Proeryptotic Activity of 4-Hydroxynonenal: A New Potential Physiopathological Role for Lipid Peroxidation Products

 ,
,  ,
,  ,
,  , ,
, , 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Red Blood Cells and Treatment
2.2. Flow Cytometry
2.2.1. Measurement of Phosphatidylserine (PS) Externalization and Forward Scatter (FSC)
2.2.2. Measurement of Intracellular Reactive Oxygen and Nitrogen Species
2.2.3. Measurement of Cytosolic Ca++
2.2.4. Measurement of Ceramide
2.3. Haemolysis
2.4. Western Blotting
2.5. Scanning Electron Microscopy (SEM)
2.6. Glutathione Measurements
2.7. PGE2 Measurements
2.8. Endothelial Cell Culture
2.8.1. Adherence Assay of Eryptotic Rbcs
2.8.2. Measurements of ICAM-1 Expression
2.9. Statistical Analysis
3. Results and Discussion
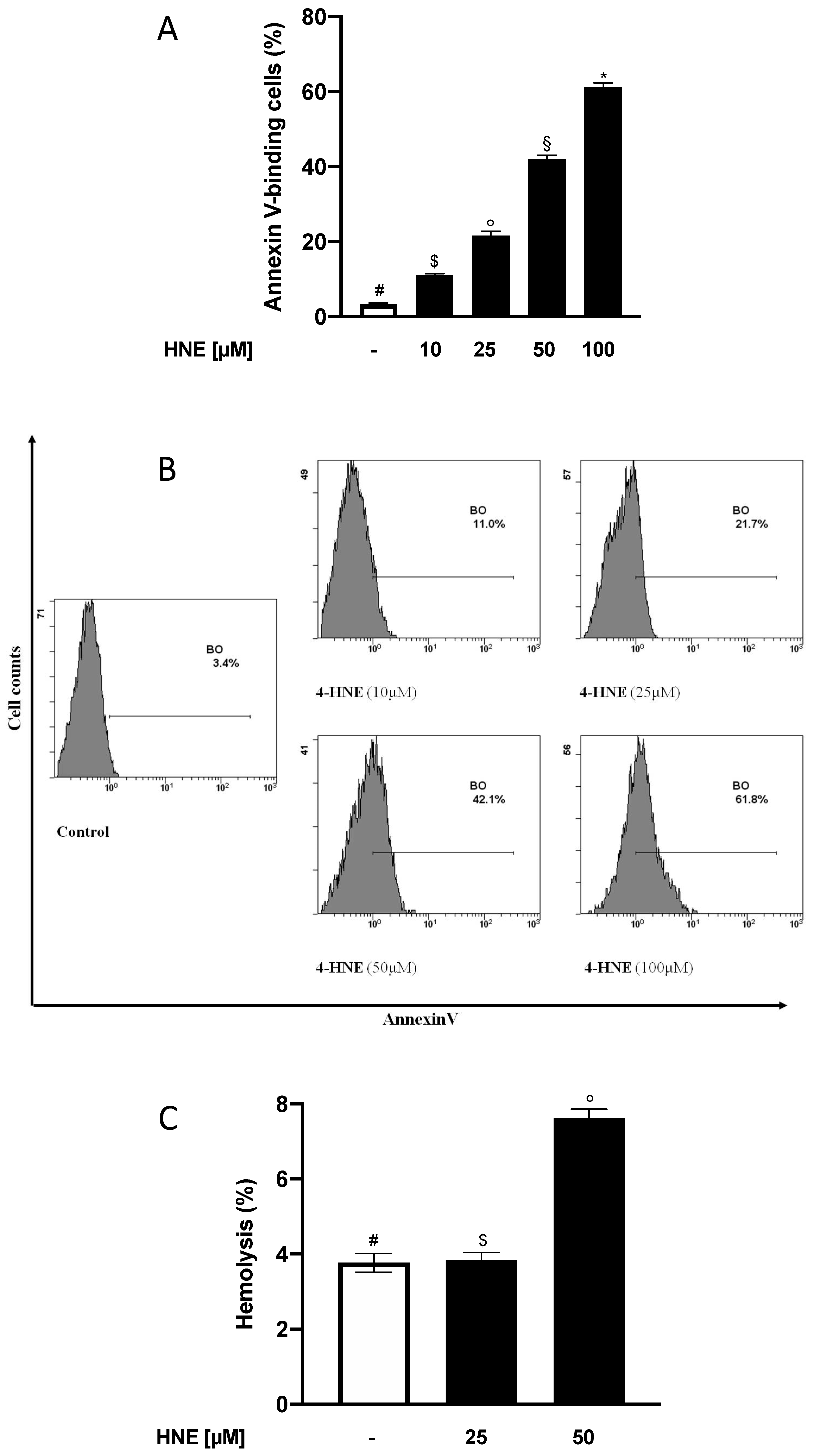
3.1. HNE Induces PS Externalization in Human RBCs
3.2. HNE Induces a Low Grade of Haemolysis in Human RBCs
3.3. HNE Induces Morphological Changes in Human RBCs
3.4. HNE Induces Cleavage of Procaspase 3 in Human RBCs
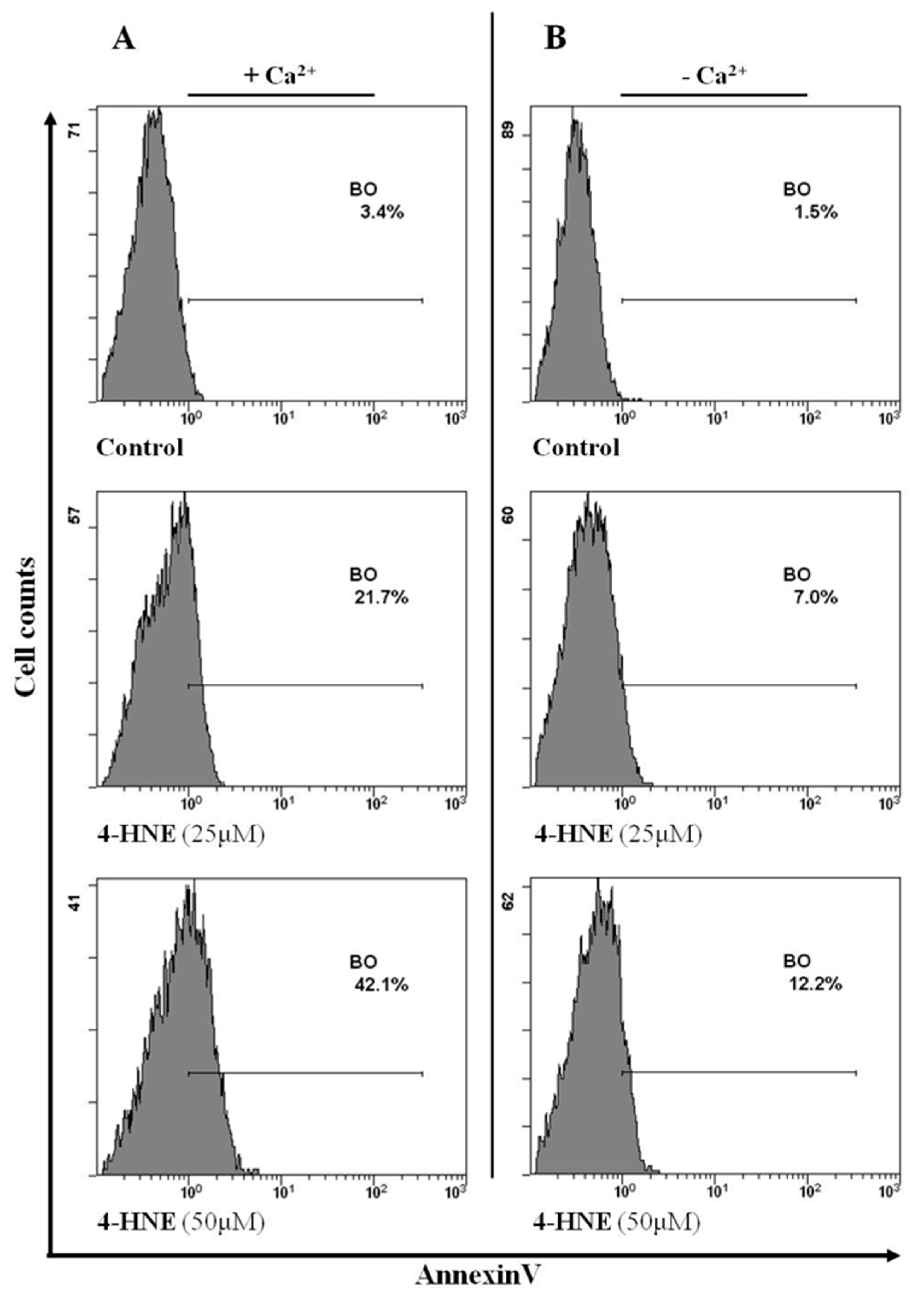
3.5. Intracellular Ca++ Variations Are Involved in the HNE-Induced Eryptosis in Human Rbcs
3.6. Endocellular Redox Variations Are Not Involved in the HNE-Induced Eryptosis in Human RBCs
3.7. PGHS Activation and PGE2 Levels Are Involved in the HNE-Induced Eryptosis in Human RBCs
3.8. Increased Levels of Endocellular Ceramide Are Involved in the HNE-Induced Eryptosis in Human RBCs
3.9. HNE-Induced Eryptosis in Human RBCs is Associated with Endothelial Cell Adhesion and Dysfunction
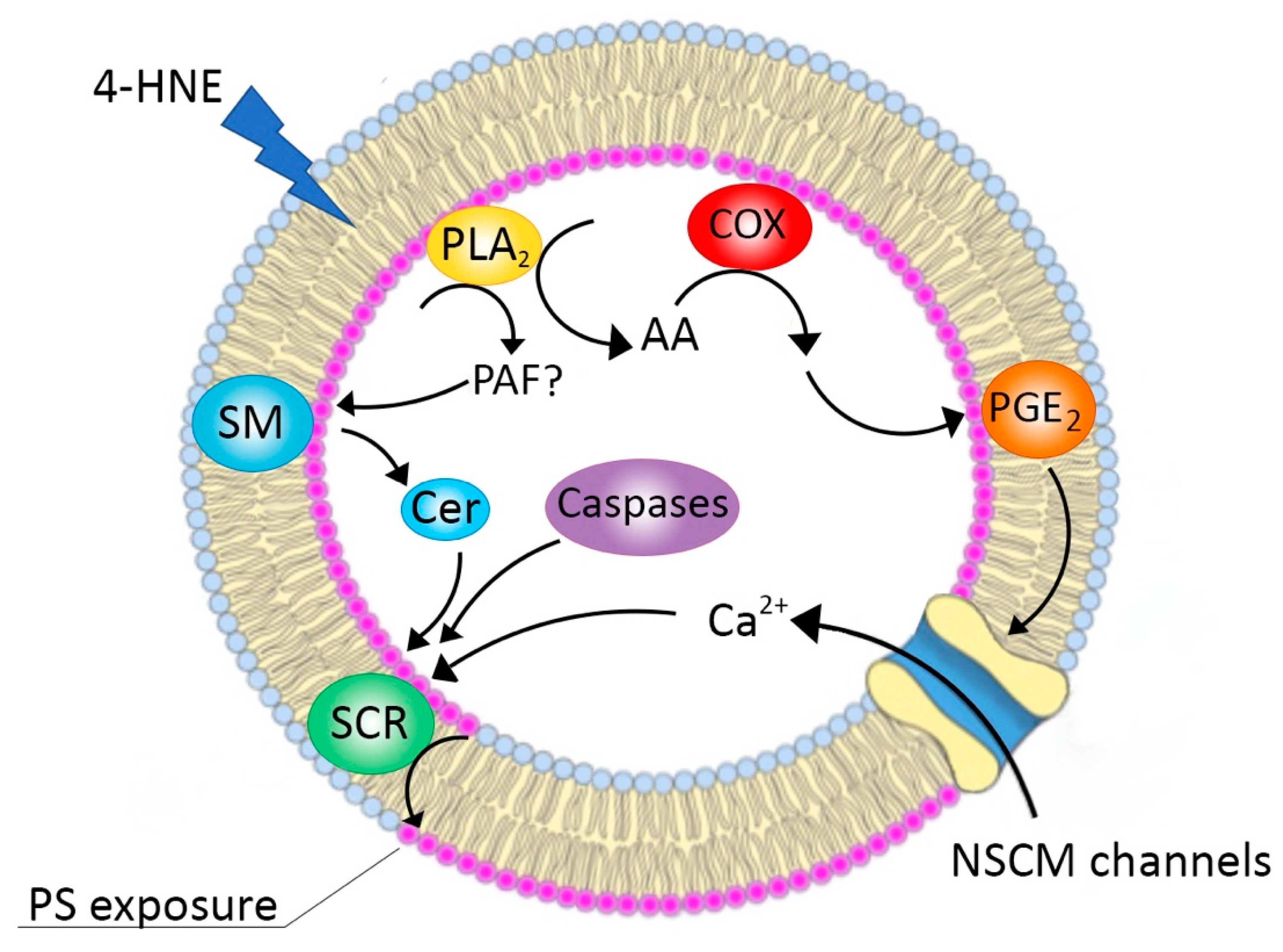
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lang, E.; Qadri, S.M.; Lang, F. Killing me softly—Suicidal erythrocyte death. Int. J. Biochem. Cell Biol. 2012, 44, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, M.C.; Wagner-Britz, L.; Nguyen, D.B.; Asanidze, S.; Mutua, J.; Mohamed, N.; Hanf, B.; Ghashghaeinia, M.; Kaestner, L.; Bernhardt, I. Novel Insights in the Regulation of Phosphatidylserine Exposure in Human Red Blood Cells. Cell. Physiol. Biochem. 2016, 39, 1941–1954. [Google Scholar] [CrossRef] [PubMed]
