
GPCRs in Intracellular Compartments: New Targets for Drug Discovery

 , ,
, , 
Abstract
1. Introduction
2. Evolution Promoted Biological Complexity by Compartmentalization of Metabolic Processes
3. G protein-Coupled Receptors (GPCRs) and Their Role in Modulating Different Types of Stimuli
4. GPCRs Are Present in Different Cellular Compartments
5. GPCR Sorting to the Plasma Membrane and to Intracellular Compartments
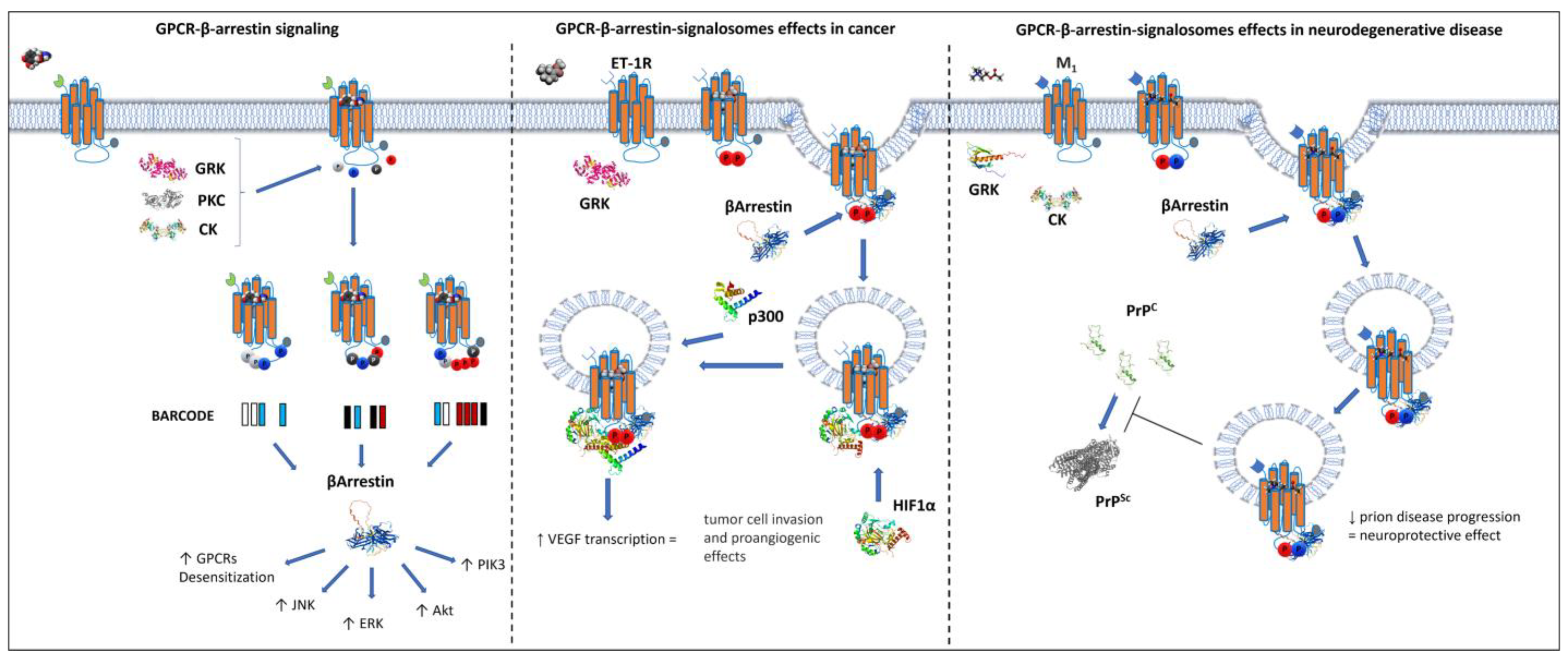
6. β-Arrestin-Mediated GPCR Compartmentalization
7. Activation of GPCRs in Internal Cell Compartments
8. Distinctive Signals Generated by GPCRs in Intracellular Compartments
9. Designing Drugs to Target GPCRs Localized in Internal Compartments
10. Concluding Remarks and Future Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brasier, M.; McLoughlin, N.; Green, O.; Wacey, D. A Fresh Look at the Fossil Evidence for Early Archaean Cellular Life. Phil. Trans. R. Soc. B 2006, 361, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G. Cell Evolution and the Problem of Membrane Topology. Nat. Rev. Mol. Cell Biol. 2007, 8, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Kerfeld, C.A.; Melnicki, M.R. Assembly, Function and Evolution of Cyanobacterial Carboxysomes. Curr. Opin. Plant Biol. 2016, 31, 66–75. [Google Scholar] [CrossRef]
- Honigmann, A.; Pralle, A. Compartmentalization of the Cell Membrane. J. Mol. Biol. 2016, 428, 4739–4748. [Google Scholar] [CrossRef] [PubMed]
- Diekmann, Y.; Pereira-Leal, J.B. Evolution of Intracellular Compartmentalization. Biochem. J. 2013, 449, 319–331. [Google Scholar] [CrossRef]
- Chomicki, G.; Werner, G.D.A.; West, S.A.; Kiers, E.T. Compartmentalization Drives the Evolution of Symbiotic Cooperation. Phil. Trans. R. Soc. B 2020, 375, 20190602. [Google Scholar] [CrossRef]
- Sommer, M.S.; Schleiff, E. Protein Targeting and Transport as a Necessary Consequence of Increased Cellular Complexity. Cold Spring Harb. Perspect. Biol. 2014, 6, a016055. [Google Scholar] [CrossRef][Green Version]
- Schmidt, V.; Willnow, T.E. Protein Sorting Gone Wrong–VPS10P Domain Receptors in Cardiovascular and Metabolic Diseases. Atherosclerosis 2016, 245, 194–199. [Google Scholar] [CrossRef]
- Gabaldón, T.; Pittis, A.A. Origin and Evolution of Metabolic Sub-Cellular Compartmentalization in Eukaryotes. Biochimie 2015, 119, 262–268. [Google Scholar] [CrossRef]
- Martin, W. Evolutionary Origins of Metabolic Compartmentalization in Eukaryotes. Phil. Trans. R. Soc. B 2010, 365, 847–855. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The Structure and Function of G-Protein-Coupled Receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Strotmann, R.; Schröck, K.; Böselt, I.; Stäubert, C.; Russ, A.; Schöneberg, T. Evolution of GPCR: Change and Continuity. Mol. Cell Endocrinol. 2011, 331, 170–178. [Google Scholar] [CrossRef]
- Scarselli, M.; Annibale, P.; Gerace, C.; Radenovic, A. Enlightening G-Protein-Coupled Receptors on the Plasma Membrane Using Super-Resolution Photoactivated Localization Microscopy. Biochem. Soc. Trans. 2013, 41, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.-Y. Regulation, Signaling, and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef] [PubMed]
- Smrcka, A.V. G Protein Βγ Subunits: Central Mediators of G Protein-Coupled Receptor Signaling. Cell. Mol. Life Sci. 2008, 65, 2191–2214. [Google Scholar] [CrossRef]
- Logothetis, D.E.; Kurachi, Y.; Galper, J.; Neer, E.J.; Clapham, D.E. The Βγ Subunits of GTP-Binding Proteins Activate the Muscarinic K+ Channel in Heart. Nature 1987, 325, 321–326. [Google Scholar] [CrossRef]
- Wickman, K.D.; Iñiguez-Lluhi, J.A.; Davenport, P.A.; Taussig, R.; Krapivinsky, G.B.; Linder, M.E.; Gilman, A.G.; Clapham, D.E. Recombinant G-Protein Βγ-Subunits Activate the Muscarinic-Gated Atrial Potassium Channel. Nature 1994, 368, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Takesono, A.; Cismowski, M.J.; Ribas, C.; Bernard, M.; Chung, P.; Hazard, S.; Duzic, E.; Lanier, S.M. Receptor-Independent Activators of Heterotrimeric G-Protein Signaling Pathways. J. Biol. Chem. 1999, 274, 33202–33205. [Google Scholar] [CrossRef] [PubMed]
- Blumer, J.B.; Smrcka, A.V.; Lanier, S.M. Mechanistic Pathways and Biological Roles for Receptor-Independent Activators of G-Protein Signaling. Pharmacol. Ther. 2007, 113, 488–506. [Google Scholar] [CrossRef]
- Heuss, C.; Gerber, U. G-Protein-Independent Signaling by G-Protein-Coupled Receptors. Trends Neurosci. 2000, 23, 469–475. [Google Scholar] [CrossRef]
- Sokolina, K.; Kittanakom, S.; Snider, J.; Kotlyar, M.; Maurice, P.; Gandía, J.; Benleulmi-Chaachoua, A.; Tadagaki, K.; Oishi, A.; Wong, V.; et al. Systematic Protein–Protein Interaction Mapping for Clinically Relevant Human GPCR s. Mol. Syst. Biol. 2017, 13, 918. [Google Scholar] [CrossRef]
- Eichel, K.; von Zastrow, M. Subcellular Organization of GPCR Signaling. Trends Pharmacol. Sci. 2018, 39, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Mendizabal-Zubiaga, J.; Melser, S.; Bénard, G.; Ramos, A.; Reguero, L.; Arrabal, S.; Elezgarai, I.; Gerrikagoitia, I.; Suarez, J.; Rodríguez De Fonseca, F.; et al. Cannabinoid CB1 Receptors Are Localized in Striated Muscle Mitochondria and Regulate Mitochondrial Respiration. Front. Physiol. 2016, 7, 476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bedigian, A.V.; Wang, W.; Eggert, U.S. G Protein-Coupled Receptors Participate in Cytokinesis. Cytoskeleton 2012, 69, 810–818. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, K.L.; Jong, Y.-J.I.; Gonchar, Y.; Burkhalter, A.; Romano, C. Activation of Metabotropic Glutamate Receptor MGlu5 on Nuclear Membranes Mediates Intranuclear Ca2+ Changes in Heterologous Cell Types and Neurons. J. Biol. Chem. 2003, 278, 28210–28219. [Google Scholar] [CrossRef]
