Abstract
The decreased hepatic drug metabolism (predominately first phase) is one of the essential reasons for numerous side effects and for increased drug toxicity during influenza virus infection (IVI). The present study aims to investigate some mechanisms of the preventive effect of a standardized polyphenol complex from the medicinal plant Geranium sanguineum L. (PPhC) (10 mg/kg nasally). A verified experimental model of IVI A/Aichi/2/68 (H3N2) (4.5 lg LD50) in male ICR (Institute of Cancer Research, USA) mice was used. Changes in hepatic monooxygenase activities as well as nicotinamide adenine dinucleotide phosphate (NADPH)-cytochrome C reductase activity and cytochtome P450 content were studied on days 2, 6, 9, 21 of the infection together with thiobarbituric acid reactive substances in the liver supernatant. Our data clearly demonstrates that IVI affects all components of the electronic chain of cytochrome P-450. N-demethylases and hydroxylases as well as the activity of cytochrome C reductase and cytochtome P-450 content were decreased in the course of the virus infection. This implies that free radicals play an important role not only in the pathogenesis of IVI, but also in the modulation of the hepatic monooxygenase activity. This is also consistent with the established polyphenol complex PPhC from the medicinal plant Geranium sanguineum L. preventive effect against increased thiobarbituric acid reactive substances (TBARS)-levels. PPhC restored most of the monooxygenase activities that were inhibited in IVI animals, even over the control levels, probably via multiple mechanisms that may entail antioxidant activity and selective antiviral and protein-binding effects. In contrast to infected animals, in healthy mice, PPhC showed moderate reversible inhibitory effect on hepatic monooxygenase activities.
1. Introduction
The inhibition of hepatic drug metabolism is one of the main reasons for a number of side effects and increased drug toxicity, observed in the course of influenza virus infection (IVI) [1,2]. Several experimental studies have shown that the first phase of drug metabolism is primarily inhibited by IVI, which is coupled with an increased production of free radicals [3,4,5,6,7,8]. However, the relationship between changed P450 monooxygenase activity and free radicals, generated during IVI remains clear. The successful amelioration of IVI via antioxidant treatment with vitamins (vitamins C, B, and E), flavonoids, and plant extracts supports the possible connection between increased free radical processes and changes in hepatic drug metabolism in the course of influenza infection [9,10]. Such naturally occurring products have shown significant restorative effects on cytochrome P450 (CYP450) monooxygenase activity, which was inhibited in virus-infected mice [3,11,12]. On the basis of these findings, we propose that in addition to the classical antiviral drugs available for the treatment of influenza, some of the investigated plant extracts with antioxidant capacity can be useful in the treatment of IVI and possibly other viral infections [13].
Geranium sanguineum L. is a widespread medicinal plant in Bulgaria that belongs to the family Geraniaceae. The aqueous and alcoholic extracts from the aerial roots are traditionally used by popular medicine in the treatments of several diseases including various infections [14,15].
It has been previously demonstrated that the polyphenolic complex (PPhC), isolated from the Bulgarian medicinal plant Geranium sanguineum L., exhibited significant antioxidant and antiviral activities in vivo and in vitro [14,15]. However, it is not clear how PPhC can modulate impaired hepatic drug metabolism in the course of IVI infection.
The aim of the present study was to evaluate the preventive effect of standardized PPhC on hepatic oxidative drug metabolism in the course of an experimental IVI in mice.
2. Materials and Methods
2.1. Plant Material and Extraction
The extracts of Geranium sanguineum L. were isolated, purified, and analyzed, as previously described [14]. Briefly, the plant was collected between June and August in the flowering stage in the Lyulin Mountain, Bulgaria. A specimen was deposited in the Herbarium of the Institute of Botany at the Bulgarian Academy of Sciences, Sofia, Bulgaria (SOM-5/96). For all in vivo studies, we used a combined (total) ethanol extract from Geranium sanguineum L. Briefly, air-dried aerial roots (500 g) were extracted by repeated extraction with ethanol (3 × 600 mL) using Soxhlet apparatus for 24 h at room temperature. The ethanol extracts were combined and lyophilized to obtain combined yields of 16.7% (flow chart A, Figure 1). In order to analyze the constituents of the standardized ethanol extract of Geranium sanguineum L., a subsequent extraction was performed with diethyl ether (Et2O; fraction F1), chloroform (CHCl3; fraction F2), dichloromethane (CH2Cl2; fraction F3), ethyl acetate (EtOAc; fraction F4), n-butanol (n-ButOH; fraction F5), and water (fraction F6). Finally, each fraction was purified by column chromatography and analyzed by reverse phase high-performance liquid chromatography RP-HPLC (flow chart B, Figure 1).
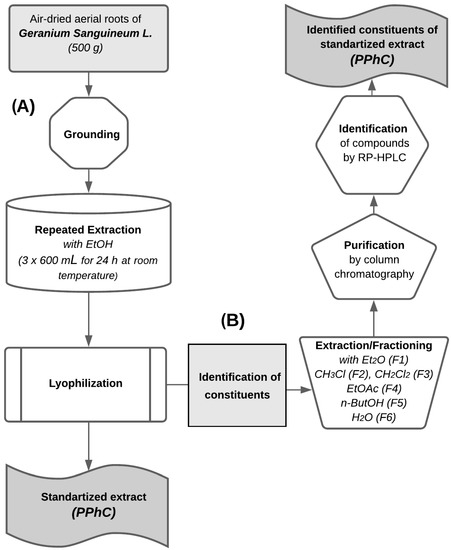
Figure 1.
Flow chart for isolation and analysis of standardized extract of Geranium sanguineum L. (A) Isolation procedure of standardized ethanol extract of Geranium sanguineum L. (B) Analytical procedure for the identification of the individual compounds of standardized extract of Geranium sanguineum L.