- Qadri, S.M.; Bissinger, R.; Solh, Z.; Oldenborg, P.A. Eryptosis in health and disease: A paradigm shift towards understanding the (patho)physiological implications of programmed cell death of erythrocytes. Blood Rev. 2017, 31, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Du Plooy, J.N.; Bester, J. A Comprehensive Review on Eryptosis. Cell. Physiol. Biochem. 2016, 39, 1977–2000. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Lang, E.; Foeller, M. Physiology and pathophysiology of eryptosis. Transfus. Med. Hemother. 2012, 39, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Lang, F. Triggers, inhibitors, mechanisms, and significance of eryptosis: The suicidal erythrocyte death. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Sarang, Z.; Mádi, A.; Koy, C.; Varga, S.; Glocker, M.O.; Ucker, D.S.; Kuchay, S.; Chishti, A.H.; Melino, G.; Fésüs, L.; et al. Tissue transglutaminase (TG2) facilitates phosphatidylserine exposure and calpain activity in calcium-induced death of erythrocytes. Cell Death Differ. 2007, 14, 1842–1844. [Google Scholar] [CrossRef][Green Version]
- Esterbauer, H.; Muskiet, F.; Horrobin, D.F. Cytotoxicity and genotoxicity of lipid-oxidation products. Am. J. Clin. Nutr. 1993, 57, 779S–786S. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Riahi, Y.; Cohen, G.; Shamni, O.; Sasson, S. Signaling and cytotoxic functions of 4-hydroxyalkenals. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E879–E886. [Google Scholar] [CrossRef]
- Subramaniam, R.; Roediger, F.; Jordan, B.; Mattson, M.P.; Keller, J.N.; Waeg, G.; Butterfield, D.A. The Lipid Peroxidation Product, 4-Hydroxy-2-trans-Nonenal, Alters the Conformation of Cortical Synaptosomal Membrane Proteins. J. Neurochem. 2002, 69, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Strohmaier, H.; Hinghofer-Szalkay, H.; Jörg Schaur, R. Detection of 4-hydroxynonenal (HNE) as a physiological component in human plasma. J. Lipid Mediat. Cell Signal. 1995, 11, 51–61. [Google Scholar] [CrossRef]
- Calzada, C.; Colas, R.; Guillot, N.; Guichardant, M.; Laville, M.; Véricel, E.; Lagarde, M. Subgram daily supplementation with docosahexaenoic acid protects low-density lipoproteins from oxidation in healthy men. Atherosclerosis 2010, 208, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Vella, R.E.; Soulère, L.; Becchi, M.; Lagarde, M.; Soulage, C.O. Structural and functional changes in human insulin induced by the lipid peroxidation byproducts 4-hydroxy-2-nonenal and 4-hydroxy-2-hexenal. Chem. Res. Toxicol. 