- Irannejad, R.; Tomshine, J.C.; Tomshine, J.R.; Chevalier, M.; Mahoney, J.P.; Steyaert, J.; Rasmussen, S.G.F.; Sunahara, R.K.; El-Samad, H.; Huang, B.; et al. Conformational Biosensors Reveal GPCR Signalling from Endosomes. Nature 2013, 495, 534–538. [Google Scholar] [CrossRef]
- Yarwood, R.E.; Imlach, W.L.; Lieu, T.; Veldhuis, N.A.; Jensen, D.D.; Klein Herenbrink, C.; Aurelio, L.; Cai, Z.; Christie, M.J.; Poole, D.P.; et al. Endosomal Signaling of the Receptor for Calcitonin Gene-Related Peptide Mediates Pain Transmission. Proc. Natl. Acad. Sci. USA 2017, 114, 12309–12314. [Google Scholar] [CrossRef]
- Gorvin, C.M.; Rogers, A.; Hastoy, B.; Tarasov, A.I.; Frost, M.; Sposini, S.; Inoue, A.; Whyte, M.P.; Rorsman, P.; Hanyaloglu, A.C.; et al. AP2σ Mutations Impair Calcium-Sensing Receptor Trafficking and Signaling, and Show an Endosomal Pathway to Spatially Direct G-Protein Selectivity. Cell Rep. 2018, 22, 1054–1066. [Google Scholar] [CrossRef]
- Kotowski, S.J.; Hopf, F.W.; Seif, T.; Bonci, A.; von Zastrow, M. Endocytosis Promotes Rapid Dopaminergic Signaling. Neuron 2011, 71, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Lyga, S.; Volpe, S.; Werthmann, R.C.; Gotz, K.; Sungkaworn, T.; Lohse, M.J.; Calebiro, D. Persistent CAMP Signaling by Internalized LH Receptors in Ovarian Follicles. Endocrinology 2016, 2016, 63–71. [Google Scholar] [CrossRef]
- Jensen, D.D.; Lieu, T.; Halls, M.L.; Veldhuis, N.A.; Imlach, W.L.; Mai, Q.N.; Poole, D.P.; Quach, T.; Aurelio, L.; Conner, J.; et al. Neurokinin 1 Receptor Signaling in Endosomes Mediates Sustained Nociception and Is a Viable Therapeutic Target for Prolonged Pain Relief. Sci. Transl. Med. 2017, 9, eaal3447. [Google Scholar] [CrossRef]
- Ferrandon, S.; Feinstein, T.N.; Castro, M.; Wang, B.; Bouley, R.; Potts, J.T.; Gardella, T.J.; Vilardaga, J.-P. Sustained Cyclic AMP Production by Parathyroid Hormone Receptor Endocytosis. Nat. Chem. Biol. 2009, 5, 734–742. [Google Scholar] [CrossRef]
- Jimenez-Vargas, N.N.; Pattison, L.A.; Zhao, P.; Lieu, T.; Latorre, R.; Jensen, D.D.; Castro, J.; Aurelio, L.; Le, G.T.; Flynn, B.; et al. Protease-Activated Receptor-2 in Endosomes Signals Persistent Pain of Irritable Bowel Syndrome. Proc. Natl. Acad. Sci. USA 2018, 115, E7438–E7447. [Google Scholar] [CrossRef]
- Feinstein, T.N.; Yui, N.; Webber, M.J.; Wehbi, V.L.; Stevenson, H.P.; King, J.D.; Hallows, K.R.; Brown, D.; Bouley, R.; Vilardaga, J.-P. Noncanonical Control of Vasopressin Receptor Type 2 Signaling by Retromer and Arrestin. J. Biol. Chem. 2013, 288, 27849–27860. [Google Scholar] [CrossRef]
- Martin, R.D.; Sun, Y.; Bourque, K.; Audet, N.; Inoue, A.; Tanny, J.C.; Hébert, T.E. Receptor- and Cellular Compartment-Specific Activation of the CAMP/PKA Pathway by A1-Adrenergic and ETA Endothelin Receptors. Cell Signal. 2018, 44, 43–50. [Google Scholar] [CrossRef]
- Cui, D.; Trier, K.; Chen, X.; Zeng, J.; Yang, X.; Hu, J.; Ge, J. Distribution of Adenosine Receptors in Human Sclera Fibroblasts. Mol. Vis. 2008, 14, 523–529. [Google Scholar]
- Morinelli, T.A.; Raymond, J.R.; Baldys, A.; Yang, Q.; Lee, M.; Luttrell, L.; Ullian, M.E. Identification of a Putative Nuclear Localization Sequence within ANG II AT 1A Receptor Associated with Nuclear Activation. Am. J. Physiol. -Cell Physiol. 2007, 292, C1398–C1408. [Google Scholar] [CrossRef]
- Lee, D.K.; Lança, A.J.; Cheng, R.; Nguyen, T.; Ji, X.D.; Gobeil, F.; Chemtob, S.; George, S.R.; O’Dowd, B.F. Agonist-Independent Nuclear Localization of the Apelin, Angiotensin AT1, and Bradykinin B2 Receptors. J. Biol. Chem. 2004, 279, 7901–7908. [Google Scholar] [CrossRef]
- Don-Salu-Hewage, A.S.; Chan, S.Y.; McAndrews, K.M.; Chetram, M.A.; Dawson, M.R.; Bethea, D.A.; Hinton, C.V. Cysteine (C)-X-C Receptor 4 Undergoes Transportin 1-Dependent Nuclear Localization and Remains Functional at the Nucleus of Metastatic Prostate Cancer Cells. PLoS ONE 2013, 8, e57194. [Google Scholar] [CrossRef]
- Di Benedetto, A.; Sun, L.; Zambonin, C.G.; Tamma, R.; Nico, B.; Calvano, C.D.; Colaianni, G.; Ji, Y.; Mori, G.; Grano, M.; et al. Osteoblast Regulation via Ligand-Activated Nuclear Trafficking of the Oxytocin Receptor. Proc. Natl. Acad. Sci. USA 2014, 111, 16502–16507. [Google Scholar] [CrossRef]
- Jong, Y.-J.I.; Harmon, S.K.; O’Malley, K.L. GPCR Signalling from within the Cell: Intracellular GPCR Signaling. Br. J. Pharmacol. 2018, 175, 4026–4035. [Google Scholar] [CrossRef] [PubMed]
- Belous, A.E.; Jones, C.M.; Wakata, A.; Knox, C.D.; Nicoud, I.B.; Pierce, J.; Chari, R.S. Mitochondrial Calcium Transport Is Regulated by P2Y1- and P2Y2-like Mitochondrial Receptors. J. Cell. Biochem. 2006, 99, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Abadir, P.M.; Walston, J.D.; Carey, R.M. Subcellular Characteristics of Functional Intracellular Renin–Angiotensin Systems. Peptides 2012, 38, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, H.; Xu, H.; Guo, D.; Shi, H.; Li, Y.; Zhang, W.; Gu, Y. 5-HTR3 and 5-HTR4 Located on the Mitochondrial Membrane and Functionally Regulated Mitochondrial Functions. Sci. Rep. 2016, 6, 37336. [Google Scholar] [CrossRef] [PubMed]
