about 75% of patients, while 5–10% of cases are due to genetic mutations in uncommon genes. The main gene associated with SQTS is KCNH2, reported in about 15% of SQTS patients; other uncommon genes were also reported [51]. Lastly, genes mainly associated with CPVT are RYR2 (60%) and CASQ2 (5%); other genes are rarely reported (≤1% each) [52]. Supplementary Table S1 shows the genes reported in the literature associated with the above mentioned cardiomyopathies and channelopathies [26].
Most previously reported genes are commonly included in diagnostic tests. Moreover, in recent years, advances in high-throughput sequencing strategies, such as next-generation sequencing (NGS), have revolutionized the diagnosis of inherited heart diseases in terms of expanding the number of involved genes and the discovery of new genes potentially associated with the diseases [5,53,54,55,56,57].
This wide genetic scenario highlights the need for a standardized method to estimate the genetic evidence and the clinical validity of gene–disease relationships. A proposed framework to evaluate relevant genetic and experimental evidence supporting or contradicting a gene–disease relationship is represented by the NIH-funded Clinical Genome Resource (ClinGen) (https://search.clinicalgenome.org/, accessed on 26 September 2022) [58,59], a standardized evidence-based framework, from expert panel curation. ClinGen is an authoritative open-access online resource that defines genes and variants on the basis of the level of evidence of disease association, which can be strong, definitive, or moderate. ClinGen also reports genes that have a gene–disease association in the literature data, but present limited or conflicting evidence to support a causal role in the disease since the time of the initial report. These genes are classified as limited, disputed, or refuted.
In this manuscript, we report the prevalence and type of genetic variants identified in patients with cardiomyopathies and channelopathies, focusing on genes with weaker evidence (limited, disputed, refuted) of disease association and genes not reported for the specific phenotype according to the ClinGen definition. Here, we define “uncommon genes” as all limited, disputed, or refuted genes, or those without a reported association with the clinical phenotype of the patients.
2. Materials and Methods
2.1. Patient Enrolment and DNA Extraction
In total, 133 unrelated patients with a clinical diagnosis or suspicion of an inherited cardiac disease (cardiomyopathy or channelopathy) were enrolled from June 2018 to date and followed up on an average period of 3 years at the Department of Cardiomyopathy and Inherited Heart Disease Clinic, UOC Cardiology, University of Campania ‘Luigi Vanvitelli’ of Naples, and at the Center for the Study of Hypertrophic Cardiomyopathies of the Cardiology Division of the Federico II University of Naples. Genetic testing was performed at the CEINGE diagnostic laboratory.
The reference clinicians of the patients required the molecular analysis to investigate the presence of a possible genetic disease. A blood sample was collected in EDTA from each study subject. Each patient gave their written informed consent to molecular analysis according to the tenets of the Helsinki Declaration [60] and the internal ethics committee (N. 77/21). Genomic DNA was extracted using a Maxwell 16 instrument (Promega, Madison, WI, USA). DNA quality was assessed with a Nanodrop (Thermo Fisher, Waltham, MA, USA) spectrophotometer and Tape Station (Agilent, Santa Clara, CA, USA) analyzers to verify their concentration, purity, and integrity before proceeding to library preparation.
2.2. Molecular Analysis
Molecular testing was carried out by analyzing a panel of target genes through an NGS-based procedure. In particular, 2 custom panels were used: (1) a first-level panel including 60 diagnostic genes associated to the different genetic cardiomyopathies, including HCM, DCM, ACM, and channelopathies, such as LQTS and BrS; and (2) an enlarged panel of 129 genes including rarer genes; this panel was used in cases of complex or unclear phenotypes and in the presence of a positive family history for SCD. In both cases, HaloPlex technology (Agilent, Santa Clara, CA, USA) was used for library preparation. In detail, each genomic DNA sample was fragmented using a pool of restriction enzymes. The obtained fragments were enriched with hybridization with the custom capture probes, and then purified and PCR-amplified to obtain a DNA library or sample. During this procedure, each genomic DNA sample was univocally tagged with a barcode sequence to allow for sample multiplexing during the subsequent sequencing step.
The obtained enriched and indexed libraries were sequenced using the MiSeq (Illumina, San Diego, CA, USA) instrument (2 × 250 PE). Alissa software (Agilent, Santa Clara, CA, USA) was used to perform sequence data analysis. This tool allows for the alignment of the sequences to the reference genome to obtain a list of genomic variants that can be prioritized using a customizable pipeline in order to highlight pathogenic mutations or potentially pathogenic variants. This pipeline allows for variant classification based on their position with regard to the gene structure (exonic or intronic), population frequency, coding effect, ClinVar classification, and functional predictions. In addition, we classified the variants according to American College of Medical Genetics and Genomics (ACMG) guidelines adapted to cardiomyopathy [61]. In detail, all variants were evaluated for their frequency in international population database gnomAD (http://gnomad.broadinstitute.org/, accessed on 26 September 2022): we excluded variants showing minor frequency alleles (MAF) below the calculated maximal tolerated allele frequency for the specific cardiomyopathy [62]. According to these criteria, the maximal credible allele frequency was 0.000084 for DCM, 0.00004 for HCM, 0.000092 for ACM, 0.00001 for BrS, and 0.0000082 for LQTS [62]. However, we kept 10 variants showing an MAF greater than the threshold because they are reported as pathogenetic in the HGMD database. The latter classifies variants on the basis of the interpretation at first reporting; thus, even if it is less valuable with respect to ACMG classification and more prone to subsequent modifications, it may provide additional data supporting the variants’ correct classification and subsequent interpretation. Moreover, bioinformatic pathogenicity evaluation for missense variants was performed by using multiple in silico tools, including Grantham distance and an Align–GVGD matrix. The splicing module from Alamut visual v.2.7.0.0 software was used to test the pathogenicity of a possible splicing variant. In some cases, we had informative families, i.e., ones having at least 3 affected relatives in which we analyzed variant cosegregation. In families with one affected member being a noncarrier, the variant is considered to be non-disease-causing. Pathogenic or doubtful variants were confirmed with Sanger sequencing.