2011, 24, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Syslová, K.; Kačer, P.; Kuzma, M.; Najmanová, V.; Fenclová, Z.; Vlčková, Š.; Lebedová, J.; Pelclová, D. Rapid and easy method for monitoring oxidative stress markers in body fluids of patients with asbestos or silica-induced lung diseases. J. Chromatogr. B 2009, 877, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Michel, P.; Sitte, N.; Eggert, W.; Albrecht-Nebe, H.; Esterbauer, H.; Siems, W.G. Increased levels of 4-hydroxynonenal modified proteins in plasma of children with autoimmune diseases. Free Radic. Biol. Med. 1997, 23, 357–360. [Google Scholar] [CrossRef]
- Smathers, R.L.; Fritz, K.S.; Galligan, J.J.; Shearn, C.T.; Reigan, P.; Marks, M.J.; Petersen, D.R. Characterization of 4-HNE modified L-FABP reveals alterations in structural and functional dynamics. PLoS ONE 2012, 7, e38459. [Google Scholar] [CrossRef]
- Forman, H.J. Reactive oxygen species and α,β-unsaturated aldehydes as second messengers in signal transduction. Ann. N. Y. Acad. Sci. 2010, 1203, 35. [Google Scholar] [CrossRef]
- Poli, G.; Schaur, R.J.; Siems, W.G.; Leonarduzzi, G. 4-Hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008, 28, 569–631. [Google Scholar] [CrossRef]
- Shoeb, M.; Ansari, N.; Srivastava, S.; Ramana, K. 4-Hydroxynonenal in the Pathogenesis and Progression of Human Diseases. Curr. Med. Chem. 2013, 21, 230–237. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 2006, 10, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Carini, M.; Yeum, K.J.; Vistoli, G. Novel molecular approaches for improving enzymatic and nonenzymatic detoxification of 4-hydroxynonenal: Toward the discovery of a novel class of bioactive compounds. Free Radic. Biol. Med. 2014, 69, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Tesoriere, L.; Attanzio, A.; Allegra, M.; Livrea, M.A. Dietary indicaxanthin from cactus pear (Opuntia ficus-indica L. Mill) fruit prevents eryptosis induced by oxysterols in a hypercholesterolaemia-relevant proportion and adhesion of human erythrocytes to endothelial cell layers. Br. J. Nutr. 2015, 114, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Herbst, U.; Toborek, M.; Kaiser, S.; Mattson, M.P.; Hennig, B. 4-hydroxynonenal induces dysfunction and apoptosis of cultured endothelial cells. J. Cell. Physiol. 1999, 181, 295–303. [Google Scholar] [CrossRef]