2.2. Used Chemicals
The reference compounds quercetin, kaempferol, myricetin, morin, apigenin, retusin, quercetin-3-O-galactoside (hyperoside), (−)-catechin, (+)-catechin, (−)-epicatechin, chlorogenic, ellagic, quinic, and caffeic acids were purchased from Sigma-Aldrich Chemie GmbH (Deisenhofen, Germany). Dr. N. Mahmood and Dr. A.J. Hay (NIMR, Mill Hill, London, UK) provided some of the polyphenol compounds. All solvents, reagents, and other chemicals were obtained from different suppliers (VWR, Sigma-Aldrich, Carl Roth, Merck, and others).
2.3. Extracts Purification and Analysis
(a) Column chromatography (CC) was performed using different sorbents and eluent systems as follows: (i) Sephadex LH-20—eluent system MeOH; (ii) Silica gel—eluent system EtOAc/MeOH in different ratio; and (iii) Polyamide S—eluent system MeOH/water 70:30.
(b) Thin layer chromatography (TLC) was achieved using different sorbents and suitable eluent systems as follows: (i) Polygram Silica gel UV254 (Merck)—eluent system EtOAc/formic acid/AcOH/water 100:11:11:27; (ii) Kieselgel 60 F254 (Merck) with eluent systems EtOAc/formic acid/AcOH 100:11:11 or CHCl3/MeOH 50:50, 75:25 or 85:15; (iii) Polyamide 11 F254 (Merck) with eluent systems EtOAc/formic acid/water 10:2:3 or CHCl3/MeOH 8:1; and (iv) Kieselgel 60 (without a fluorescent indicator) with eluent systems CHCl3/MeOH 10:50 or 85:15. For determination of the different constituents were used the following spray reagents: (i) natural substance-polyethylene glycol (NST-PEG, as a 1.0% solution of diphenylborate aminoethanol in MeOH) for flavonoids and (ii) 0.5% Echtblausalz in MeOH for tannins. The individual compounds were identified by comparison with authentic samples on different sorbents using diverse eluent systems and observed at UV364 before and after spray with NST-PEG for flavonoids and Echtblausalz for catechins. Authentic samples were provided by Prof. E. Wollenweber (Institute of Botany, Darmstadt, Germany).
(c) Reverse phase high-performance liquid chromatography (RP-HPLC) was performed using the following solvents, standards, and chemicals: acetonitrile (HPLC-gradient grade), MeOH (HPLC-grade), formic acid and ortho-phosphoric acid (analytical grade), and bi-distilled water (HPLC-grade). All standards and chemicals were purchased from Merck (Darmstadt, Germany). The reference standards of the flavone aglycones were supplied by Carl Roth GmbH & Co. KG (Karlsruhe, Germany).
The samples were prepared as previously described [14]. Briefly, 0.2 g dried extracts from plant were dissolved in 20 mL MeOH and treated in an ultrasonic bath for 30 min. After re-dissolving in 1.5 M HCl (20 mL), hydrolysis was achieved on a water bath under reflux for 20 min at a temperature of 100 °C. The samples were then cooled at room temperature and diluted with MeOH (1/50).
The chromatographic analysis was performed on a Shimadzu 4A (Schimadzu Corporation, Kyoto, Japan) chromatographic system equipped with a tertiary pump and a Rheodyne injector (100 µL sample loop) coupled with an UV–Vis detector. Knauer Chromatography software and workstation were used for controlling the system and collecting the data. Preparative reversed phase chromatography was carried out on an Atlima C18 Rocket column (57 mm × 7 mm, particle size 3.0 μm) from Alltech Inc. (Lancashire, UK) at room temperature.
For the extract analysis, were used two RP-HPLC methods. A gradient elution (analytical method I) was carried out using bi-distilled water/methanol/formic acid (74.7:25:0.3; v/v/v) as a mobile phase A and acetonitrile/formic acid (99.7:0.3; v/v) as a mobile phase B. The linear gradient elution and run setting were as follows: 100% mobile phase A for 5 min to 100% mobile phase B after an additional 10 min, standing at 100% B for 5 min, and returning to 100% A after another 5 min. The flow rate was 1.0 mL/min and the quantification of catechins was performed at 270 nm. The samples were sonicated until completely dissolved and filtered through a Chromafil filter (0.45 µm from Millipore Ireland B.V., County Cork, Ireland) into a 2.0 mL HPLC glass vial in order to separate the dissolved from particulate material directly prior to injection. Then, 20 µL of each sample was injected into the Atlima C18 Rocket analytical HPLC column. The results obtained from the linear gradient elution are presented in Figure 2A.
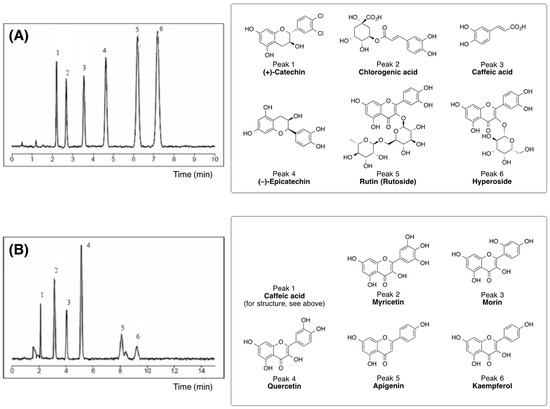
Figure 2.
(A) Standard reverse phase high performance liquid chromatography (RP-HPLC) analysis of Geranium sanguineum L. extract using analytical method I with a linear gradient elution system (mobile phase A: water/methanol/formic acid, 74.7:25:0.3, v/v/v; mobile phase B: acetonitrile/formic acid, 99.7:0.3, v/v) at 270 nm and flow rate 1.0 mL/min [14]. (B) RP-HPLC separation of Geranium sanguineum L. extracts (obtained from ethyl acetate and n-butanol extraction) using separation method II with isocratic gradient elution system (mobile phase: water/acetonitrile (65:35, v/v, adjusted to pH 2.3 with ortho-phosphoric acid) at 254 nm and flow rate 1.0 mL/min [14]. The structures of the identified compounds for each peak and method are respectively indicated.