3. Results
In total, 133 independent subjects underwent genetic testing because of a clinical suspicion of inherited cardiomyopathy or channelopathy over a period of enrollment of 3 years (2018–2020). The average sequencing coverage of the target regions was in the range of 150–250X with a 50X minimal acceptable threshold. In detail, 60/133 patients showing a phenotype, and/or laboratory or instrumental data supporting a clinical diagnosis of a specific inherited cardiomyopathy according to international guidelines were analyzed using a first-level panel including 60 main diagnostic genes that had been associated with different cardiomyopathies/channelopathies. The remaining 73/133 subjects, showing unclear cardiological clinical signs and/or a positive family history for SCD, were analyzed using an enlarged 129 gene panel. In our cohort of 133 patients, we detected a total of 82 variants, excluding polymorphisms. The variants, shown in Table 1, were reported following Human Genome Variation Society (HGVS) nomenclature (http://www.HGVS.org/varnomen, accessed on 26 September 2022), and annotated according to the Human Gene Mutation Database (HGMD) Professional 2020.4, (http://www.hgmd.cf.ac.uk/, accessed on 26 September 2022), NCBI SNP Database (http://www.ncbi.nlm.nih.gov, accessed on 26 September 2022), and ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar, accessed on 26 September 2022).

Table 1.
List of genetic variants (n = 82) identified in the 133 patients of the study cohort.
Among the 82 detected variants, 27 were novel as they were missing in the reference SNP, ClinVar, and HGMD databases. The ClinVar database reported 39/82 variants. Of these variants, 20 were classified as pathogenic or likely pathogenic (P/LP), 13 as conflicting (CI), and 6 of uncertain or unknown significance (VUS). The remaining 43 variants were not reported (NR) in ClinVar. HGMD reported 38/82 variants as pathogenic or likely pathogenic, while the remaining 44 were not reported. According to ACMG interpretation criteria, 39/82 variants were classified as P/LP (48%), of which 6 were novel (without reference SNP ID number), and 41/82 as VUSs (50%), of which 26 were novel ones. Furthermore, two of the identified variants (2%) were classified by ACMG guideline as benign or likely benign (B/LB). According to ACMG criteria, we globally identified P/LP variants in 53 patients (40% of the analyzed population), VUSs in 38 patients (29%), and B/LB variants in 4 patients (3%). In addition, 11 patients carried multiple variants. We did not identify any pathogenic mutations or VUSs in the remaining 38 patients (29%), who showed only known polymorphisms and/or LB variants in the analyzed genes. If we separately analyzed the two cohorts of patients, among the 60 patients admitted to the first-level test, 24 carried a P/LP variant (41%, 7 channelopathy and 17 cardiomyopathy patients) and 24 carried a VUS (41%, 7 channelopathy and 17 cardiomyopathy patients). Similarly, among the 73 patients analyzed using the enlarged panel, 29 carried a P/LP variant (40%, 4 channelopathy and 25 cardiomyopathy patients) and 14 carried a VUS (19%, 6 channelopathy and 8 cardiomyopathy patients).
Figure 1 shows the distribution of P/LP and VUS variants among channelopathy (Figure 1A,C) and cardiomyopathy (Figure 1B,D) patients analyzed via the two genes panels. The percentage of patients with an unknown causal gene included subjects without pathogenic mutations or VUSs, carrying known polymorphisms and/or LB variants in the analyzed genes.

Figure 1.
Percentage of channelopathy and cardiomyopathy patients carrying pathogenic/likely pathogenic (P/LP) and unknown significant (VUS) variants, as identified using (A,B) first-level and (C,D) extended panels. Variants were classified according to ACMG guideline classification with respect to patients without a known causative gene.
In total, 11 patients (8.3%) carried multiple variants: in particular, 3 patients carried two different P/LP mutations, 3 patients carried 1 P/LP mutation and a VUS, and 5 patients carried more than one VUS. We analyzed 5/11 patients with the first-level test, and 6/11 with the enlarged one. Furthermore, 7/11 patients showed double mutations in uncommon genes with respect to their phenotype, 2 patients were doubly mutated in main genes, and 2 patients showed one mutation in an uncommon and the other in a main gene.
Globally, among the 82 variants, 50 (61%) were localized in genes without reported evidence, or classified with limited or disputed evidence for a role in cardiomyopathies or channelopathies in ClinGen (uncommon genes), while 32 (39%) were had a definitive or moderate association with the disease.
Among the 50 identified variants in uncommon genes, 48 were P/LP or VUS (13 as P/LP and 35 as VUSs) and 2 as B/LB according to ACMG classification. In addition, 11/13 P/LP and 14/35 VUSs were identified among patients who had undergone extended molecular analysis.
In particular, 9 ACM patients carried variants in the CACNB2 (n = 2), HCN4 (n = 1), KCNE3 (n = 1), LDB3 (n = 1), MYH6 (n = 1), PLN (n = 1), POLG (n = 1), and RYR2 (n = 1) genes. One ACM patient was a compound heterozygous (confirmed with segregation analysis) carrier of two missense variants (c.752C>T and c.1760C>T) in the POLG gene. Furthermore, c.40_42delAGA (p.Arg14del) in the PLN gene was present in both a patient with ACM and an independent DCM patient. ClinGen reports a definitive association between the PLN gene and the DCM but not ACM phenotype [63].
In addition, 17 independent HCM patients carried pathogenic variants or VUSs in the following uncommon genes: ABCC9, ANK2, CAV3, CTNNA3, DSP, KCNQ1, FHL1, OBSCN, PKP2, RYR2, TGFB3 (reported in one patient each); RAF1 (n = 2), SCN10A (n = 2), and TTN (n = 2).
In total, 14 independent BrS patients carried P/LP or VUSs in the following uncommon genes: ANK2, CACNA1D, CACNA2D1, CASQ2 (n = 2), CAV3, DSC2, KCNE3, KRAS, MYBPC3, POLG, PRDM16, SGCD, and TGFB3.
In 3 DCM patients, we identified 3 VUSs in uncommon genes KCNQ1, TBX1, and TRPM4. Lastly, in LQTS patients, we identified 3 VUSs in uncommon genes KRT17, LAMA3, and POLG.
The detailed distribution of all identified variants divided by clinical phenotype is reported in Table 1.
Overall, in this study group, we detected 39 pathogenic or likely pathogenic mutations, of which 26 were identified in main genes (67%) and 13 (33%) in uncommon genes. Furthermore, we identified 41 VUSs, of which 35 (85%) in uncommon genes and 6 (15%) in main genes. Our findings indicate that P/LP variants are more likely to be identified in main genes, while VUSs are mostly identified in uncommon genes. When considering the two analyzed cohorts separately, we found that the number of variants (both P/LP and VUSs) in the main genes was almost the same within the two different panels; the number of P/LP variants identified in uncommon genes increased when the enlarged panel was used (Figure 2).