- Liu, W.; Kato, M.; Akhand, A.A.; Hayakawa, A.; Suzuki, H.; Miyata, T.; Kurokawa, K.; Hotta, Y.; Ishikawa, N.; Nakashima, I. 4-hydroxynonenal induces a cellular redox status-related activation of the caspase cascade for apoptotic cell death. J. Cell Sci. 2000, 113, 635–641. [Google Scholar] [PubMed]
- Awasthi, Y.C.; Sharma, R.; Cheng, J.Z.; Yang, Y.; Sharma, A.; Singhal, S.S.; Awasthi, S. Role of 4-hydroxynonenal in stress-mediated apoptosis signaling. Mol. Asp. Med. 2003, 24, 219–230. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, R.; Chaudhary, P.; Vatsyayan, R.; Pearce, V.; Jeyabal, P.V.S.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. 4-Hydroxynonenal induces p53-mediated apoptosis in retinal pigment epithelial cells. Arch. Biochem. Biophys. 2008, 480, 85–94. [Google Scholar] [CrossRef]
- Sharma, R.; Sharma, A.; Dwivedi, S.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. 4-Hydroxynonenal self-limits Fas-mediated DISC-independent apoptosis by promoting export of Daxx from the nucleus to the cytosol and its binding to Fas. Biochemistry 2008, 47, 143–156. [Google Scholar] [CrossRef]
- Awasthi, Y.C.; Sharma, R.; Sharma, A.; Yadav, S.; Singhal, S.S.; Chaudhary, P.; Awasthi, S. Self-regulatory role of 4-hydroxynonenal in signaling for stress-induced programmed cell death. Free Radic. Biol. Med. 2008, 45, 111–118. [Google Scholar] [CrossRef]
- Biswas, D.; Sen, G.; Biswas Tuli, T. Reduced cellular redox status induces 4-hydroxynonenal-mediated caspase 3 activation leading to erythrocyte death during chronic arsenic exposure in rats. Toxicol. Appl. Pharmacol. 2010, 244, 315–327. [Google Scholar] [CrossRef]
- Vatsyayan, R.; Kothari, H.; Pendurthi, U.R.; Rao, L.V.M. 4-hydroxy-2-nonenal enhances tissue factor activity in human monocytic cells via p38 mitogen-activated protein kinase activation-dependent phosphatidylserine exposure. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1601–1611. [Google Scholar] [CrossRef] [PubMed]