For isocratic RP-HPLC separation (separation method II), a mobile phase of water/acetonitrile in a ratio of 65:35 (v/v) adjusted to pH 2.3 with ortho-phosphoric acid was used. The mobile phase was filtered through a Chromafil filter (0.45 μm) and de-gassed before sonication. The flow rate was 1.0 mL/min and the chromatograms were recorded at 254 nm selected on the specific UV absorption maximum of the assayed compounds. The results from the isocratic HPLC separation are shown in Figure 2B.
Identification of flavanols and phenolic acids was carried out by comparison of the retention times (tr) and the respective UV absorbance (UVmax in arbitrary units, a.u.) of the unknown peaks with those of the standards [14]. For this purpose, a standard mixture containing ten reference compounds diluted in bi-distilled water/formic acid 99.7:0.3 (v/v) was prepared and analyzed. As references, we used the following compounds: (+)-catechin (0.14 mg/mL), (D)-epicatechin (0.38 mg/mL), chlorogenic acid (0.6 mg/mL), caffeic acid (0.14 mg/mL), rutin (0.8 mg/mL), hyperoside (0.8 mg/mL), myricetin (0.9 mg/mL), morin (0.8 mg/mL), apigenin (0.7 mg/mL), and kaempferol (0.8 mg/mL)). Then, calibration curves were obtained using standard solutions in concentrations between 25% and 125%. All sample preparations from dried extracts and standard solutions were prepared in duplicate and analyzed by HPLC-C18 in triplicate [14].
2.4. Quantification and Qualification
Qualitative determination of polyphenols and quantitative determination of tannins, flavonoids, catechins, and proanthocyanidins were performed as previously described and in accordance with the standards of the European Pharmacopoeia 4.8 [14].
In Vivo Experiments
The experimental model of influenza virus infection A/Aichi/2/68 (H3N2) (4.5 lg LD50) was developed and verified in male albino ICR (Institute of Cancer Research, USA) mice (12 per group). Infected and healthy animals were divided and kept separately in different divisions of standard conditions of the vivarium. All in vivo experiments were performed in compliance with good laboratory practice (GLP). Mice were allocated in groups of 12, housed in single cages under standard vivarium conditions in rooms with standard humidity, heat, and in light regime 12/12 with food and water ad libitum, and opportunities for free movement. Healthy and infected mice were accommodated in separate sections of the vivarium. The animals were under daily care and under constant veterinary supervision with an option for quarantine. The experiments with animals were conducted according to the ethical principles and professional responsibility of scientists established by the Ethics Commission at the Institute of Neurobiology and the Institute of Microbiology at the Bulgarian Academy of Sciences. Four different groups were created. Two of them consisted of healthy and influenza virus infected animals) were pre-treated with standardized PPhC nasally at the dose of 10 mg/kg (in 0.05 mL) 30 min before influenza virus inoculation. The other two different groups (healthy and influenza virus infected) received an equal volume saline instead of PPhC. The animals were sacrificed via decapitation under light ether anesthesia on the 2, 6, 9, and 21 days after viral inoculation. Livers were perfused on ice with 0.1 M Na phosphate buffer. Livers were homogenized in phosphate buffer at a 10-fold dilution with 20% Na phosphate buffer and centrifuged at 9000× g (at 4 °C) to obtain the supernatant (20%). The supernatant was used for the determination of the following parameters: monooxygenase N-demethylase activities (with substrates ethylmorphine (EMND), amidopyrine (APND), and analgin (ANND) according to the method of Nash [16] with the modification by Klinger and Muller [17], and hydroxylase activity using p-nitrophenol (pNF) and aniline (AN) as substrates in accordance with the method of Mazel [18]. NADPH-cytochrome P450 reductase (with substrate cytochrome C, CCR) was conducted according to Roering et al. [19] and cytochrome P450 content (CYP450) according to Matsubara et al. [20] using a modified protocol by Klinger and Muller [16]. The protein content in the blood plasma and homogenates was measured following a known protocol [21]. Thiobarbituric acid reactive substances (TBARS) in liver were determined following the method of Buege and Aust [22], while the total antioxidant activity (TAA) in blood serum was measured using the known procedure of Kovacevic [23].
2.5. Statistical Data Analysis
The Student–Fisher t-test was used for the statistical evaluation of the data. Some correlation coefficients between the parameters of oxidative stress and drug metabolism were calculated according to Brave and Pearson [24].
3. Results
3.1. Biologically Active Constituents of the Standardized Polyphenolic Complex (PPhC)
To investigate the in vivo effects of the biologically active constituents of PPhC, we prepared ethanol extracts from the aerial roots of the medicinal plant Geranium sanguineum L. following a known procedure [14]. In general, the main extract contained 16.15% tannins, 0.126% flavonoids, and 2.1 mg/kg catechins and proanthocyanidins [14]. In addition to the above-mentioned content, we found 12% free sugar moieties and a small amount of amino acids. It was also found (by using the Folin–Ciocalteu reagent) that the total percentage of soluble phenolic constituents of the extract was 34.60% (w/w). The total extract was fractionated by organic solvents with increasing polarity starting with diethyl ether, chloroform, 1,2-dichloroethane, ethyl acetate, n-butanol, and water. Each fraction was purified by CC on silica gel and purified by repeated chromatography on Polyamide S as described previously [14]. The phytochemical characterization of the total ethanol extract and its fractions is presented in Table 1.

Table 1.
Polyphenol content of the total ethanol extract isolated from Geranium sanguineum L. and its fractions.
3.2. Effects of the Viral Infection on Hepatic Monooxygenase Activity in Laboratory Mice
In general, we found that the key components of the electronic chain in the hepatic endoplasmic reticulum of infected animals (the content of cytochrome P450 and CCR activity as well as the most monooxygenase activity) were significantly inhibited by the viral infection. The most pronounced inhibiting effect was observed between the second and the ninth day of IVI for CYP450 content and between the ninth and 21st day for CCR activity. These results are presented in Figure 3 (for CYP450 and CCR) and Figure 4 (for monooxygenases).