- Suofu, Y.; Li, W.; Jean-Alphonse, F.G.; Jia, J.; Khattar, N.K.; Li, J.; Baranov, S.V.; Leronni, D.; Mihalik, A.C.; He, Y.; et al. Dual Role of Mitochondria in Producing Melatonin and Driving GPCR Signaling to Block Cytochrome c Release. Proc. Natl. Acad. Sci. USA 2017, 114, E7997–E8006. [Google Scholar] [CrossRef] [PubMed]
- Hebert-Chatelain, E.; Desprez, T.; Serrat, R.; Bellocchio, L.; Soria-Gomez, E.; Busquets-Garcia, A.; Pagano Zottola, A.C.; Delamarre, A.; Cannich, A.; Vincent, P.; et al. A Cannabinoid Link between Mitochondria and Memory. Nature 2016, 539, 555–559. [Google Scholar] [CrossRef]
- Boivin, B.; Lavoie, C.; Vaniotis, G.; Baragli, A.; Villeneuve, L.; Ethier, N.; Trieu, P.; Allen, B.; Hebert, T. Functional β-Adrenergic Receptor Signalling on Nuclear Membranes in Adult Rat and Mouse Ventricular Cardiomyocytes. Cardiovasc. Res. 2006, 71, 69–78. [Google Scholar] [CrossRef]
- Mullershausen, F.; Zecri, F.; Cetin, C.; Billich, A.; Guerini, D.; Seuwen, K. Persistent Signaling Induced by FTY720-Phosphate Is Mediated by Internalized S1P1 Receptors. Nat. Chem. Biol. 2009, 5, 428–434. [Google Scholar] [CrossRef]
- Calebiro, D.; Nikolaev, V.O.; Gagliani, M.C.; de Filippis, T.; Dees, C.; Tacchetti, C.; Persani, L.; Lohse, M.J. Persistent CAMP-Signals Triggered by Internalized G-Protein–Coupled Receptors. PLoS Biol. 2009, 7, e1000172. [Google Scholar] [CrossRef]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef]
- Kwon, S.-H.; Liu, K.D.; Mostov, K.E. Intercellular Transfer of GPRC5B via Exosomes Drives HGF-Mediated Outward Growth. Curr. Biol. 2014, 24, 199–204. [Google Scholar] [CrossRef]
- Albert, A.; Boeszebattaglia, K. The Role of Cholesterol in Rod Outer Segment Membranes. Prog. Lipid Res. 2005, 44, 99–124. [Google Scholar] [CrossRef]
- Palczewski, K. G Protein–Coupled Receptor Rhodopsin. Annu. Rev. Biochem. 2006, 75, 743–767. [Google Scholar] [CrossRef]
- Daaka, Y.; Luttrell, L.M.; Ahn, S.; Della Rocca, G.J.; Ferguson, S.S.G.; Caron, M.G.; Lefkowitz, R.J. Essential Role for G Protein-Coupled Receptor Endocytosis in the Activation of Mitogen-Activated Protein Kinase. J. Biol. Chem. 1998, 273, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Ferguson, S.S.G.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.-T.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. β-Arrestin-Dependent Formation of β 2 Adrenergic Receptor-Src Protein Kinase Complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef]
- Morisco, C.; Marrone, C.; Galeotti, J.; Shao, D.; Vatner, D.E.; Vatner, S.F.; Sadoshima, J. Endocytosis Machinery Is Required for Β1-Adrenergic Receptor-Induced Hypertrophy in Neonatal Rat Cardiac Myocytes. Cardiovasc. Res. 2008, 78, 36–44. [Google Scholar] [CrossRef]
- Irannejad, R.; Pessino, V.; Mika, D.; Huang, B.; Wedegaertner, P.B.; Conti, M.; von Zastrow, M. Functional Selectivity of GPCR-Directed Drug Action through Location Bias. Nat. Chem. Biol. 2017, 13, 799–806. [Google Scholar] [CrossRef]
- Godbole, A.; Lyga, S.; Lohse, M.J.; Calebiro, D. Internalized TSH Receptors En Route to the TGN Induce Local Gs-Protein Signaling and Gene Transcription. Nat. Commun. 2017, 8, 443. [Google Scholar] [CrossRef] [PubMed]
- Branco, A.F.; Allen, B.G. G Protein–Coupled Receptor Signaling in Cardiac Nuclear Membranes. J. Cardiovasc. Pharmacol. 2015, 65, 101–109. [Google Scholar] [CrossRef]
- Campden, R.; Audet, N.; Hébert, T.E. Nuclear G Protein Signaling: New Tricks for Old Dogs. J. Cardiovasc. Pharmacol. 2015, 65, 110–122. [Google Scholar] [CrossRef]
- Vaniotis, G.; Allen, B.G.; Hébert, T.E. Nuclear GPCRs in Cardiomyocytes: An Insider’s View of β-Adrenergic Receptor Signaling. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H1754–H1764. [Google Scholar] [CrossRef] [PubMed]
- Tadevosyan, A.; Maguy, A.; Villeneuve, L.R.; Babin, J.; Bonnefoy, A.; Allen, B.G.; Nattel, S. Nuclear-Delimited Angiotensin Receptor-Mediated Signaling Regulates Cardiomyocyte Gene Expression. J. Biol. Chem. 2010, 285, 22338–22349. [Google Scholar] [CrossRef] [PubMed]
- Vaniotis, G.; Del Duca, D.; Trieu, P.; Rohlicek, C.V.; Hébert, T.E.; Allen, B.G. Nuclear β-Adrenergic Receptors Modulate Gene Expression in Adult Rat Heart. Cell. Signal. 2011, 23, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular Vesicles: Exosomes, Microvesicles, and Friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- EL Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J.A. Extracellular Vesicles: Biology and Emerging Therapeutic Opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Longoni, B.; Fasciani, I.; Kolachalam, S.; Pietrantoni, I.; Marampon, F.; Petragnano, F.; Aloisi, G.; Coppolino, M.F.; Rossi, M.; Scarselli, M.; et al. Neurotoxic and Neuroprotective Role of Exosomes in Parkinson’s Disease. CPD 2020, 25, 4510–4522. [Google Scholar] [CrossRef] [PubMed]
- Bebelman, M.P.; Crudden, C.; Pegtel, D.M.; Smit, M.J. The Convergence of Extracellular Vesicle and GPCR Biology. Trends Pharmacol. Sci. 2020, 41, 627–640. [Google Scholar] [CrossRef]
- Joseph, K.; Spicer, E.K.; Tholanikunnel, B.G. Regulatory Mechanism of G Protein-Coupled Receptor Trafficking to the Plasma Membrane. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 521, pp. 131–150. ISBN 9780123918628. [Google Scholar]
- Blobel, G. Protein Targeting (Nobel Lecture). ChemBioChem 2000, 1, 86–102. [Google Scholar] [CrossRef]