- Chapple, S.J.; Cheng, X.; Mann, G.E. Effects of 4-hydroxynonenal on vascular endothelial and smooth muscle cell redox signaling and function in health and disease. Redox Biol. 2013, 1, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Z.; Li, J.; Gu, D.; Li, S.; Shen, C.; Song, Z. Increased 4-Hydroxynonenal Formation Contributes to Obesity-Related Lipolytic Activation in Adipocytes. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Croze, M.L.; Vella, R.E.; Soulère, L.; Lagarde, M.; Soulage, C.O. The lipid peroxidation by-product 4-hydroxy-2-nonenal (4-HNE) induces insulin resistance in skeletal muscle through both carbonyl and oxidative stress. Endocrinology 2012, 153, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- Tesoriere, L.; Attanzio, A.; Allegra, M.; Cilla, A.; Gentile, C.; Livrea, M.A. Oxysterol mixture in hypercholesterolemia-relevant proportion causes oxidative stress-dependent eryptosis. Cell. Physiol. Biochem. 2014, 34, 1075–1089. [Google Scholar] [CrossRef] [PubMed]
- Rother, R.P.; Bell, L.; Hillmen, P.; Gladwin, M.T. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: A novel mechanism of human disease. J. Am. Med. Assoc. 2005, 293, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Qadri, S.M. Mechanisms and significance of eryptosis, the suicidal death of erythrocytes. Blood Purif. 2012, 33, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Polliack, A. The contribution of scanning electron microscopy in haematology: Its role in defining leucocyte and erythrocyte disorders. J. Microsc. 1981, 123, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.L.; Sahai, E.A.; Yeo, M.; Bosch, M.; Dewar, A.; Olson, M.F. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 2001, 3, 339–345. [Google Scholar] [CrossRef]
- Arashiki, N.; Otsuka, Y.; Ito, D.; Yang, M.; Komatsu, T.; Sato, K.; Inaba, M. The covalent modification of spectrin in red cell membranes by the lipid peroxidation product 4-hydroxy-2-nonenal. Biochem. Biophys. Res. Commun. 2010, 391, 1543–1547. [Google Scholar] [CrossRef]