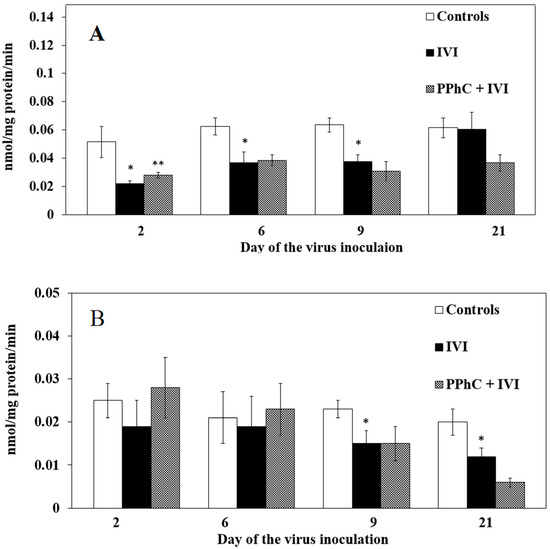
Figure 3.
Panel of liver cytochrome P450 (CYP450) (A) content and NADPH-cytochrome P450 reductase activity with substrate cytochrome C (CCR) (B) activity characterizing the effect of PPhC in IVI-infected mice. Statistical significance (p < 0.05) between the mean healthy groups enzymatic activities vs. IVI models is indicated via the asterisk (*) and between the mean IVI group’s enzymatic activities vs. those treated with the PPhC IVI groups is indicated via a double asterisk (**).
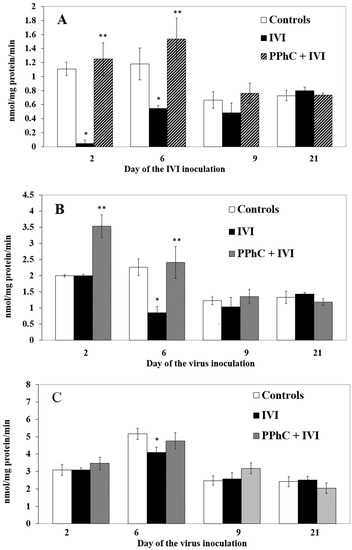
Figure 4.
Panel of liver enzyme of monooxygenase N-demethylase (EMND) (A), APND (B), and ANND (C) activities characterizing the effect of PPhC in IVI-infected mice. Statistical significance (p < 0.05) between the mean healthy group’s enzymatic activities vs. IVI models is indicated via the asterisk (*), and between the mean IVI group’s enzymatic activities vs. those treated with the PPhC IVI groups is indicated via a double asterisk (**).
The monooxygenase activities with different substrates were decreased by IVI. From N-demethylases, most effectively affected EMND on the second and sixth day (by 90% and by 55%, respectively) (cf. Figure 4A), APNN on the 6th day (by 60%) (cf. Figure 4B), and less affected with the substrate analgin, significant only on the sixth day (cf. Figure 4C) (by 16%).
Hepatic hydroxylases with substrates of p-nitrophenol (pNF) and aniline (AN) also exhibited a trend to modification during the course of the infection (cf. Figure 5A,B, respectively). p-NF hydroxylase increased by IVI on the second day even insignificantly, while on the sixth day and 21st day, it remained unchanged. The AN hydroxylase activity decreased significantly on the second (by 60%) and sixth days (by 50%) as well as the 21st day of the IVI (by 33%).
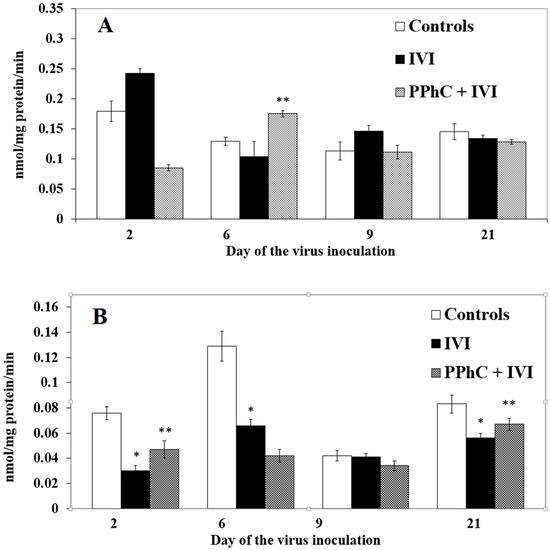
Figure 5.
Panel of the liver enzyme of hydroxylase (with substrates pNF (A) and aniline-AN (B) activities characterizing the effect of PPhC in IVI-infected mice. Statistical significance (p < 0.05) between the mean healthy group’s enzymatic activities vs. IVI models is indicated via the asterisk (*), and between the mean IVI group’s enzymatic activities vs. those treated with PPhC IVI models is indicated with a double asterisk (**).
3.3. Effect of Standardized PPhC on Monooxygenase Activity in Healthy ICR Mice
On the sixth day after pre-treatment, PPhC tended to insignificantly decrease the monooxygenase activities when compared to the untreated controls. This trend was significant only for the CYP450 content and the AN hydroxylase activity (Table 2). The observed effect was accompanied by a moderate pro-oxidant effect of PPhC alone in healthy animals and for TBARS and TAA on the sixth day after PPhC pre-treatment (Table 2).
Table 2.
Differences (in %) between the average scores of the main parameters of oxidative stress (TBARS and TAA) and drug metabolism (CYP450 content and aniline hydroxylase, ANH) achieved in animals treated with PPhC (* p < 0.05) vs. healthy untreated animals used as a control group (100%).
3.4. Preventive Effect of Standardized PPhC Pre-Treatment on Hepatic Drug Metabolism in IVI-Infected Mice
Relative to healthy animals, the opposite effect of PPhC in infected mice was established in hepatic drug metabolism. In infected animals, all parameters of the decreased oxidative drug metabolism were significantly restored differently by a PPhC treatment (cf. Figure 2, Figure 3 and Figure 4).