- Köchl, R.; Alken, M.; Rutz, C.; Krause, G.; Oksche, A.; Rosenthal, W.; Schülein, R. The Signal Peptide of the G Protein-Coupled Human Endothelin B Receptor Is Necessary for Translocation of the N-Terminal Tail across the Endoplasmic Reticulum Membrane. J. Biol. Chem. 2002, 277, 16131–16138. [Google Scholar] [CrossRef]
- Shirvani, H.; Gätà, G.; Marullo, S. Regulated GPCR Trafficking to the Plasma Membrane: General Issues and the CCR5 Chemokine Receptor Example. In GPCR Signalling Complexes–Synthesis, Assembly, Trafficking and Specificity; Dupré, D.J., Hébert, T.E., Jockers, R., Eds.; Springer: Dordrecht, The Netherlands, 2012; Volume 63, pp. 97–111. ISBN 9789400747647. [Google Scholar]
- McLatchie, L.M.; Fraser, N.J.; Main, M.J.; Wise, A.; Brown, J.; Thompson, N.; Solari, R.; Lee, M.G.; Foord, S.M. RAMPs Regulate the Transport and Ligand Specificity of the Calcitonin-Receptor-like Receptor. Nature 1998, 393, 333–339. [Google Scholar] [CrossRef]
- Hay, D.L.; Walker, C.S.; Gingell, J.J.; Ladds, G.; Reynolds, C.A.; Poyner, D.R. Receptor Activity-Modifying Proteins; Multifunctional G Protein-Coupled Receptor Accessory Proteins. Biochem. Soc. Trans. 2016, 44, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.L.; Pioszak, A.A. Receptor Activity-Modifying Proteins (RAMPs): New Insights and Roles. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 469–487. [Google Scholar] [CrossRef]
- Bouschet, T.; Martin, S.; Henley, J.M. Receptor-Activity-Modifying Proteins Are Required for Forward Trafficking of the Calcium-Sensing Receptor to the Plasma Membrane. J. Cell Sci. 2005, 118, 4709–4720. [Google Scholar] [CrossRef]
- Harikumar, K.G.; Simms, J.; Christopoulos, G.; Sexton, P.M.; Miller, L.J. Molecular Basis of Association of Receptor Activity-Modifying Protein 3 with the Family B G Protein-Coupled Secretin Receptor. Biochemistry 2009, 48, 11773–11785. [Google Scholar] [CrossRef] [PubMed]
- Lenhart, P.M.; Broselid, S.; Barrick, C.J.; Leeb-Lundberg, L.M.F.; Caron, K.M. G-Protein-Coupled Receptor 30 Interacts with Receptor Activity-Modifying Protein 3 and Confers Sex-Dependent Cardioprotection. J. Mol. Endocrinol. 2013, 51, 191–202. [Google Scholar] [CrossRef]
- Wootten, D.; Lindmark, H.; Kadmiel, M.; Willcockson, H.; Caron, K.; Barwell, J.; Drmota, T.; Poyner, D. Receptor Activity Modifying Proteins (RAMPs) Interact with the VPAC 2 Receptor and CRF 1 Receptors and Modulate Their Function: RAMP Interactions with VPAC 2 and CRF 1 Receptors. Br. J. Pharmacol. 2013, 168, 822–834. [Google Scholar] [CrossRef]
- Björk, S.; Hurt, C.M.; Ho, V.K.; Angelotti, T. REEPs Are Membrane Shaping Adapter Proteins That Modulate Specific G Protein-Coupled Receptor Trafficking by Affecting ER Cargo Capacity. PLoS ONE 2013, 8, e76366. [Google Scholar] [CrossRef]
- Free, R.B.; Hazelwood, L.A.; Cabrera, D.M.; Spalding, H.N.; Namkung, Y.; Rankin, M.L.; Sibley, D.R. D1 and D2 Dopamine Receptor Expression Is Regulated by Direct Interaction with the Chaperone Protein Calnexin. J. Biol. Chem. 2007, 282, 21285–21300. [Google Scholar] [CrossRef]
- Achour, L.; Scott, M.G.H.; Shirvani, H.; Thuret, A.; Bismuth, G.; Labbé-Jullié, C.; Marullo, S. CD4-CCR5 Interaction in Intracellular Compartments Contributes to Receptor Expression at the Cell Surface. Blood 2009, 113, 1938–1947. [Google Scholar] [CrossRef]
- Rosemond, E.; Rossi, M.; McMillin, S.M.; Scarselli, M.; Donaldson, J.G.; Wess, J. Regulation of M 3 Muscarinic Receptor Expression and Function by Transmembrane Protein 147. Mol. Pharmacol. 2011, 79, 251–261. [Google Scholar] [CrossRef]
- Wei, Z.; Xu, X.; Fang, Y.; Khater, M.; Naughton, S.X.; Hu, G.; Terry, A.V.; Wu, G. Rab43 GTPase Directs Postsynaptic Trafficking and Neuron-Specific Sorting of G Protein–Coupled Receptors. J. Biol. Chem. 2021, 296, 100517. [Google Scholar] [CrossRef]
- Watson, P.H.; Fraher, L.J.; Natale, B.V.; Kisiel, M.; Hendy, G.N.; Hodsman, A.B. Nuclear Localization of the Type 1 Parathyroid Hormone/Parathyroid Hormone-Related Peptide Receptor in MC3T3-E1 Cells: Association with Serum-Induced Cell Proliferation. Bone 2000, 26, 221–225. [Google Scholar] [CrossRef]
- Savard, M.; Barbaz, D.; Bélanger, S.; Müller-Esterl, W.; Bkaily, G.; D’orléans-Juste, P.; Coté, J.; Bovenzi, V.; Gobeil, F. Expression of Endogenous Nuclear Bradykinin B2 Receptors Mediating Signaling in Immediate Early Gene Activation. J. Cell. Physiol. 2008, 216, 234–244. [Google Scholar] [CrossRef]
- Jong, Y.-J.I.; Kumar, V.; O’Malley, K.L. Intracellular Metabotropic Glutamate Receptor 5 (MGluR5) Activates Signaling Cascades Distinct from Cell Surface Counterparts. J. Biol. Chem. 2009, 284, 35827–35838. [Google Scholar] [CrossRef]
- Saito, T.; Giaid, A. Cyclooxygenase-2 and Nuclear Factor-KappaB in Myocardium of End Stage Human Heart Failure. Congest. Heart Fail. 1999, 5, 222–227. [Google Scholar]
- Lusk, C.P.; Blobel, G.; King, M.C. Highway to the Inner Nuclear Membrane: Rules for the Road. Nat. Rev. Mol. Cell Biol. 2007, 8, 414–420. [Google Scholar] [CrossRef]
- Zuleger, N.; Kerr, A.R.W.; Schirmer, E.C. Many Mechanisms, One Entrance: Membrane Protein Translocation into the Nucleus. Cell. Mol. Life Sci. 2012, 69, 2205–2216. [Google Scholar] [CrossRef] [PubMed]
- Wing, C.E.; Fung, H.Y.J.; Chook, Y.M. Karyopherin-Mediated Nucleocytoplasmic Transport. Nat. Rev. Mol. Cell Biol. 2022, 23, 307–328. [Google Scholar] [CrossRef]
- Favre, N.; Camps, M.; Arod, C.; Chabert, C.; Rommel, C.; Pasquali, C. Chemokine Receptor CCR2 Undergoes Transportin1-Dependent Nuclear Translocation. Proteomics 2008, 8, 4560–4576. [Google Scholar] [CrossRef]