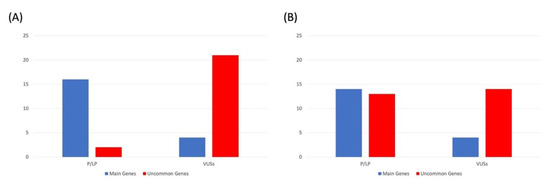
Figure 2.
Number of pathogenic/likely pathogenic (P/LP) variants and VUSs according to ACMG guidelines, identified in the patients analyzed using (A) first-level and (B) enlarged panels and divided by main and uncommon genes.
As shown in Figure 3A, P/LP mutations in both main and uncommon genes (accordingly with association with the reported cardiomyopathy in Clinical Genome Resource) were mainly detected in HCM patients (57% of the P/LP variants), followed by ACM (20%), LQTS, DCM, and BrS patients (12%, 6% and 6%, respectively). Among ACM, HCM, BrS, DCM, and LQTS patients carrying P or LP mutations, 4/10 (40%), 7/29 (24%), 2/3 (67%), 2/3 (67%), 1/6 (17%) were detected in uncommon genes, respectively (Figure 3B). Only 3 (19%) of these variants were identified in patients who had undergone the first-level test, while the remaining 13 (81%) were identified with the enlarged-gene panel.

Figure 3.
(A) Percentage and number of pathogenic or likely pathogenic variants collectively identified in the study cohort and (B) divided by main and uncommon genes according to clinical phenotype.
Furthermore, among the 41 identified VUSs, 6 (15%) were detected in ACM, 6 (15%) in DCM, 13 (32%) in HCM, 13 (32%) in BrS, and 3 (7%) in LQTS patients (Figure 4A). Interestingly, 35/41 VUSs (85%) fell into the uncommon-gene group, with 22 (63%) identified in patients who had undergone the first-level test and 13 (37%) in those who performed the enlarged one. When we analyzed the distribution of the VUSs in uncommon and main genes per each disease, we found a higher percentage of VUSs in common genes in all the diseases with the exception of DCM (Figure 4B).
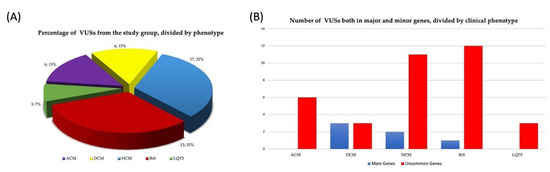
Figure 4.
(A) Percentage and number of VUSs collectively identified in the study cohort and (B) divided by main and uncommon genes according to clinical phenotype.
In the enrolled patients among 133 analyzed subjects, we detected 53 patients 40%) carrying at least one P or LP variants. In particular, 15 out of 53 patients (28%) carried P or LP variants in uncommon genes and 38 (72%) in main genes (Figure 5). Table 2 shows the demographic, clinical, and instrumental data of eight patients carrying pathogenic or likely pathogenic variants detected in uncommon genes with respect to the clinical diagnosis or suspicion for the inherited cardiac disease (cardiomyopathy or channelopathy). All except 1 patient (NA04) were analyzed with the enlarged panel of 129 genes, including rarer genes.
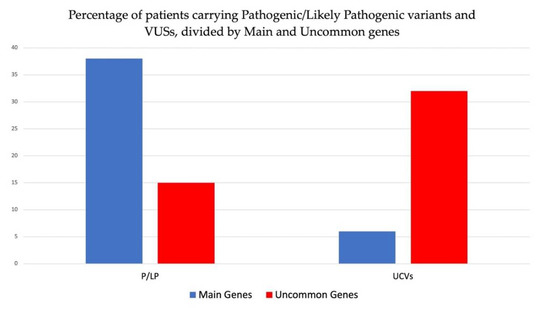
Figure 5.
Percentage of patients carrying pathogenic or likely pathogenic variants and VUSs collectively identified in study cohort and divided by main and uncommon genes.
Table 2.
Demographic, clinical, and instrumental data of patients carrying pathogenic or likely pathogenic variants in uncommon genes with respect to the clinical diagnosis or suspicion of an inherited cardiac disease (cardiomyopathy or channelopathy).
These eight patients were recruited according to the physician’s clinical suspicion for an inherited cardiomyopathy or channelopathy. In detail, four HCM patients carried mutations in uncommon genes ABCC9, KCNQ1, PKP2 and RAF1, and two BrS patients in uncommon genes MYBPC3 and POLG. Furthermore, we identified one ACM patient carrying a mutation in uncommon gene KCNE3 and one ACM patient in trans carrying two variants in uncommon gene POLG (c.752C>T and c.1760C>T), classified as LP/P according to the novel ACMG criteria.
Furthermore, VUSs were identified in 38 out of 133 patients (29%), of which 32 (84%) showed VUSs in uncommon genes and 6 (16%) in main genes (Figure 5).
4. Discussion
In this study, we report the results of a targeted-NGS-based genetic screening carried out on 133 unrelated patients from South Italy diagnosed with cardiomyopathy or channelopathy. We used a large panel of 129 cardiomyopathy-related genes to increase the yield of genetic testing considering the genetic heterogeneity and variable and overlapping phenotypes, which is a characteristic of hereditary structural and electric cardiac diseases. Using the clinical genome resource approach to assess the strength of gene–disease association, we focused on variants in genes without definitive or strong evidence for an association with cardiomyopathies or channelopathies, which we defined as uncommon causal genes.
Globally, our NGS panel test demonstrated a yield of 40% if we considered the genetic test to be positive in the presence of pathogenic or likely pathogenic variants in both main and uncommon genes. When we included the VUSs, the global yield of the test increased up to 69%.
We detected 39 pathogenic or likely pathogenic variants according to the ACMG classification in 53 patients (44 cardiomyopathies and 9 channelopathies), including 13 patients carrying P or LP variants in uncommon genes. The role of these uncommon genes as causal genes should be evaluated in the context of the clinical manifestations and the evidence of the functional effects of the variant on the encoded protein; moreover, when large pedigrees are available, the analysis of the segregation of the genetic variant with the phenotype allows for confirming the phenotype–genotype association. Furthermore, we verified whether similar gene–disease associations to those identified in our study were reported in the literature. For example, in our study cohort, we detected a mutation in sarcomeric gene MYBPC3 (c.906-1G>C; rs587776700) in a BrS patient. Experimental studies showed that this variant disrupts mRNA splicing, leading to a loss of protein function [64]. Some studies reported a MYBPC3 mutation in patients showing the Brugada phenotype [65,66]. In addition, the literature reported cases of Brugada phenocopies in patients carrying a pathogenic mutation in the MYBPC3 gene [67], speculating that Brugada type 1 ECG could be an early sign of an occult structural heart disease. Brugada syndrome is generally considered to be a disease of the ion channels. However, recent studies have also identified structural heart impairment, i.e., epicardial surface and interstitial fibrosis, increased collagen, and reduced contractility in BrS patients. These observations agree with the finding of causal mutations in genes that are molecular causes of cardiomyopathies, such as sarcomeric genes [66]. These data suggest that structural and electrical cardiac alterations may be present in BrS patients, which may develop from the same molecular alteration.