The CYP450 content was significantly restored by PPhC (up to 40%) only on the second day when compared to the infected controls. The restoring effect of PPhC on CCR activity was maximal on the crucial days of the infection, on the second (by 46%) and on the sixth day (by 20%) and disappeared on the 21st day (Figure 2A,B). The levels of monooxygenases were restored by PPhC, even above the control, being most pronounced on the second and sixth day for EMND (by 108% and by 77%, respectively) (cf. Figure 3A), and for APND (by 76% and by 68%, respectively) (cf. Figure 3B). The restoring effect of PPhC on hydroxylases toward pNF activity was highest on the sixth day (by 55%) (Figure 4A) and toward aniline-AN activity on the second day (by 24%) and 21st day (by 17%) (Figure 4B).
3.5. Correlation between the Changes in the Oxidative Stress Parameters and Liver Monooxygenase Activity in Infected Mice
In general, high TBARS concentrations and decreased TAA levels were found during the course of infection (Figure 6A,B). The observed dynamics of the viral infection revealed that the effect of IVI on TBARS and TAA persisted until day 21st post-infection.
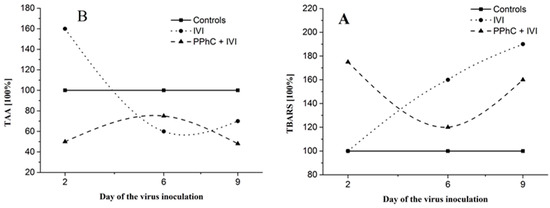
Figure 6.
The effects of IVI on the lipid peroxidation measured by thiobarbituric acid reactive substances (TBARS) levels (A), and on TAA (B). The TBARS and TAA levels in the control plasma (without IVI) are expressed as 100%.
PPhC pre-treatment demonstrated a significant preventive effect toward increased TBARS levels and decreased TAA established in IVI animals (cf. Figure 5B and Figure 6A). The preventive effect of PPhC reached a maximal activity on the second and ninth day (Figure 5B and Figure 6A) in comparison to untreated infected controls. At the same time, a similar dynamic of the preventive effect of PPhC on the oxidative drug metabolism was established, which is presented in Figure 7.
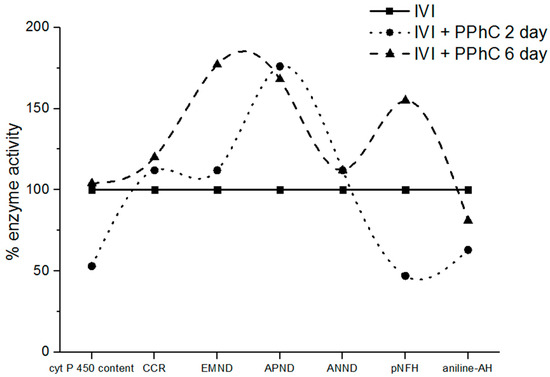
Figure 7.
Panel of liver monooxygenase enzyme activities displaying the recovering effect of PPhC on the second and sixth day of IVI.
3.6. Correlation Coefficients between Parameters of Drug Metabolism and Oxidative Stress
Pearson’s correlation coefficients (R) between the parameters of the oxidative stress and drug metabolism were calculated (Table 3). Our results show a high negative correlation between the oxidative stress (increased TBARS levels) and the decreased drug metabolism (both the content of CYP450 and the activity of CCR) in the infected animals on the sixth day after virus inoculation (see Table 3).
Table 3.
Correlation coefficients between the parameters of oxidative stress (TBARS) and some parameters of drug metabolism in infected animals treated with PPhC (* p < 0.001).
In the PPhC-treated group, the correlation coefficients decreased, especially describing the relation TBARS/CCR. This is probably due to a significant decrease in the oxidative stress level in the PPhC treated group.
4. Discussion
Both clinical evidence and the literature data indicate that influenza infection induces significant changes in pharmacological effects and toxicity of several symptomatic medications used for its management including antipyrine, theophylline, paracetamol, and warfarine [25,26,27]. Our present experimental data also demonstrate that the possible negative consequences of altered effects, higher toxicity, and changed metabolism of some drugs (substrates of monooxygenases) may be due to changes in hepatic monooxygenase activity by IVI and evidence has been generated for decreased drug metabolism during the course of the viral infection. As a result of the total inhibition of the CYP450 chain in the liver, the metabolism of several monooxygenase substrates such as ethylmorphine, amidopyrin, analgin, aniline, and p-nitrophenol were impaired at different degrees of severity. Similar results about other monooxygenase substrates (р-nitroanisole, 7-ethoxycoumarin, cytochrome С) were also reported in other experimental models of IVI [28].
The present study confirms the important role of free radicals in the development of IVI [5,7,12,29,30] in accordance with other studies [31,32,33]. Furthermore, we noticed that the drug metabolism inhibition correlated to established higher free radical production at different time points during IVI. Cytochrome P450 monooxygenase activity with different substrates was decreased and correlated well to higher free radical production in the liver during IVI. Calculated high inverse correlation coefficients between both processes, increased TBARS, and decreased monooxygenase activity were confirmed. Increased TBARS in the result of the infection is comparable to the inhibited enzyme activities, more pronounced on the second and sixth day of IVI. The dynamics of IVI are similar to the dynamics of the modified parameters of drug metabolism and supports the concept that both processes are related. It is likely that the inhibition of drug metabolism is partly due to the activation of free radical processes in the liver and deep damage of the membranes and membrane-bound enzymes as drug metabolizing monooxygenases.
Calculated correlation coefficients between the parameters of the oxidative stress and drug metabolism activity demonstrated a high level of significant relationship between the two processes. Moreover, free radicals play an important role not only in the pathogenesis of IVI, but also in the unspecific modulation of the hepatic monooxygenase activity, confirmed by established high correlation coefficients. We assume that the key points in this setting are the Cytochrome C reductase activity (R = −0.836) and P450 content (R = −0.914) (cf. Table 2).