- Joyal, J.-S.; Nim, S.; Zhu, T.; Sitaras, N.; Rivera, J.C.; Shao, Z.; Sapieha, P.; Hamel, D.; Sanchez, M.; Zaniolo, K.; et al. Subcellular Localization of Coagulation Factor II Receptor-like 1 in Neurons Governs Angiogenesis. Nat. Med. 2014, 20, 1165–1173. [Google Scholar] [CrossRef]
- Sergin, I.; Jong, Y.-J.I.; Harmon, S.K.; Kumar, V.; O’Malley, K.L. Sequences within the C Terminus of the Metabotropic Glutamate Receptor 5 (MGluR5) Are Responsible for Inner Nuclear Membrane Localization. J. Biol. Chem. 2017, 292, 3637–3655. [Google Scholar] [CrossRef]
- Bhosle, V.K.; Rivera, J.C.; Chemtob, S. New Insights into Mechanisms of Nuclear Translocation of G-Protein Coupled Receptors. Small GTPases 2017, 10, 254–263. [Google Scholar] [CrossRef]
- Mizuno-Yamasaki, E.; Rivera-Molina, F.; Novick, P. GTPase Networks in Membrane Traffic. Annu. Rev. Biochem. 2012, 81, 637–659. [Google Scholar] [CrossRef]
- Bhosle, V.K.; Rivera, J.C.; Zhou, T.; Omri, S.; Sanchez, M.; Hamel, D.; Zhu, T.; Rouget, R.; Rabea, A.A.; Hou, X.; et al. Nuclear Localization of Platelet-Activating Factor Receptor Controls Retinal Neovascularization. Cell Discov. 2016, 2, 16017. [Google Scholar] [CrossRef]
- Williams, C.L. The Polybasic Region of Ras and Rho Family Small GTPases: A Regulator of Protein Interactions and Membrane Association and a Site of Nuclear Localization Signal Sequences. Cell Signal. 2003, 15, 1071–1080. [Google Scholar] [CrossRef]
- Belous, A.; Wakata, A.; Knox, C.D.; Nicoud, I.B.; Pierce, J.; Anderson, C.D.; Pinson, C.W.; Chari, R.S. Mitochondrial P2Y-Like Receptors Link Cytosolic Adenosine Nucleotides to Mitochondrial Calcium Uptake. J. Cell. Biochem. 2004, 92, 1062–1073. [Google Scholar] [CrossRef]
- Gbahou, F.; Cecon, E.; Viault, G.; Gerbier, R.; Jean-Alphonse, F.; Karamitri, A.; Guillaumet, G.; Delagrange, P.; Friedlander, R.M.; Vilardaga, J.-P.; et al. Design and Validation of the First Cell-Impermeant Melatonin Receptor Agonist: Cell-Impermeant Melatonin Receptor Agonist. Br. J. Pharmacol. 2017, 174, 2409–2421. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, S.; Zhang, X.; Liu, H. QR Code Model: A New Possibility for GPCR Phosphorylation Recognition. Cell Commun. Signal. 2022, 20, 23. [Google Scholar] [CrossRef]
- Tobin, A.B.; Butcher, A.J.; Kong, K.C. Location, Location, Location…site-Specific GPCR Phosphorylation Offers a Mechanism for Cell-Type-Specific Signalling. Trends Pharmacol. Sci. 2008, 29, 413–420. [Google Scholar] [CrossRef]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.-Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct Phosphorylation Sites on the β 2 -Adrenergic Receptor Establish a Barcode That Encodes Differential Functions of β-Arrestin. Sci. Signal. 2011, 4, ra51. [Google Scholar] [CrossRef]
- Rabalski, A.J.; Gyenis, L.; Litchfield, D.W. Molecular Pathways: Emergence of Protein Kinase CK2 (CSNK2) as a Potential Target to Inhibit Survival and DNA Damage Response and Repair Pathways in Cancer Cells. Clin. Cancer Res. 2016, 22, 2840–2847. [Google Scholar] [CrossRef]
- Rossi, M.; Ruiz de Azua, I.; Barella, L.F.; Sakamoto, W.; Zhu, L.; Cui, Y.; Lu, H.; Rebholz, H.; Matschinsky, F.M.; Doliba, N.M.; et al. CK2 Acts as a Potent Negative Regulator of Receptor-Mediated Insulin Release in Vitro and in Vivo. Proc. Natl. Acad. Sci. USA 2015, 112, E6818–E6824. [Google Scholar] [CrossRef]
- Torrecilla, I.; Spragg, E.J.; Poulin, B.; McWilliams, P.J.; Mistry, S.C.; Blaukat, A.; Tobin, A.B. Phosphorylation and Regulation of a G Protein–Coupled Receptor by Protein Kinase CK2. J. Cell. Biol. 2007, 177, 127–137. [Google Scholar] [CrossRef]
- Luo, J.; Busillo, J.M.; Stumm, R.; Benovic, J.L. G Protein-Coupled Receptor Kinase 3 and Protein Kinase C Phosphorylate the Distal C-Terminal Tail of the Chemokine Receptor CXCR4 and Mediate Recruitment of β-Arrestin. Mol. Pharmacol. 2017, 91, 554–566. [Google Scholar] [CrossRef]
- Bagnato, A.; Rosanò, L. New Routes in GPCR/β-Arrestin-Driven Signaling in Cancer Progression and Metastasis. Front. Pharmacol. 2019, 10, 114. [Google Scholar] [CrossRef]
- Marampon, F.; Di Nisio, V.; Pietrantoni, I.; Petragnano, F.; Fasciani, I.; Scicchitano, B.M.; Ciccarelli, C.; Gravina, G.L.; Festuccia, C.; Del Fattore, A.; et al. Pro-Differentiating and Radiosensitizing Effects of Inhibiting HDACs by PXD-101 (Belinostat) in in Vitro and in Vivo Models of Human Rhabdomyosarcoma Cell Lines. Cancer Lett. 2019, 461, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Petragnano, F.; Pietrantoni, I.; Di Nisio, V.; Fasciani, I.; Del Fattore, A.; Capalbo, C.; Cheleschi, S.; Tini, P.; Orelli, S.; Codenotti, S.; et al. Modulating the Dose-Rate Differently Affects the Responsiveness of Human Epithelial Prostate- and Mesenchymal Rhabdomyosarcoma-Cancer Cell Line to Radiation. Int. J. Radiat. Biol. 2020, 96, 823–835. [Google Scholar] [CrossRef]
- Wang, W.; Han, T.; Tong, W.; Zhao, J.; Qiu, X. Overexpression of GPR35 Confers Drug Resistance in NSCLC Cells by β-Arrestin/Akt Signaling. Onco. Targets Ther. 2018, 11, 6249–6257. [Google Scholar] [CrossRef]
- Scarpa, M.; Molloy, C.; Jenkins, L.; Strellis, B.; Budgett, R.F.; Hesse, S.; Dwomoh, L.; Marsango, S.; Tejeda, G.S.; Rossi, M.; et al. Biased M1 Muscarinic Receptor Mutant Mice Show Accelerated Progression of Prion Neurodegenerative Disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2107389118. [Google Scholar] [CrossRef]