- Alviz-Amador, A.; Galindo-Murillo, R.; Pérez-González, H.; Rodríguez-Cavallo, E.; Vivas-Reyes, R.; Méndez-Cuadro, D. Effect of 4-HNE Modification on ZU5-ANK Domain and the Formation of Their Complex with β-Spectrin: A Molecular Dynamics Simulation Study. J. Chem. Inf. Model. 2020. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Hasui, Y.; Osawa, T. Covalent attachment of 4-hydroxy-2-nonenal to erythrocyte proteins. J. Biochem. 1997, 122, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Berg, C.P.; Engels, I.H.; Rothbart, A.; Lauber, K.; Renz, A.; Schlosser, S.F.; Schulze-Osthoff, K.; Wesselborg, S. Human mature red blood cells express caspase-3 and caspase-8, but are devoid of mitochondrial regulators of apoptosis. Cell Death Differ. 2001, 8, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Carelli-Alinovi, C.; Pirolli, D.; Giardina, B.; Misiti, F. Protein kinase C mediates caspase 3 activation: A role for erythrocyte morphology changes. Clin. Hemorheol. Microcirc. 2015, 59, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Carelli-Alinovi, C.; Dinarelli, S.; Sampaolese, B.; Misiti, F.; Girasole, M. Morphological changes induced in erythrocyte by amyloid beta peptide and glucose depletion: A combined atomic force microscopy and biochemical study. Biochim. Biophys. Acta Biomembr. 2019, 186, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Grey, J.L.; Kodippili, G.C.; Simon, K.; Low, P.S. Identification of contact sites between ankyrin and band 3 in the human erythrocyte membrane. Biochemistry 2012, 51, 6838–6846. [Google Scholar] [CrossRef]
- Lang, K.S.; Lang, P.A.; Bauer, C.; Duranton, C.; Wieder, T.; Huber, S.M.; Lang, F. Mechanisms of suicidal erythrocyte death. Cell. Physiol. Biochem. 2005, 15, 195–202. [Google Scholar] [CrossRef]
- Föller, M.; Kasinathan, R.S.; Koka, S.; Lang, C.; Shumilina, E.; Birnbaumer, L.; Lang, F.; Huber, S.M. TRPC6 contributes to the Ca 2+ leak of human erythrocytes. Cell. Physiol. Biochem. 2008, 21, 183–192. [Google Scholar] [CrossRef]
- Schwarzer, E.; Arese, P.; Skorokhod, O.A. Role of the lipoperoxidation product 4-hydroxynonenal in the pathogenesis of severe malaria anemia and malaria immunodepression. Oxid. Med. Cell. Longev. 2015. [Google Scholar] [CrossRef]
- Tozoni, S.S.; Dias, G.F.; Bohnen, G.; Grobe, N.; Pecoits-Filho, R.; Kotanko, P.; Moreno-Amaral, A.N. Uremia and hypoxia independently induce eryptosis and erythrocyte redox imbalance. Cell. Physiol. Biochem. 2019, 53, 794–804. [Google Scholar]