Similarly, although HCM is commonly considered to be a disease of the sarcomere, recent literature data reported an association of the HCM phenotype with mutations in the ABCC9 [48,68], PKP2 [69,70,71], and RAF1 [72] genes, and in other nonsarcomeric genes [73]. The pathogenic mechanisms leading to the development of the HCM phenotype need to be elucidated.
Furthermore, in our study, we identified (L)P variants in the POLG gene in two patients with BrS and ACM, respectively. Our data are in accordance with recently published data in which the POLG gene was associated with the ACM phenotype [74].
Overall, in our cohort, the prevalence of (L)P variants in uncommon genes was about 8% (3% in the first-level and 15% in the enlarged panels). This result supports the need for extended genetic testing including uncommon genes for such heterogeneous diseases as cardiomyopathies and channelopathies. Indeed, by limiting the genetic test to just phenotype-related (main) genes, we would have failed in identifying 13 mutations and the corresponding carrying patients (8% of the screened population). On the other hand, our approach was able to increase the diagnostic sensitivity of the test identifying these additional patients.
Biotechnological research is always looking for new diagnostic strategies [75]. As NGS-based applications are currently widely used in diagnostic laboratories, and their costs are decreasing progressively, their use is desirable to enlarge the spectrum of the genes related to a specific disease. Indeed, accumulating data from different studies may allow for reconsidering the genotype–phenotype associations for some genes now considered to be uncommon.
In addition to pathogenic and likely pathogenic mutations, as expected in this kind of extended molecular screening, we identified many VUSs (41/82; 50% of the total number of identified variants) according to ACMG classification. Of the VUSs, 85% (35/41) were detected in uncommon genes. We detected variant c.764G>C in an HCM patient in the FHL1 gene, which is classifiable as a VUS according to ACMG criteria. However, it is reported as pathogenic by HGMD database, showing strong evidence for a primary pathogenic role in HCM [73,76]. The FHL1 gene was classified without evidence for a HCM association by ClinGen. The ACMG rule for classification is hypothesized to minimize the risk for false positive interpretations; however, it is also well-accepted that these rigorous criteria result in undercalling pathogenic variants in well-established cardiomyopathy genes.
Certainly, VUS interpretation and the consequent clinical management of their carriers is a key challenge following NGS-based molecular testing [77]. The possibility to study a specific VUS in the context of a large pedigree by allowing for the robust analysis of the variant’s segregation with the clinical phenotype may be decisive to establish its pathogenic role; however, this is often unfeasible [78]. To definitively assess the molecular consequence of an identified VUS and thus establish its functional effects, the most proper approach should include in vitro functional assays [79,80,81,82]. Nevertheless, these functional evaluations are often difficult to be realized, especially in a routine setting and on a large scale, since different cellular models and different assays need to be used on the basis of the gene involved and on the type of molecular defect. The more that we enlarge the analyzed genomic region, the more the number of VUSs per patient increases. Nevertheless, even if VUS communication may be challenging and frustrating for both patients and clinicians, it offers a great opportunity for the re-evaluation of cases over time. Indeed, monitoring these variants over time may allow for their reclassification, and hence provide novel clues for the management of carriers and their relatives. Moreover, some of the VUSs may act as modifier genes, influencing a patient’s clinical phenotype, and may contribute to also explaining the observed clinical heterogeneity within families.
5. Conclusions
Considering all the above, extensive genetic test approaches are required to unravel the molecular bases of such a complex group of diseases as cardiomyopathies and channelopathies. NGS-based approaches, by allowing for the simultaneous analysis of multiple genes, increase the detection rate of causal mutations in inherited structural and electric hereditary cardiac diseases, and provide epidemiological data regarding the prevalence of causative mutations in uncommon genes. In addition, several uncertain or unknown variants (VUSs) are being identified and require more efforts in the near future to assess their pathogenicity.
The use of an NGS-based approach in diagnostics also increases the yield of mutations carriers’ identification in the rarest and uncommon genes, as shown in our study. Indeed, our data are a parameter of the increased diagnostic yield of such extended molecular analyses that also enable screening for rare and uncommon genes in a simple, reliable, and cost-saving manner.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biom12101417/s1. Table S1: genes reported in the literature associated with cardiomyopathies and channelopathies.
Author Contributions
C.M., G.F. and V.D. conceived and designed the study; V.D., F.U. and M.V.E. performed experimental data; F.B., B.M., C.M., G.F. and V.D. analyzed the data; C.M., G.F., V.D. and R.L. wrote and revised the paper; G.L., M.A.L., R.L., M.C. and E.M. collected the clinical data. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Italian Ministry of University and Research, grant numbers PON03PE_00060_7 and PRIN-20173ZWACS (R.L. and G.L.).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by an internal institutional review board (N. 77/21).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data are reported within the text.