- Bradley, S.J.; Molloy, C.; Valuskova, P.; Dwomoh, L.; Scarpa, M.; Rossi, M.; Finlayson, L.; Svensson, K.A.; Chernet, E.; Barth, V.N.; et al. Biased M1-Muscarinic-Receptor-Mutant Mice Inform the Design of next-Generation Drugs. Nat. Chem. Biol. 2020, 16, 240–249. [Google Scholar] [CrossRef]
- Yang, N.J.; Hinner, M.J. Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. In Site-Specific Protein Labeling; Gautier, A., Hinner, M.J., Eds.; Springer: New York, NY, USA, 2015; Volume 1266, pp. 29–53. ISBN 9781493922710. [Google Scholar]
- Yu, H.; Dickson, E.J.; Jung, S.-R.; Koh, D.-S.; Hille, B. High Membrane Permeability for Melatonin. J. Gen. Physiol. 2016, 147, 63–76. [Google Scholar] [CrossRef]
- Giorgi, E.P.; Stein, W.D. The Transport of Steroids into Animal Cells in Culture. Endocrinology 1981, 108, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Nicolussi, S.; Gertsch, J. Endocannabinoid Transport Revisited. In Vitamins & Hormones; Elsevier: Amsterdam, The Netherlands, 2015; Volume 98, pp. 441–485. ISBN 9780128030080. [Google Scholar]
- Schuster, V.L. Prostaglandin Transport. Prostaglandins Other Lipid Mediat. 2002, 68–69, 633–647. [Google Scholar] [CrossRef]
- Goto, H.; Miyamoto, M.; Kihara, A. Direct Uptake of Sphingosine-1-Phosphate Independent of Phospholipid Phosphatases. J. Biol. Chem. 2021, 296, 100605. [Google Scholar] [CrossRef] [PubMed]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB1 Receptors Regulate Neuronal Energy Metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef]
- Jong, Y.-J.I.; Kumar, V.; Kingston, A.E.; Romano, C.; O’Malley, K.L. Functional Metabotropic Glutamate Receptors on Nuclei from Brain and Primary Cultured Striatal Neurons. J. Biol. Chem. 2005, 280, 30469–30480. [Google Scholar] [CrossRef]
- Jong, Y.-J.I.; Schwetye, K.E.; O’Malley, K.L. Nuclear Localization of Functional Metabotropic Glutamate Receptor MGlu1 in HEK293 Cells and Cortical Neurons: Role in Nuclear Calcium Mobilization and Development. J. Neurochem. 2007, 101, 458–469. [Google Scholar] [CrossRef]
- Gründemann, D.; Schechinger, B.; Rappold, G.; Schömig, E. Molecular Identification of the Corticosterone-Sensitive Extraneuronal Catecholamine Transporter. Nat. Neurosci. 1998, 1, 349–351. [Google Scholar] [CrossRef]
- Eisenhofer, G. The Role of Neuronal and Extraneuronal Plasma Membrane Transporters in the Inactivation of Peripheral Catecholamines. Pharmacol. Ther. 2001, 91, 35–62. [Google Scholar] [CrossRef]
- Buu, N.T.; Hui, R.; Falardeau, P. Norepinephrine in Neonatal Rat Ventricular Myocytes: Association with the Cell Nucleus and Binding to Nuclear A1- and β-Adrenergic Receptors. J. Mol. Cell. Cardiol. 1993, 25, 1037–1046. [Google Scholar] [CrossRef]
- Jonker, J.W.; Schinkel, A.H. Pharmacological and Physiological Functions of the Polyspecific Organic Cation Transporters: OCT1, 2, and 3 (SLC22A1-3). J. Pharmacol. Exp. Ther. 2004, 308, 2–9. [Google Scholar] [CrossRef]
- Wright, C.D.; Chen, Q.; Baye, N.L.; Huang, Y.; Healy, C.L.; Kasinathan, S.; O’Connell, T.D. Nuclear A1-Adrenergic Receptors Signal Activated ERK Localization to Caveolae in Adult Cardiac Myocytes. Circ. Res. 2008, 103, 992–1000. [Google Scholar] [CrossRef]
- Benton, K.C.; Wheeler, D.S.; Kurtoglu, B.; Ansari, M.B.Z.; Cibich, D.P.; Gonzalez, D.A.; Herbst, M.R.; Khursheed, S.; Knorr, R.C.; Lobner, D.; et al. Norepinephrine Activates β 1 -adrenergic Receptors at the Inner Nuclear Membrane in Astrocytes. Glia 2022, 70, 1777–1794. [Google Scholar] [CrossRef]
- Zhu, T.; Gobeil, F.; Vazquez-Tello, A.; Leduc, M.; Rihakova, L.; Bossolasco, M.; Bkaily, G.; Peri, K.; Varma, D.R.; Orvoine, R.; et al. Intracrine Signaling through Lipid Mediators and Their Cognate Nuclear G-Protein-Coupled Receptors: A Paradigm Based on PGE 2, PAF, and LPA 1 ReceptorsThis Paper Is One of a Selection of Papers Published in This Special Issue, Entitled The Nucleus: A Cell Within A Cell. Can. J. Physiol. Pharmacol. 2006, 84, 377–391. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, K.P.; Rhee, H.J.; Das, S.; Rafter, J.D.; Oh, Y.S.; Cho, W. Internalized Group V Secretory Phospholipase A2 Acts on the Perinuclear Membranes. J. Biol. Chem. 2002, 277, 9358–9365. [Google Scholar] [CrossRef]
- Vazquez-Tello, A.; Fan, L.; Hou, X.; Joyal, J.-S.; Mancini, J.A.; Quiniou, C.; Clyman, R.I.; Gobeil, F.; Varma, D.R.; Chemtob, S. Intracellular-Specific Colocalization of Prostaglandin E 2 Synthases and Cyclooxygenases in the Brain. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2004, 287, R1155–R1163. [Google Scholar] [CrossRef][Green Version]
- Rubio-Aliaga, I.; Daniel, H. Peptide Transporters and Their Roles in Physiological Processes and Drug Disposition. Xenobiotica 2008, 38, 1022–1042. [Google Scholar] [CrossRef]
- Groneberg, D.A.; Döring, F.; Nickolaus, M.; Daniel, H.; Fischer, A. Expression of PEPT2 Peptide Transporter MRNA and Protein in Glial Cells of Rat Dorsal Root Ganglia. Neurosci. Lett. 2001, 304, 181–184. [Google Scholar] [CrossRef]
- Pacheco, M.F.; Woodward, D.J.; McKelvy, J.F.; Griffin, W.S.T. TRH in the Rat Cerebellum: II. Uptake by Cerebellar Slices. Peptides 1981, 2, 283–288. [Google Scholar] [CrossRef]
- Baratti-Elbaz, C.; Ghinea, N.; Lahuna, O.; Loosfelt, H.; Pichon, C.; Milgrom, E. Internalization and Recycling Pathways of the Thyrotropin Receptor. Mol. Endocrinol. 1999, 13, 1751–1765. [Google Scholar] [CrossRef]