- Bissinger, R.; Bhuyan, A.A.M.; Qadri, S.M.; Lang, F. Oxidative stress, eryptosis and anemia: A pivotal mechanistic nexus in systemic diseases. FEBS J. 2019, 286, 826–854. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Lang, F. Mechanisms and pathophysiological significance of eryptosis, the suicidal erythrocyte death. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Cheng, J.Z.; Sharma, R.; Yang, Y.; Singhal, S.S.; Sharma, A.; Saini, M.K.; Singh, S.V.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. Accelerated Metabolism and Exclusion of 4-Hydroxynonenal through Induction of RLIP76 and hGST5.8 Is an Early Adaptive Response of Cells to Heat and Oxidative Stress. J. Biol. Chem. 2001, 276, 41213–41223. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Kempe, D.S.; Myssina, S.; Tanneur, V.; Birka, C.; Laufer, S.; Lang, F.; Wieder, T.; Huber, S.M. PGE2 in the regulation of programmed erythrocyte death. Cell Death Differ. 2005, 12, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, S.; Rossin, D.; Testa, G.; Gamba, P.; Staurenghi, E.; Biasi, F.; Poli, G.; Leonarduzzi, G. Up-regulation of COX-2 and mPGES-1 by 27-hydroxycholesterol and 4-hydroxynonenal: A crucial role in atherosclerotic plaque instability. Free Radic. Biol. Med. 2018, 129, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Zarrouki, B.; Soares, A.F.; Guichardant, M.; Lagarde, M.; Géloën, A. The lipid peroxidation end-product 4-HNE induces COX-2 expression through p38MAPK activation in 3T3-L1 adipose cell. FEBS Lett. 2007, 581, 2394–2400. [Google Scholar] [CrossRef] [PubMed]
- Ramana, K.V.; Fadl, A.A.; Tammali, R.; Reddy, A.B.M.; Chopra, A.K.; Srivastava, S.K. Aldose reductase mediates the lipopolysaccharide-induced release of inflammatory mediators in RAW264.7 murine macrophages. J. Biol. Chem. 2006, 281, 33019–33029. [Google Scholar] [CrossRef] [PubMed]
- Honma, S.; Takahashi, N.; Shinohara, M.; Nakamura, K.; Mitazaki, S.; Abe, S.; Yoshida, M. Amelioration of cisplatin-induced mouse renal lesions by a cyclooxygenase (COX)-2 selective inhibitor. Eur. J. Pharmacol. 2013, 715, 181–188. [Google Scholar] [CrossRef]