Acknowledgments
The authors thank Serena Conato for the technical support to the experimental work.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| NGS | Next-generation sequencing |
| VUS | Variants of unknown significance |
| SCD | Sudden cardiac death |
| HCM | Hypertrophic cardiomyopathy |
| ACM | Arrhythmogenic cardiomyopathy |
| DCM | Dilated cardiomyopathy |
| LQTS | Long QT syndrome |
| BrS | Brugada syndrome |
| SQTS | Short QT syndrome |
| CPVT | Catecholaminergic polymorphic ventricular tachycardia |
| MYBPC3 | Cardiac myosin binding protein 3 |
| MYH7 | β-myosin heavy chain |
| TNNT2 | Cardiac troponin T |
| TNNI3 | Cardiac troponin I |
| TPM1 | α-tropomyosin |
| ACTC1 | Cardiac α-actin |
| MYL2 | Myosin regulatory light chain |
| MYL3 | Myosin essential light chain |
| MYOZ2 | Myozenin 2 gene |
| ACTN2 | α-actinin-2 |
| TTN | Titin |
| MYOM1 | Myomesin 1 |
| ANKRD1 | Cardiac ankyrin repeat protein 1 |
| TCAP | Telethonin |
| PKP2 | Plakophilin |
| DSP | Desmoplakin |
| DSC2 | Desmocollin 2 |
| DSG2 | Desmoglein |
| TMEM43 | Transmembrane protein 43 |
| PLN | Phospholamban |
| CDH2 | Cadherin-2 |
| CTNNA3 | Catenin alpha 3 |
| FLNC | Filamin C |
| TJP1 | Tight junction protein 1 |
| ANK2 | Ankyrin 2 |
| TP63 | Tumor protein P63 |
| SCN10A | sodium voltage-gated channel alpha subunit 10 |
| CACNA1C | Calcium voltage-gated channel subunit alpha1 c |
| ABCC9 | ATP binding cassette subfamily c member 9 |
| SCN1B | Sodium voltage-gated channel beta subunit 1 |
| KCNH2 | Potassium voltage-gated channel subfamily h member 2 |
| CACNB2 | Calcium voltage-gated channel auxiliary subunit beta 2 |
| TRPM4 | Transient receptor potential cation channel subfamily m member 4 |
| ANK3 | Ankyrin 3 |
| CACNA2D1 | Calcium voltage-gated channel auxiliary subunit alpha2delta 1 |
| FGF12 | Fibroblast growth factor 12 |
| GPD1L | Glycerol-3-phosphate dehydrogenase 1 like |
| HCN4 | Hyperpolarization activated cyclic nucleotide gated potassium channel 4 |
| KCNQ1 | Potassium voltage-gated channel subfamily q member 1 |
| KCNH2 | Potassium voltage-gated channel subfamily h member 2 |
| SCN5A | Sodium voltage-gated channel alpha subunit 5 |
| AKAP9 | a-Kinase anchoring protein 9 |
| RYR2 | Ryanodine receptor 2 |
| CASQ2 | Calsequestrin 2 |
| CALM1 | Calmodulin 1 |
| CALM2 | Calmodulin 2 |
| CALM3 | Calmodulin 3 |
| KCNJ2 | Potassium inwardly rectifying channel subfamily j member 2 |
| TRDN | Triadin |
| ACMG | American College of Medical Genetics and Genomics |
| RAF1 | RAF-1 proto-oncogene, serine/threonine kinase |
| LDLR | Low-density lipoprotein receptor |
| GAA | Glucosidase alpha, acid |
| LP | Likely pathogenic |
| FHL1 | Four and a half LIM domains 1 |
| P | Pathogenic |
| LP | Likely pathogenic |
| B | Benign |
| LB | Likely benign |
| ESC | European Society of Cardiology |
| AHA | American Heart Association |
| ACC | American College of Cardiology |
References
- Schaufelberger, M. Cardiomyopathy and pregnancy. Heart 2019, 105, 1543–1551. [Google Scholar] [CrossRef]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.A.; Cook, S.A.; Seidman, J.G.; Seidman, C.E. Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2871–2886. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies. Heart Rhythm 2011, 8, 1308–1339. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Mirra, B.; Barretta, F.; Lombardo, B.; Scudiero, O.; Frisso, G. Sudden cardiac death in young athletes: Literature review of molecular basis. Cardiogenetics 2020, 10, 8860. [Google Scholar] [CrossRef]
- Limongelli, G.; Nunziato, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Mazzaccara, C.; Caiazza, M.; D’Aponte, A.; D’Andrea, A.; Bossone, E.; et al. Yield and clinical significance of genetic screening in elite and amateur athletes. Eur. J. Prev. Cardiol. 2021, 28, 1081–1090. [Google Scholar] [CrossRef]
- D’Argenio, V.; Esposito, M.V.; Nunziato, M.; De Simone, A.; Buono, P.; Salvatore, F.; Frisso, G. Molecular diagnosis of Brugada syndrome via next-generation sequencing of a multigene panel in a young athlete. Med. Sport 2018, 71, 27–34. [Google Scholar]
- Lombardo, B.; Izzo, V.; Terracciano, D.; Ranieri, A.; Mazzaccara, C.; Fimiani, F.; Cesaro, A.; Gentile, L.; Leggiero, E.; Pero, R.; et al. Laboratory medicine: Health evaluation in elite athletes. Clin. Chem. Labo. Med. 2019, 57, 1450–1473. [Google Scholar] [CrossRef] [PubMed]
- Monda, E.; Sarubbi, B.; Russo, M.G.; Caiazza, M.; Mazzaccara, C.; Magrelli, J.; Rubino, M.; Esposito, A.; Perna, A.; Passariello, A.; et al. Unexplained sudden cardiac arrest in children: Clinical and genetic characteristics of survivors. Eur. J. Prev. Cardiol. 2021, 28, 1134–1137. [Google Scholar] [CrossRef]
- Jacoby, D.; McKenna, W.J. Genetics of inherited cardiomyopathy. Eur. Heart J. 2012, 33, 296–304. [Google Scholar] [CrossRef]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef]
- Norton, N.; Li, D.; Rampersaud, E.; Morales, A.; Martin, E.R.; Zuchner, S.; Guo, S.; Gonzalez, M.; Hedges, D.J.; Robertson, P.D.; et al. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 144–153. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Siegfried, J.D. Update 2011: Clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2011, 57, 1641–1649. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Amin, A. Channelopathies, genetic testing and risk stratification. Int. J. Cardiol. 2017, 237, 53–55. [Google Scholar] [CrossRef]