- Conti, F.; Sertic, S.; Reversi, A.; Chini, B. Intracellular Trafficking of the Human Oxytocin Receptor: Evidence of Receptor Recycling via a Rab4/Rab5 “Short Cycle”. Am. J. Physiol. -Endocrinol. Metab. 2009, 296, E532–E542. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, C.G.; Bussolati, G.; Bosco, M.; Kimura, T.; Pizzorno, M.C.; Chernin, M.I.; Cassoni, P.; Novak, J.F. Constitutive and Ligand-Induced Nuclear Localization of Oxytocin Receptor. J. Cell. Mol. Med. 2007, 11, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Smit, M.J.; Vischer, H.F.; Bakker, R.A.; Jongejan, A.; Timmerman, H.; Pardo, L.; Leurs, R. Pharmacogenomic and Structural Analysis of Constitutive G Protein–Coupled Receptor Activity. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 53–87. [Google Scholar] [CrossRef] [PubMed]
- Boivin, B.; Vaniotis, G.; Allen, B.G.; Hébert, T.E. G Protein-Coupled Receptors in and on the Cell Nucleus: A New Signaling Paradigm? J. Recept. Signal Transduct. 2008, 28, 15–28. [Google Scholar] [CrossRef]
- Jakubík, J.; El-Fakahany, E.E. Allosteric Modulation of GPCRs of Class A by Cholesterol. Int. J. Mol. Sci. 2021, 22, 1953. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Ercan, B.; Krshnan, L.; Triebl, A.; Koh, D.H.Z.; Wei, F.-Y.; Tomizawa, K.; Torta, F.T.; Wenk, M.R.; Saheki, Y. Movement of Accessible Plasma Membrane Cholesterol by the GRAMD1 Lipid Transfer Protein Complex. eLife 2019, 8, e51401. [Google Scholar] [CrossRef]
- Ango, F.; Prézeau, L.; Muller, T.; Tu, J.C.; Xiao, B.; Worley, P.F.; Pin, J.P.; Bockaert, J.; Fagni, L. Agonist-Independent Activation of Metabotropic Glutamate Receptors by the Intracellular Protein Homer. Nature 2001, 411, 962–965. [Google Scholar] [CrossRef]
- Tripp, E.; O’Brien, S.; Calebiro, D. Modern Methods to Explore GPCR Signalling in Live Cells. Authorea 2022. [Google Scholar] [CrossRef]
- Adams, S.R.; Harootunian, A.T.; Buechler, Y.J.; Taylor, S.S.; Tsien, R.Y. Fluorescence Ratio Imaging of Cyclic AMP in Single Cells. Nature 1991, 349, 694–697. [Google Scholar] [CrossRef]
- Werthmann, R.C.; Volpe, S.; Lohse, M.J.; Calebiro, D. Persistent CAMP Signaling by Internalized TSH Receptors Occurs in Thyroid but Not in HEK293 Cells. FASEB J. 2012, 26, 2043–2048. [Google Scholar] [CrossRef]
- Dean, T.; Vilardaga, J.-P.; Potts, J.T.; Gardella, T.J. Altered Selectivity of Parathyroid Hormone (PTH) and PTH-Related Protein (PTHrP) for Distinct Conformations of the PTH/PTHrP Receptor. Mol. Endocrinol. 2008, 22, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Gulyás, G.; Tóth, J.T.; Tóth, D.J.; Kurucz, I.; Hunyady, L.; Balla, T.; Várnai, P. Measurement of Inositol 1,4,5-Trisphosphate in Living Cells Using an Improved Set of Resonance Energy Transfer-Based Biosensors. PLoS ONE 2015, 10, e0125601. [Google Scholar] [CrossRef]
- Sato, M.; Ueda, Y.; Umezawa, Y. Imaging Diacylglycerol Dynamics at Organelle Membranes. Nat. Methods 2006, 3, 797–799. [Google Scholar] [CrossRef]
- Thestrup, T.; Litzlbauer, J.; Bartholomäus, I.; Mues, M.; Russo, L.; Dana, H.; Kovalchuk, Y.; Liang, Y.; Kalamakis, G.; Laukat, Y.; et al. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat. Methods 2014, 11, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-F.; Mehta, S.; Zhang, J. Signaling Microdomains in the Spotlight: Visualizing Compartmentalized Signaling Using Genetically Encoded Fluorescent Biosensors. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 587–608. [Google Scholar] [CrossRef]
- Nash, C.A.; Wei, W.; Irannejad, R.; Smrcka, A.V. Golgi Localized Β1-Adrenergic Receptors Stimulate Golgi PI4P Hydrolysis by PLCε to Regulate Cardiac Hypertrophy. eLife 2019, 8, e48167. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, A.R.B.; Plouffe, B.; Cahill, T.J.; Shukla, A.K.; Tarrasch, J.T.; Dosey, A.M.; Kahsai, A.W.; Strachan, R.T.; Pani, B.; Mahoney, J.P.; et al. GPCR-G Protein-β-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 2016, 166, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Jean-Alphonse, F.G.; Wehbi, V.L.; Chen, J.; Noda, M.; Taboas, J.M.; Xiao, K.; Vilardaga, J.-P. β2-Adrenergic Receptor Control of Endosomal PTH Receptor Signaling via Gβγ. Nat. Chem. Biol. 2017, 13, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, E.C.; Mehta, S.; Zhang, J. Genetically Encoded Fluorescent Biosensors Illuminate the Spatiotemporal Regulation of Signaling Networks. Chem. Rev. 2018, 118, 11707–11794. [Google Scholar] [CrossRef]
- Barbier, P.; Colelli, A.; Maggio, R.; Bravi, D.; Corsini, G.U. Pergolide Binds Tightly to Dopamine D2 Short Receptors and Induces Receptor Sequestration. J. Neural Transm. 1997, 104, 867–874. [Google Scholar] [CrossRef]
- Shioda, N.; Yabuki, Y.; Wang, Y.; Uchigashima, M.; Hikida, T.; Sasaoka, T.; Mori, H.; Watanabe, M.; Sasahara, M.; Fukunaga, K. Endocytosis Following Dopamine D2 Receptor Activation Is Critical for Neuronal Activity and Dendritic Spine Formation via Rabex-5/PDGFRβ Signaling in Striatopallidal Medium Spiny Neurons. Mol. Psychiatry 2017, 22, 1205–1222. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, D.; Laifenfeld, D. Mitochondria, Synaptic Plasticity, And Schizophrenia. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2004; Volume 59, pp. 273–296. ISBN 9780123668608. [Google Scholar]
- Carli, M.; Aringhieri, S.; Kolachalam, S.; Longoni, B.; Grenno, G.; Rossi, M.; Gemignani, A.; Fornai, F.; Maggio, R.; Scarselli, M. Is Adult Hippocampal Neurogenesis Really Relevant for the Treatment of Psychiatric Disorders? CN 2021, 19, 1640–1660. [Google Scholar] [CrossRef] [PubMed]