Antioxidant prevention of the monooxygenase activity in IVI-infected mice and rats was reported with some vitamins, flavonoids, and plant extracts [5,6,7,8,12,29,34,35]. Some plant polyphenols also exhibit protective antioxidant effects during IVI induced oxidative stress [36,37]. We presently confirm the antioxidant effect of standardized PPhC from Geranium sanguineum L. toward increased TBARS levels in infected animals, found in our previous studies [30,37,38]. At the same time, PPhC pre-treatment significantly restored all inhibited hepatic P450 monooxygenase activities in different days of the infection. The modulating effect of PPhC on drug metabolism is probably partly due to its antioxidant potential [38]. PPhC can alleviate the course of the infection via elimination of the destroying effect of free radicals on membrane-bound enzymes (e.g., P450 monooxygenases). Hence, from the findings of our correlation study, it can be suggested that PPhC significantly contributes to the reduction of the oxidative stress produced by an influenza virus infection.
Supported by recent scientific findings, we suggest that PPhC can also act as a membrane-protector, as previously reported by us [30,39,40]. Considering that hepatic P-450 monooxygenases are membrane-bound enzymes, we hypothesize that the membrane protective properties of PPhC are also involved in the protection of drug metabolism in infected animals. It should be noted that a specific antiviral activity of PPhC has been previously reported [39,41,42,43]. PPhC inhibited the reproduction of IVI types A and B in vitro and in vivo and protected mice from mortality in the IVI model [39,41,42,43]. Hence, PPhC may be endowed with a dual beneficial mode of action during IVI infection, entailing direct antiviral action and pharmacological action directed to the positive modulation of metabolic pathways of symptomatic antiviral drugs. It will be interesting to study (i) whether the antiviral effects of PPhC are also exerted in other viral infections; (ii) whether modulation of the decreased hepatic drug metabolism is also induced by other viruses, and furthermore, (iii) how this can modulate the therapeutic responses to the tailored therapeutic agents.
In addition, PPhC possesses additional pleiotropic biological and pharmacological effects that should be taken into account when considering it as a potential antiviral remedy. These include anti-inflammatory and anti-cancer effects, preventive action on neurodegenerative diseases [44,45,46], a stimulating effect on the phagocytic activity of blood polymorphonuclear lymphocytes, and beneficial effect on the spontaneous macrophage NO production, etc. [47,48]. We concluded that the strong protective effect on drug metabolism by PPhC during IVI is due to the combination of the biological activities of the polyphenol compounds. Therefore, we assume that the mechanism of the preventive effect of PPhC on IVI is likely related to its specific antiviral activity as well as to its antioxidant capacity. In addition, the possible protein-binding effect of PPhC on membrane proteins and enzymes (e.g., monooxygenases) reported in previous studies [30,39,40,49] should not be excluded. Established complex biological effects including synergistic combined effects are due to the rich content of the constituents of PPhC, namely tannins (16.15%), flavonoids (0.126%), catechins, and proanthocyanidines (0.105%). Difference in the effects of PPhC in infected versus healthy mice can be explained by its protein binding ability toward the membrane-bond monooxygenase enzymes in the liver. Similar differences in the effects of some antioxidants in healthy and infected animals have been established in several of our previous studies [5].
5. Conclusions
The mechanism of the protective effect of standardized PPhC on inhibited monooxygenase activities in the course of an influenza virus infection (IVI) appears to be complex, involving its antioxidant activity, selective antiviral, and protein-binding effects. We propose that PPhC can be useful in the prevention and treatment of IVI in ameliorating oxidative damage in the liver and restoration of inhibited drug metabolism.
Author Contributions
Conceptualization, L.T. and S.A.; methodology, J.S., N.T.T. and L.T.; software, S.A. and E.P.; validation, N.T.T., J.S. and R.N.; formal analysis, S.A. and E.P.; investigation, R.N.; resources, J.S.; data curation N.T.T.; writing—original draft preparation, S.A. and L.T.; writing—review and editing, F.N. and N.T.T.; visualization, S.A., L.T. and N.T.T.; supervision, L.T.; project administration, J.S.; funding acquisition, A.B. and F.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AN | hydroxylase activity using aniline as substrate |
| ANND | monooxygenase N-demethylase activity with substrate analgin |
| APND | monooxygenase N-demethylase activity with substrate amidopyrine |
| CCR | NADPH-cytochrome P450 reductase activity with substrate cytochrome C |
| CYP450 | cytochrome P450 |
| EMND | monooxygenase N-demethylase activity with substrate ethylmorphine |
| GLP | good laboratory practice |
| IVI | influenza virus infection |
| pNF | hydroxylase activity using p-nitrophenol as a substrate |
| PPhC | polyphenol complex from the medicinal plant Geranium sanguineum L. |
| TAA | total antioxidant activity |
| TBARS | thiobarbituric acid reactive substances |
References
- Enkirch, T.; Sauber, S.; Anderson, D.E.; Gan, E.S.; Gan, E.S.; Kenanov, D.; Maurer-Stroh, S.; von Messling, V. Identification and in vivo efficacy assessment of approved orally bioavailable human host protein-targeting drugs with broad anti-influenza A activity. Front. Immunol. 2019, 10, 1097. [Google Scholar] [CrossRef] [PubMed]
- Pizzomo, A.; Terrier, O.; de Lamballerie, C.N.; Julien, T.; Padey, B.; Traversier, A.; Roche, M.; Hamelin, M.-E.; Rhéaume, C.; Croze, S.; et al. Repurposing of drugs as novel influenza inhibitors from clinical gene expression infection signatures. Front. Immunol. 2019, 10, 60. [Google Scholar]
- Noboru, U.; Toyoda, H. Antioxidant Therapy as a Potential Approach to Severe Influenza-Associated Complications. Molecules 2011, 16, 2032–2052. [Google Scholar]
- Rabovsky, J.; Judy, D.J.; Rodak, D.J.; Petersen, M. Influenza virus-induced alterations of cytochrome P-450 enzyme activities following exposure of mice to coal and diesel particulates. Environ. Res. 1986, 40, 136–144. [Google Scholar] [CrossRef]
- Mileva, M.; Tancheva, L.; Ribarov, S. Determination of processes of lipid peroxidation and liver monooxygenase activity i white mice after treatment with different doses of alpha-tocopheryl-acetate. Eur. J. Pharm. Sci. 1998, 6, 333. [Google Scholar] [CrossRef]
- Mileva, M.; Tantcheva, L.; Bakalova, R.; Galabov, A.S.; Savov, V.; Ribarov, S. Effect of vitamin E on lipid peroxidation and liver monooxygenase activity in experimental influenza virus infection. Toxicol. Lett. 2000, 114, 39–45. [Google Scholar] [CrossRef]
- Mileva, M.; Bakalova, R.; Tancheva, L.; Galabov, A.S. Effect of immobilization, cold and cold-restrain stress on liver monooxygenase activity and lipid peroxidation in influenza-infected mice. Arch. Toxicol. 2002, 76, 96–103. [Google Scholar] [PubMed]
- Tantcheva, L.P.; Stoeva, E.S.; Galabov, A.S.; Braykova, A.A.; Savov, V.M.; Mileva, M.M. Effect of vitamin E and vitamin C combination on experimental influenza virus infection. Method Find. Exp. Clin. Pharmacol. 2003, 25, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.uptodate.com/contents/treatment-of-seasonal-influenza-in-adults/abstract/1-6 (accessed on December 09, 2019).