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C.; Ruppert, V.; Arbustini, E.; Wichter, T.; Germain, M.; et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 2011, 32, 1065–1076. [Google Scholar] [CrossRef]
- Detta, N.; Frisso, G.; Limongelli, G.; Marzullo, M.; Calabro, R.; Salvatore, F. Genetic analysis in a family affected by sick sinus syndrome may reduce the sudden death risk in a young aspiring competitive athlete. Int. J. Cardiol. 2014, 170, E63–E65. [Google Scholar] [CrossRef]
- D’Argenio, V.; Frisso, G.; Precone, V.; Boccia, A.; Fienga, A.; Pacileo, G.; Limongelli, G.; Paolella, G.; Calabro, R.; Salvatore, F. DNA Sequence Capture and Next-Generation Sequencing for the Molecular Diagnosis of Genetic Cardiomyopathies. J. Mol. Diagn. 2014, 16, 32–44. [Google Scholar] [CrossRef]
- Frisso, G.; Limongelli, G.; Pacileo, G.; Del Giudice, A.; Forgione, L.; Calabro, P.; Iacomino, M.; Detta, N.; Di Fonzo, L.M.; Maddaloni, V.; et al. A child cohort study from southern Italy enlarges the genetic spectrum of hypertrophic cardiomyopathy. Clin. Genet. 2009, 76, 91–101. [Google Scholar] [CrossRef]
- Jarcho, J.A.; McKenna, W.; Pare, J.A.; Solomon, S.D.; Holcombe, R.F.; Dickie, S.; Levi, T.; Donis-Keller, H.; Seidman, J.G.; Seidman, C.E. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N. Engl. J. Med. 1989, 321, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Meder, B.; Ruhle, F.; Weis, T.; Homuth, G.; Keller, A.; Franke, J.; Peil, B.; Bermejo, J.L.; Frese, K.; Huge, A.; et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur. Heart J. 2014, 35, 1069–1077. [Google Scholar] [CrossRef]
- Mazzanti, A.; Underwood, K.; Nevelev, D.; Kofman, S.; Priori, S.G. The new kids on the block of arrhythmogenic disorders: Short QT syndrome and early repolarization. J. Cardiovasc. Electr. 2017, 28, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar]
- Lee, H.H.; Ching, C.K. Practical Aspects in Genetic Testing for Cardiomyopathies and Channelopathies. Clin. Biochem. Rev. 2019, 40, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Biagini, E.; Olivotto, I.; Iascone, M.; Parodi, M.I.; Girolami, F.; Frisso, G.; Autore, C.; Limongelli, G.; Cecconi, M.; Maron, B.J.; et al. Significance of sarcomere gene mutations analysis in the end-stage phase of hypertrophic cardiomyopathy. Am. J. Cardiol. 2014, 114, 769–776. [Google Scholar] [CrossRef]
- Mazzaccara, C.; D’Argenio, V.; Nunziato, M.; Esposito, M.V.; Salvatore, F.; Frisso, G. Clinical molecular biology in the assessment and prevention of cardiological risk in case of participation in sports activity and intense physical activity. Biochim. Clin. 2019, 43, 24–43. [Google Scholar]
- Girolami, F.; Frisso, G.; Benelli, M.; Crotti, L.; Iascone, M.; Mango, R.; Mazzaccara, C.; Pilichou, K.; Arbustini, E.; Tomberli, B.; et al. Contemporary genetic testing in inherited cardiac disease: Tools, ethical issues, and clinical applications. J. Cardiovasc. Med. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Lopes, L.R.; Garcia-Hernandez, S.; Lorenzini, M.; Futema, M.; Chumakova, O.; Zateyshchikov, D.; Isidoro-Garcia, M.; Villacorta, E.; Escobar-Lopez, L.; Garcia-Pavia, P.; et al. Alpha-protein kinase 3 (ALPK3) truncating variants are a cause of autosomal dominant hypertrophic cardiomyopathy. Eur. Heart J. 2021, 42, 3063–3073. [Google Scholar] [CrossRef]
- Chiu, C.; Bagnall, R.D.; Ingles, J.; Yeates, L.; Kennerson, M.; Donald, J.A.; Jormakka, M.; Lind, J.M.; Semsarian, C. Mutations in Alpha-Actinin-2 Cause Hypertrophic Cardiomyopathy A Genome-Wide Analysis. J. Am. Coll. Cardiol. 2010, 55, 1127–1135. [Google Scholar] [CrossRef]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Tabish, A.M.; Azzimato, V.; Alexiadis, A.; Buyandelger, B.; Knoll, R. Genetic epidemiology of titin-truncating variants in the etiology of dilated cardiomyopathy. Biophys. Rev. 2017, 9, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Lakdawala, N.K.; Funke, B.H.; Baxter, S.; Cirino, A.L.; Roberts, A.E.; Judge, D.P.; Johnson, N.; Mendelsohn, N.J.; Morel, C.; Care, M.; et al. Genetic testing for dilated cardiomyopathy in clinical practice. J. Card. Fail. 2012, 18, 296–303. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Japp, A.G.; Gulati, A.; Cook, S.A.; Cowie, M.R.; Prasad, S.K. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Favalli, V.; Serio, A.; Grasso, M.; Arbustini, E. Genetic causes of dilated cardiomyopathy. Heart 2016, 102, 2004–2014. [Google Scholar] [CrossRef]
- Zhao, Y.; Feng, Y.; Zhang, Y.M.; Ding, X.X.; Song, Y.Z.; Zhang, A.M.; Liu, L.; Zhang, H.; Ding, J.H.; Xia, X.S. Targeted Next-Generation Sequencing Reveals Hot Spots and Doubly Heterozygous Mutations in Chinese Patients with Familial Cardiomyopathy. Biomed. Res. Int. 2015, 2015, 561819. [Google Scholar] [CrossRef]
- Spezzacatene, A.; Sinagra, G.; Merlo, M.; Barbati, G.; Graw, S.L.; Brun, F.; Slavov, D.; Di Lenarda, A.; Salcedo, E.E.; Towbin, J.A.; et al. Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias. J. Am. Heart Assoc. 2015, 4, e002149. [Google Scholar] [CrossRef] [PubMed]
- Zegkos, T.; Panagiotidis, T.; Parcharidou, D.; Efthimiadis, G. Emerging concepts in arrhythmogenic dilated cardiomyopathy. Heart Fail. Rev. 2021, 26, 1219–1229. [Google Scholar] [CrossRef]