- Capannolo, M.; Fasciani, I.; Romeo, S.; Aloisi, G.; Rossi, M.; Bellio, P.; Celenza, G.; Cinque, B.; Cifone, M.G.; Scarselli, M.; et al. The Atypical Antipsychotic Clozapine Selectively Inhibits Interleukin 8 (IL-8)-Induced Neutrophil Chemotaxis. Eur. Neuropsychopharmacol. 2015, 25, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Carli, M.; Kolachalam, S.; Longoni, B.; Pintaudi, A.; Baldini, M.; Aringhieri, S.; Fasciani, I.; Annibale, P.; Maggio, R.; Scarselli, M. Atypical Antipsychotics and Metabolic Syndrome: From Molecular Mechanisms to Clinical Differences. Pharmaceuticals 2021, 14, 238. [Google Scholar] [CrossRef]
- Jutila, A.; Söderlund, T.; Pakkanen, A.L.; Huttunen, M.; Kinnunen, P.K.J. Comparison of the Effects of Clozapine, Chlorpromazine, and Haloperidol on Membrane Lateral Heterogeneity. Chem. Phys. Lipids 2001, 112, 151–163. [Google Scholar] [CrossRef]
- Steinkopf, S.; Schelderup, A.K.; Gjerde, H.L.; Pfeiffer, J.; Thoresen, S.; Gjerde, A.U.; Holmsen, H. The Psychotropic Drug Olanzapine (Zyprexa®) Increases the Area of Acid Glycerophospholipid Monolayers. Biophys. Chem. 2008, 134, 39–46. [Google Scholar] [CrossRef]
- Liu, X. Overview: Role of Drug Transporters in Drug Disposition and Its Clinical Significance. In Drug Transporters in Drug Disposition, Effects and Toxicity; Liu, X., Pan, G., Eds.; Springer: Singapore, 2019; Volume 1141, pp. 1–12. ISBN 9789811376467. [Google Scholar]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC Transporters in Cancer: More than Just Drug Efflux Pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef]
- Teodori, E.; Dei, S.; Martelli, C.; Scapecchi, S.; Gualtieri, F. The functions and structure of ABC transporters: Implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance (MDR). Curr. Drug Targets 2006, 7, 893–909. [Google Scholar] [CrossRef]
- Thomsen, A.R.B.; Jensen, D.D.; Hicks, G.A.; Bunnett, N.W. Therapeutic Targeting of Endosomal G-Protein-Coupled Receptors. Trends Pharmacol. Sci. 2018, 39, 879–891. [Google Scholar] [CrossRef]
- Schrader, J.M.; Irving, C.M.; Octeau, J.C.; Christian, J.A.; Aballo, T.J.; Kareemo, D.J.; Conti, J.; Camberg, J.L.; Lane, J.R.; Javitch, J.A.; et al. The Differential Actions of Clozapine and Other Antipsychotic Drugs on the Translocation of Dopamine D2 Receptors to the Cell Surface. J. Biol. Chem. 2019, 294, 5604–5615. [Google Scholar] [CrossRef]
- Seeman, P. Clozapine, a Fast-Off-D2 Antipsychotic. ACS Chem. Neurosci. 2014, 5, 24–29. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subcellular Localization | Receptor | Reference |
|---|---|---|
| Early endosomes | β2-adrenergic receptor (β2AR) | [26] |
| Calcitonin-gene-related-peptide receptor (CGRPR) | [27] | |
| Calcium-sensing receptor (CaSR) | [28] | |
| Dopamine receptor type 1 (D1R) | [29] | |
| Luteinizing hormone receptor (LHR) | [30] | |
| Neurokinin type 1 receptor (NK1R) | [31] | |
| Parathyroid hormone receptor (PTHR) | [32] | |
| Protease activated receptor 2 (PAR2) | [33] | |
| Vasopressin type 2 receptor (V2R) | [34] | |
| Nucleus | α1A-adrenergic receptor (α1A-AR) | [35] |
| α1B-adrenergic receptor (α1B-AR) | [35] | |
| Adenosine A1 receptor (ADORA1) | [36] | |
| Adenosine A2B (ADORA2B) | [36] | |
| Angiotensin AT1A receptor (AT1AR) | [37] | |
| Apelin receptor (APJ) | [38] | |
| Bradykinin B2 receptor (BKR2) | [38] | |
| Cysteine (C)-x-C receptor 4 (CXCR4) | [39] | |
| Oxytocin receptors (OXTR) | [40] | |
| C-C chemokine receptor type 2 (CCR2) | [41] | |
| Arginine vasopressin receptor 1α (AVPR1a) | [41] | |
| Sphingosine 1-phosphate receptor 1(S1P1) | [41] | |
| Mitochondria | Purinoceptor 1 like receptor (P2Y1) | [42] |
| Purinoceptor 2 like receptor (P2Y2) | [42] | |
| Angiotensin II receptor type 1 (AT1R) | [43] | |
| Angiotensin II receptor type 2 (AT2R) | [43] | |
| 5-hydroxytrptamine receptor (5-HTR3 and 5-HTR4) | [44] | |
| Melatonin MT1 receptor (MT1R) | [45] | |
| Cannabinoid type 1 receptor 1 (CB1R) | [46] | |
| Golgi | β1-adrenergic receptor (β1AR) | [47] |
| Sphingosine-1-phosphate 1 receptor (S1P1R) | [48] | |
| Thyroid stimulating hormone receptor (TSHR) | [49] | |
| ER | G Protein-Coupled Estrogen Receptor 1 (GPR30) | [50] |
| Metabotropic glutamate receptor 5 (mGluR5) | [25] | |
| Exosomes | G Protein-Coupled Receptor Class C Group 5 Member B (GPRC5B) | [51] |
| Spindle poles | Olfactory receptor 2A4 (OR2A4) | [24] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fasciani, I.; Carli, M.; Petragnano, F.; Colaianni, F.; Aloisi, G.; Maggio, R.; Scarselli, M.; Rossi, M. GPCRs in Intracellular Compartments: New Targets for Drug Discovery. Biomolecules 2022, 12, 1343. https://doi.org/10.3390/biom12101343
Fasciani I, Carli M, Petragnano F, Colaianni F, Aloisi G, Maggio R, Scarselli M, Rossi M. GPCRs in Intracellular Compartments: New Targets for Drug Discovery. Biomolecules. 2022; 12(10):1343. https://doi.org/10.3390/biom12101343
Chicago/Turabian StyleFasciani, Irene, Marco Carli, Francesco Petragnano, Francesco Colaianni, Gabriella Aloisi, Roberto Maggio, Marco Scarselli, and Mario Rossi. 2022. "GPCRs in Intracellular Compartments: New Targets for Drug Discovery" Biomolecules 12, no. 10: 1343. https://doi.org/10.3390/biom12101343
APA StyleFasciani, I., Carli, M., Petragnano, F., Colaianni, F., Aloisi, G., Maggio, R., Scarselli, M., & Rossi, M. (2022). GPCRs in Intracellular Compartments: New Targets for Drug Discovery. Biomolecules, 12(10), 1343. https://doi.org/10.3390/biom12101343