- Sgarbanti, R.; Amatore, D.; Celestino, I.; Marcocci, M.E.; Fraternale, A.; Ciriolo, M.R.; Magnani, M.; Raffaele, S.; Garaci, E.; Palamara, A.T.; et al. Intracellular Redox State as Target for Anti-Influenza Therapy: Are Antioxidants Always Effective? Curr. Top. Med. Chem. 2014, 14, 2529–2541. [Google Scholar] [CrossRef]
- Mileva, M.; Bakalova, R.; Tancheva, L.; Galabov, A.S.; Ribarov, S. Comparative immunology, microbiology and infectious diseases. Comp. Immunol. Microbiol. Infect. Dis. 2002, 25, 1–11. [Google Scholar] [CrossRef]
- Shumyantseva, V.V.; Shich, E.V.; Machova, A.A.; Bulko, T.V.; Kukes, V.G.; Sizova, O.S.; Ramenskaya, G.V.; Usanov, S.A.; Archakov, A.I. The influence of B-group vitamins on monooxygenase activity of cytochrome P450 3A4: Pharmacokinetics and electro analysis of the catalytic properties. Biochem. Mosc. Suppl. Ser. B Biomed. Chem. 2012, 6, 87–93. [Google Scholar] [CrossRef]
- Serkedjieva, J.; Ivancheva, S. Antiviral activity of species in Geraniaceae. Polyphen. Commun. 1996, 2, 449–450. [Google Scholar]
- Pantev, A.; Ivancheva, S.; Staneva, L.; Serkedjieva, J. Biologically active constituents of a polyphenol extract from Geranium sanguineum L. with anti-influenza activity. Zeitschrift Für Naturforschung C 2006, 61, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Ivancheva, S.; Manolova, N.; Serkedjieva, J.; Dimov, V.; Ivanovska, N. Polyphenols from Bulgarian Medicinal Plants with Anti-Infectious Activity. In Plant Polyphenols. Basic Life Sciences; Hemingway, R.W., Laks, P.E., Eds.; Springer: Boston, MA, USA, 1992; Volume 59. [Google Scholar]
- Nash, T. The colorimetric estimation of formaldehyde by means of the Hantzsch reaction. J. Biol. Chem. 1953, 5, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Klinger, W.; Muller, D. Ethylmorphine N-demethylation by liver homogenate of newborn and adult rats: Enzyme kinetics and age course of Vmax and Km. Acta Biol. Med. Ger. 1977, 36, 1149–1159. [Google Scholar] [PubMed]
- Mazel, P. Fundamentals of Drug Metabolism and Drug Disposition; La, D.B., Mandel, H.G., Way, E.L., Eds.; The Willkins Co: Baltimore, MD, USA, 1971; pp. 546–550. [Google Scholar]
- Roering, D.L.; Mascaro, L.; Aust, S. Microsomal electron transport: Tetrafolium reduction by rat liver microsomal NADP.H-cytochrome c-reductase. Arch. Biochem. 1972, 153, 475. [Google Scholar] [CrossRef]
- Matsubara, T.; Touchi, A.; Tochino, Y.; Sugeno, K. Quantitative determination of cytochrome P-450 in rat liver homogenate. Anal. Biochem. 1976, 75, 596–603. [Google Scholar] [CrossRef]
- Lowry, O.; Rosenbrough, N.; Farr, A.; Randall, R. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–272. [Google Scholar]
- Buege, J.A.; Aust, S.D. Methods in Enzymology; Fleisher, S., Packer, L., Eds.; Academic Press: New York, NY, USA, 1978; Volume 65, pp. 302–310. [Google Scholar]
- Kovacevic, D.; Kovacevic, G.; Djordjevic, V.; Andrejevic, S.; Cosic, V. Method for the measurement of antioxidant activity in human fluids. J. Clin. Pathol. 2001, 54, 356–361. [Google Scholar] [CrossRef]
- Zaytzev, G.N. Brave and Pearson. In Methodology of Biometric Calculations; Nauka Publishing Group: Moscow, Russia, 1973; Volume 256. [Google Scholar]
- Sonne, J.; Bossing, M.; Loft, S.; Andreasen, P.B. Antipyrine clearance in pneumonia. Clin. Pharmacol. Ther. 1985, 337, 701–704. [Google Scholar] [CrossRef]
- Kraemer, M.J.; Furukava, C.T.; Koup, J.R.; Shapiro, G.G.; Pierson, W.E.; Bierman, C.W. Altered theophyline clearance during an influenza B outbreak. Pediatrics 1982, 69, 476–480. [Google Scholar] [PubMed]
- Crump, C.E.; Rollins, B.S.; Hayden, F.G. In vitro Antiviral Activity and Cytotoxicity of Aspirin: Lack of Selective Activity against Influenza a Virus or Rhinovirus. Antivir. Chem. Chemother. 1990, 1, 217–221. [Google Scholar] [CrossRef]
- Gorbunov, N.; Volgarev, A.; Bykova, N.; Prozorovskaia, M. Microsomal hydroxylating system of the mouse liver in toxic forms of influenza infection. Bull. Exp. Biol. Med. 1992, 114, 44–71. [Google Scholar] [CrossRef]
- Krishnaiah, D.; Sarbatly, R.; Nithyanandam, R. A review of the antioxidant potential of medicinal plant species. Food Bioprod. Process. 2011, 89, 217–233. [Google Scholar] [CrossRef]
- Murzakhmetova, M.; Moldakarimov, S.; Tancheva, L.; Abarova, S.; Serkedjieva, J. Рolyphenol-rich extract from Geranium sanguineum- аntioxidant and prooxidant properties. Phytother. Res. 2008, 22, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Peterhans, E. Reactive Oxigen Species and Nitric Oxide in viral diseases. Biol. Trace. Elem. Res. 1997, 56, 107–115. [Google Scholar] [CrossRef]