- Peters, S.; Kumar, S.; Elliott, P.; Kalman, J.M.; Fatkin, D. Arrhythmic Genotypes in Familial Dilated Cardiomyopathy: Implications for Genetic Testing and Clinical Management. Heart Lung Circ. 2019, 28, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 Mutations Induce an Arrhythmogenic Dilated Cardiomyopathy Related to Disturbed Calcium Handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Limongelli, G.; Petretta, M.; Vastarella, R.; Pacileo, G.; Bonaduce, D.; Salvatore, F.; Frisso, G. A common polymorphism in the SCN5A gene is associated with dilated cardiomyopathy. J. Cardiovasc. Med. 2018, 19, 344–350. [Google Scholar]
- James, C.A.; Syrris, P.; van Tintelen, J.P.; Calkins, H. The role of genetics in cardiovascular disease: Arrhythmogenic cardiomyopathy. Eur. Heart J. 2020, 41, 1393–1400. [Google Scholar] [CrossRef]
- Stevens, T.L.; Wallace, M.J.; Refaey, M.E.; Roberts, J.D.; Koenig, S.N.; Mohler, P.J. Arrhythmogenic Cardiomyopathy: Molecular Insights for Improved Therapeutic Design. J. Cardiovasc. Dev. Dis. 2020, 7, 21. [Google Scholar]
- De Bortoli, M.; Postma, A.V.; Poloni, G.; Calore, M.; Minervini, G.; Mazzotti, E.; Rigato, I.; Ebert, M.; Lorenzon, A.; Vazza, G.; et al. Whole-Exome Sequencing Identifies Pathogenic Variants in TJP1 Gene Associated With Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e002123. [Google Scholar] [CrossRef] [PubMed]
- Poloni, G.; Calore, M.; Rigato, I.; Marras, E.; Minervini, G.; Mazzotti, E.; Lorenzon, A.; Li Mura, I.E.A.; Telatin, A.; Zara, I.; et al. A targeted next-generation gene panel reveals a novel heterozygous nonsense variant in the TP63 gene in patients with arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, 773–780. [Google Scholar] [CrossRef]
- Vio, R.; Angelini, A.; Basso, C.; Cipriani, A.; Zorzi, A.; Melacini, P.; Thiene, G.; Rampazzo, A.; Corrado, D.; Calore, C. Hypertrophic Cardiomyopathy and Primary Restrictive Cardiomyopathy: Similarities, Differences and Phenocopies. J. Clin. Med. 2021, 10, 1954. [Google Scholar] [PubMed]
- Waldmuller, S.; Schroeder, C.; Sturm, M.; Scheffold, T.; Imbrich, K.; Junker, S.; Frische, C.; Hofbeck, M.; Bauer, P.; Bonin, M.; et al. Targeted 46-gene and clinical exome sequencing for mutations causing cardiomyopathies. Mol. Cell. Probes 2015, 29, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Barrios, E.; Cesar, S.; Cruzalegui, J.; Hernandez, C.; Arbelo, E.; Fiol, V.; Brugada, J.; Brugada, R.; Campuzano, O.; Sarquella-Brugada, G. Clinical Genetics of Inherited Arrhythmogenic Disease in the Pediatric Population. Biomedicines 2022, 10, 106. [Google Scholar] [CrossRef]
- Brugada, R.; Campuzano, O.; Sarquella-Brugada, G.; Brugada, J.; Brugada, P. Brugada syndrome. Methodist DeBakey Cardiovasc. J. 2014, 10, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Adler, A.; Amin, A.S.; Abiusi, E.; Care, M.; Bikker, H.; Amenta, S.; Feilotter, H.; Nannenberg, E.A.; Mazzarotto, F.; et al. Evaluation of gene validity for CPVT and short QT syndrome in sudden arrhythmic death. Eur. Heart J. 2022, 43, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Roston, T.M.; Yuchi, Z.; Kannankeril, P.J.; Hathaway, J.; Vinocur, J.M.; Etheridge, S.P.; Potts, J.E.; Maginot, K.R.; Salerno, J.C.; Cohen, M.I.; et al. The clinical and genetic spectrum of catecholaminergic polymorphic ventricular tachycardia: Findings from an international multicentre registry. Europace 2018, 20, 541–547. [Google Scholar] [CrossRef]
- Janin, A.; Januel, L.; Cazeneuve, C.; Deliniere, A.; Chevalier, P.; Millat, G. Molecular Diagnosis of Inherited Cardiac Diseases in the Era of Next-Generation Sequencing: A Single Center’s Experience Over 5 Years. Mol. Diagn. Ther. 2021, 25, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Kalayinia, S.; Goodarzynejad, H.; Maleki, M.; Mahdieh, N. Next generation sequencing applications for cardiovascular disease. Ann. Med. 2018, 50, 91–109. [Google Scholar] [CrossRef]
- D’Argenio, V.; Esposito, M.V.; Barretta, F.; Mirra, B.; Caiazza, M.; Losi, M.A.; Limongelli, G.; Mazzaccara, C.; Frisso, G. Next-generation sequencing gene panels in inheritable cardiomyopathies and channelopaties: Yield of pathogenetic variants and variants of unknown significance in uncommon genes. Eur. Heart J. Suppl. 2020, 22, N83–N84. [Google Scholar]
- Mazzaccara, C.; Mirra, B.; Barretta, F.; Caiazza, M.; Lombardo, B.; Scudiero, O.; Tinto, N.; Limongelli, G.; Frisso, G. Molecular Epidemiology of Mitochondrial Cardiomyopathy: A Search Among Mitochondrial and Nuclear Genes. Int. J. Mol. Sci. 2021, 22, 5742. [Google Scholar] [CrossRef]
- Lombardo, B.; D’Argenio, V.; Monda, E.; Vitale, A.; Caiazza, M.; Sacchetti, L.; Pastore, L.; Limongelli, G.; Frisso, G.; Mazzaccara, C. Genetic analysis resolves differential diagnosis of a familial syndromic dilated cardiomyopathy: A new case of Alstrom syndrome. Mol. Genet. Genom. Med. 2020, 8, e1260. [Google Scholar] [CrossRef]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen—The Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef]
- Strande, N.T.; Riggs, E.R.; Buchanan, A.H.; Ceyhan-Birsoy, O.; DiStefano, M.; Dwight, S.S.; Goldstein, J.; Ghosh, R.; Seifert, B.A.; Sneddon, T.P.; et al. Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. Am. J. Hum. Genet. 2017, 100, 895–906. [Google Scholar]
- Assoc, W.M. World Medical Association Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects. JAMA 2013, 310, 2191–2194. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Lin, Y.; Williams, N.; Wang, D.; Coetzee, W.; Zhou, B.; Eng, L.S.; Um, S.Y.; Bao, R.; Devinsky, O.; McDonald, T.V.; et al. Applying High-Resolution Variant Classification to Cardiac Arrhythmogenic Gene Testing in a Demographically Diverse Cohort of Sudden Unexplained Deaths. Circ. Cardiovasc. Genet. 2017, 10, e001839. [Google Scholar] [CrossRef]