- Chen, S.H.; Fahmi, H.; Shi, Q.; Benderdour, M. Regulation of microsomal prostaglandin E2synthase-1 and 5-lipoxygenase-activating protein/5-lipoxygenase by 4-hydroxynonenal in human osteoarthritic chondrocytes. Arthritis Res. Ther. 2010, 12. [Google Scholar] [CrossRef]
- Lang, F.; Gulbins, E.; Lang, P.A.; Zappulla, D.; Föller, M. Ceramide in suicidal death of erythrocytes. Cell. Physiol. Biochem. 2010, 26, 21–28. [Google Scholar] [CrossRef]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Föller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via CXCL16/SR-PSOX. Am. J. Physiol. Cell Physiol. 2012, 302, C644–C651. [Google Scholar] [CrossRef]
- Wautier, J.L.; Wautier, M.P. Molecular basis of erythrocyte adhesion to endothelial cells in diseases. Clin. Hemorheol. Microcirc. 2013, 53, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.; Wolf, S. ICAM-1 signaling in endothelial cells. Pharmacol. Rep. 2009, 61, 22–32. [Google Scholar] [CrossRef]













© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegra, M.; Restivo, I.; Fucarino, A.; Pitruzzella, A.; Vasto, S.; Livrea, M.A.; Tesoriere, L.; Attanzio, A. Proeryptotic Activity of 4-Hydroxynonenal: A New Potential Physiopathological Role for Lipid Peroxidation Products. Biomolecules 2020, 10, 770. https://doi.org/10.3390/biom10050770
Allegra M, Restivo I, Fucarino A, Pitruzzella A, Vasto S, Livrea MA, Tesoriere L, Attanzio A. Proeryptotic Activity of 4-Hydroxynonenal: A New Potential Physiopathological Role for Lipid Peroxidation Products. Biomolecules. 2020; 10(5):770. https://doi.org/10.3390/biom10050770
Chicago/Turabian StyleAllegra, Mario, Ignazio Restivo, Alberto Fucarino, Alessandro Pitruzzella, Sonya Vasto, Maria Antonia Livrea, Luisa Tesoriere, and Alessandro Attanzio. 2020. "Proeryptotic Activity of 4-Hydroxynonenal: A New Potential Physiopathological Role for Lipid Peroxidation Products" Biomolecules 10, no. 5: 770. https://doi.org/10.3390/biom10050770
APA StyleAllegra, M., Restivo, I., Fucarino, A., Pitruzzella, A., Vasto, S., Livrea, M. A., Tesoriere, L., & Attanzio, A. (2020). Proeryptotic Activity of 4-Hydroxynonenal: A New Potential Physiopathological Role for Lipid Peroxidation Products. Biomolecules, 10(5), 770. https://doi.org/10.3390/biom10050770