- Jacoby, D.B.; Augustine, M.K. Choi. Influenza virus induces expression of antioxidant genes in human epithelial cells. Free Radic. Biol. Med. 1994, 16, 821–824. [Google Scholar] [CrossRef]
- Christen, S.; Peterhans, E.; Stocker, R. Antioxidant activities of some tryptophan metabolites: Possible implication for inflammatory diseases. Proc. Natl. Acad. Sci. USA 1990, 87, 2506–2510. [Google Scholar] [CrossRef]
- Akaike, T.; Ando, M.; Oda, T.; Doi, T.; Ijiri, S.; Araki, S.; Maeda, H. Dependence on O2– generation by xanthine oxidase of pathogenesis of influenza virus infection in mice. J. Clin. Investig. 1990, 85, 739–745. [Google Scholar] [CrossRef]
- Kumar, P.; Sharma, S.; Khanna, M.; Raj, H.G. Effect of Quercetin on lipid peroxidation and changes in lung morphology in experimental influenza virus infection. Int. J. Exp. Pathol. 2003, 84, 127–133. [Google Scholar] [CrossRef]
- Savov, V.; Galabov, A.; Tancheva, L.; Mileva, M.; Pavlova, E.; Stoeva, E.; Braykova, A. Effects of rutin and quercetin on monooxygenase activities in experimental influenza virus infection. Exp. Toxicol. Pathol. 2006, 58, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, E.; Simeonova, L.; Serkedjieva, J. Antioxidant activities of Geranium sanguineum L. polyphenolic extract in chemiluminescent model systems. Inorg. Chem. Commun. 2019, 108, 107518. [Google Scholar]
- Sökmen, M.; Angelova, M.; Krumova, E.; Pashova, S.; Ivancheva, S.; Sokmen, A.; Serkedjieva, J. In vitro antioxidant activity of polyphenol extracts with antiviral properties from Geranium sanguineum L. Life Sci. 2005, 76, 2981–2993. [Google Scholar] [CrossRef] [PubMed]
- Serkedjieva, J.; Hay, A.J. In vitro anti-influenza virus activity of a plant preparation from Geranium sanguineum L. Antivir. Res. 1998, 37, 121–130. [Google Scholar] [CrossRef]
- Serkedjieva, J. Antioxidant effects of plant polyphenols: A case study of a polyphenol-rich extract from Geranium sanguineum L. In Reactive Oxygen Species and Antioxidants in Higher Plants; Gupta, S.D., Ed.; Science Publishers: Enfield, UK, 2011. [Google Scholar]
- Serkedjieva, J.; Manolova, N. Plant polyphenol complex inhibits the reproduction of influenza and herpes simplex viruses. Basic Life Sci. 1992, 59, 705–715. [Google Scholar]
- Serkedzhieva, J.; Manolova, N. The antiviral action of a polyphenol complex isolated from the medicinal plant Geranium sanguineum L. VI. Reproduction of the influenza virus pretreated with the polyphenol complex. Acta Microbiol. Bulg. 1988, 22, 16–21. [Google Scholar]
- Serkedjieva, J. Influenza virus variants with reduced susceptibility to inhibition by a polyphenol extract from Geranium sanguineum L. Die Pharm. 2003, 58, 53–57. [Google Scholar]
- Elmann, A.; Mordechay, S.; Rindner, M.; Ravid, U. Anti-neuroinflammatory effects of geranium oil in microglial cells. J. Funct. Foods 2010, 2, 17–22. [Google Scholar] [CrossRef]
- Ren, P.; Ren, X.; Cheng, L.; Xu, L. Frankincense, pine needle and geranium essential oils suppress tumor progression through the regulation of the AMPK/mTOR pathway in breast cancer. Oncol. Rep. 2018, 39, 129–137. [Google Scholar] [CrossRef]
- El-Garawani, I.; El Nabi, S.H.; Nafie, E.; Almeldin, S. Foeniculum Vulgare and Pelargonium Graveolens Essential Oil Mixture Triggers the Cell Cycle Arrest and Apoptosis in MCF-7 Cells. Anticancer Agents Med. Chem. 2019, 19, 1103–1113. [Google Scholar] [CrossRef]
- Toshkova, R.; Nikolova, N.; Ivanova, E.; Ivancheva, S.; Serkedjieva, J. In vitro investigation on the effect of a plant preparation with antiviral activity on the functions of mice phagocyte cells. Die Pharm. 2004, 59, 150–154. [Google Scholar]
- Antonova-Nikolova, S.; Ivanova, I.; Ivancheva, S.; Tsvetkova, R.; Serkedjieva, J. Protease-inhibitory activity of a plant preparation with anti-influenza virus effect. In Proceedings of the Xth Congress of Bulgarian Microbiologists, Plovdiv, Bulgaria, 9–12 October 2002; Volume 1, pp. 358–362. [Google Scholar]
- Sekowski, S.; Ionov, M.; Kaszuba, M.; Mavlyanov, S.; Bryszewska, M.; Zamaraeva, M. Biophysical studies of interaction between hydrolysable tannins isolated from Oenothera gigas and Geranium sanguineum with human serum albumin. Colloids Surf. B Biointerfaces 2014, 123, 623–628. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).