- Van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Frank-Hansen, R.; Page, S.P.; Syrris, P.; McKenna, W.J.; Christiansen, M.; Andersen, P.S. Micro-exons of the cardiac myosin binding protein C gene: Flanking introns contain a disproportionately large number of hypertrophic cardiomyopathy mutations. Eur. J. Hum. Genet. 2008, 16, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Brito, D.; Magalhaes, A.; Cortez-Dias, N.; Miltenberger-Miltenyi, G. Rare Association of two Genetic Causes of Sudden Death in a Young Survivor. Arq. Bras. Cardiol. 2017, 108, 184–186. [Google Scholar] [CrossRef]
- Pappone, C.; Monasky, M.M.; Micaglio, E.; Ciconte, G. Right ventricular electromechanical abnormalities in Brugada syndrome: Is this a cardiomyopathy? Eur. Heart J. Suppl. 2020, 22, E101–E104. [Google Scholar] [CrossRef]
- De Maria, E.; Borghi, A.; Tonelli, L.; Selvatici, R.; Cappelli, S.; Gualandi, F. Brugada ECG pattern in hypertrophic cardiomyopathy: Brugada phenocopy or overlapping syndrome? J. Electrocardiol. 2021, 69, 132. [Google Scholar] [CrossRef]
- Fernlund, E.; Kissopoulou, A.; Green, H.; Karlsson, J.E.; Ellegard, R.; Arstrand, H.K.; Jonasson, J.; Gunnarsson, C. Hereditary Hypertrophic Cardiomyopathy in Children and Young Adults-The Value of Reevaluating and Expanding Gene Panel Analyses. Genes 2020, 11, 1472. [Google Scholar] [CrossRef]
- Novelli, V.; Malkani, K.; Cerrone, M. Pleiotropic Phenotypes Associated With PKP2 Variants. Front. Cardiovasc. Med. 2018, 5, 184. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.X.; Liu, J.; Ruan, J.Y.; Yu, S.Q.; Wang, L.M.; Zhao, S.H.; Wang, S.Y.; Kang, L.M.; Wang, J.Z.; Song, L. Deleterious Rare Desmosomal Variants Contribute to Hypertrophic Cardiomyopathy and Are Associated With Distinctive Clinical Features. Can. J. Cardiol. 2022, 38, 41–48. [Google Scholar] [CrossRef]
- Bainbridge, M.N.; Li, L.L.; Tan, Y.L.; Cheong, B.Y.; Marian, A.J. Identification of established arrhythmogenic right ventricular cardiomyopathy mutation in a patient with the contrasting phenotype of hypertrophic cardiomyopathy. BMC Med. Genet. 2017, 18, 24. [Google Scholar] [CrossRef]
- Chen, H.; Li, X.; Liu, X.; Wang, J.; Zhang, Z.; Wu, J.; Huang, M.; Guo, Y.; Li, F.; Wang, X.; et al. Clinical and mutation profile of pediatric patients with RASopathy-associated hypertrophic cardiomyopathy: Results from a Chinese cohort. Orphanet. J. Rare Dis. 2019, 14, 29. [Google Scholar] [CrossRef]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: Re-evaluating the role of non-sarcomeric genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef] [PubMed]
- Spracklen, T.F.; Kasher, P.R.; Kraus, S.; Botha, T.L.; Page, D.J.; Kamuli, S.; Booi, Z.; Chin, A.; Laing, N.; Keavney, B.D.; et al. Identification of a POLG Variant in a Family With Arrhythmogenic Cardiomyopathy and Left Ventricular Fibrosis. Circ. Genom. Precis. Med. 2021, 14, e003138. [Google Scholar] [CrossRef] [PubMed]
- Carsana, A.; Frisso, G.; Tremolaterra, M.R.; Ricci, E.; De Rasmo, D.; Salvatore, F. A larger spectrum of intragenic short tandem repeats improves linkage analysis and localization of intragenic recombination detection in the dystrophin gene: An analysis of 93 families from southern Italy. J. Mol. Diagn. 2007, 9, 64. [Google Scholar] [CrossRef][Green Version]
- Giuca, A.; Mitu, C.; Popescu, B.O.; Bastian, A.E.; Capsa, R.; Mursa, A.; Radoi, V.; Popescu, B.A.; Jurcut, R. Novel FHL1 mutation variant identified in a patient with nonobstructive hypertrophic cardiomyopathy and myopathy—A case report. BMC Med. Genet. 2020, 21, 188. [Google Scholar] [CrossRef]
- D’Argenio, V. The High-Throughput Analyses Era: Are We Ready for the Data Struggle? High Throughput 2018, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Nunziato, M.; Mazzaccara, C.; Intrieri, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Di Maggio, F.; Frisso, G.; Salvatore, F. Genotype-Phenotype Correlation: A Triple DNA Mutational Event in a Boy Entering Sport Conveys an Additional Pathogenicity Risk. Genes 2020, 11, 524. [Google Scholar] [CrossRef] [PubMed]
- Frisso, G.; Detta, N.; Coppola, P.; Mazzaccara, C.; Pricolo, M.R.; D’Onofrio, A.; Limongelli, G.; Calabro, R.; Salvatore, F. Functional Studies and In Silico Analyses to Evaluate Non-Coding Variants in Inherited Cardiomyopathies. Int. J. Mol. Sci. 2016, 17, 1883. [Google Scholar] [CrossRef]
- Pricolo, M.R.; Herrero-Galan, E.; Mazzaccara, C.; Losi, M.A.; Alegre-Cebollada, J.; Frisso, G. Protein Thermodynamic Destabilization in the Assessment of Pathogenicity of a Variant of Uncertain Significance in Cardiac Myosin Binding Protein C. J. Cardiovasc. Transl. Res. 2020, 13, 867–877. [Google Scholar] [CrossRef]
- Esposito, M.V.; Minopoli, G.; Esposito, L.; D’Argenio, V.; Di Maggio, F.; Sasso, E.; D’Aiuto, M.; Zambrano, N.; Salvatore, F. A Functional Analysis of the Unclassified Pro2767Ser BRCA2 Variant Reveals Its Potential Pathogenicity that Acts by Hampering DNA Binding and Homology-Mediated DNA Repair. Cancers 2019, 11, 1454. [Google Scholar] [CrossRef]
- Esposito, M.V.; Nunziato, M.; Starnone, F.; Telese, A.; Calabrese, A.; D’Aiuto, G.; Pucci, P.; D’Aiuto, M.; Baralle, F.; D’Argenio, V.; et al. A Novel Pathogenic BRCA1 Splicing Variant Produces Partial Intron Retention in the Mature Messenger RNA. Int. J. Mol. Sci. 2016, 17, 2145. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).