Abstract
The crystal structure of RU60358, C20H21NO3, has been determined using X-ray diffraction to establish the configuration and stereochemistry of the molecule around the C15-C16 triple bond. The compound crystallises in the orthorhombic space group P212121, a = 7.7575, b = 11.3182, c = 21.3529Å, V = 1874.80Å3 and Z = 4. The structure has been refined to a final R = 0.068 for the observed structure factors with I ≥ 3σ (I). The refined structure was found to be significantly non planar. A comparative study, using the ab initio calculations of the structure at B3LYP/6-31G** levels of theory, shows good geometrical agreement with the X-ray diffraction data. Standard deviations between the calculated and experimental bond values have been shown to be 0.01 Å and 0.5° for bond angles. Vibrational wavenumbers were obtained from a normal mode analysis using the ab initio calculations.
1. Introduction
The structures of pyrethroids compounds obtained from experimental X-ray diffraction data has been investigated by several authors [1,2,3,4]. Considerable progress has been made in relating the structure of pyrethroids with their biological activity, but improvement of such concepts requires reliable information on molecular shape (configuration, bond lengths and angles and conformation). Biological activity in pyrethroids is related to molecular structure and depends strongly on the stereochemistry adopted by the asymmetric centers [5,6,7]. In this context the compound RU60358 appears as an useful intermediate in the synthesis of some pyrethroid insecticides [8]. The study of the conformation of this molecule may yield information about the mechanism of its biological activity [9,10]. In this paper, we propose a comparative study between the experimental X-ray diffraction data and the optimized geometry predicted from ab initio molecular orbital calculations performed on the compound RU60358. Vibrational wavenumbers were then consequently predicted and correct assignments obtained.
2. Results and discussion
2.1. Description of the crystal structure
The displacement ellipsoid plot with the numbering scheme for the title compound is shown in Figure 1. Figure 2 shows a perspective view of the crystal packing in the unit cell. Selected X-ray diffraction data summarize bond lengths in Table 1 and bond angles in Table 2.
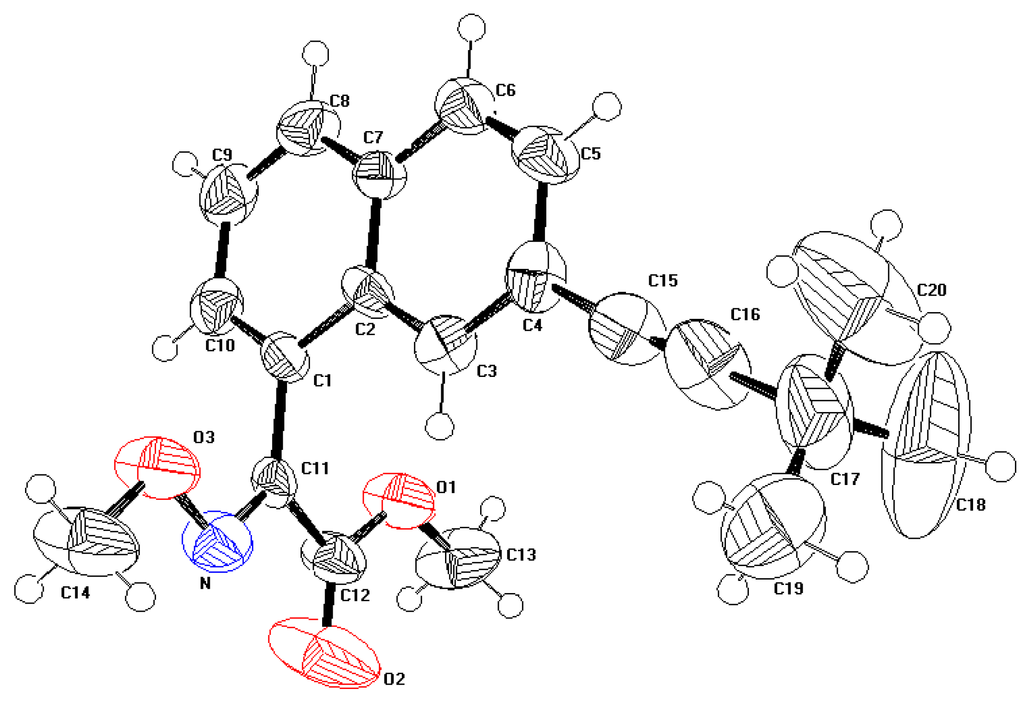
Figure 1.
Perspective view of the molecule showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 50% probability level and H atoms are shown as small spheres of arbitrary radii.
Figure 1.
Perspective view of the molecule showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 50% probability level and H atoms are shown as small spheres of arbitrary radii.
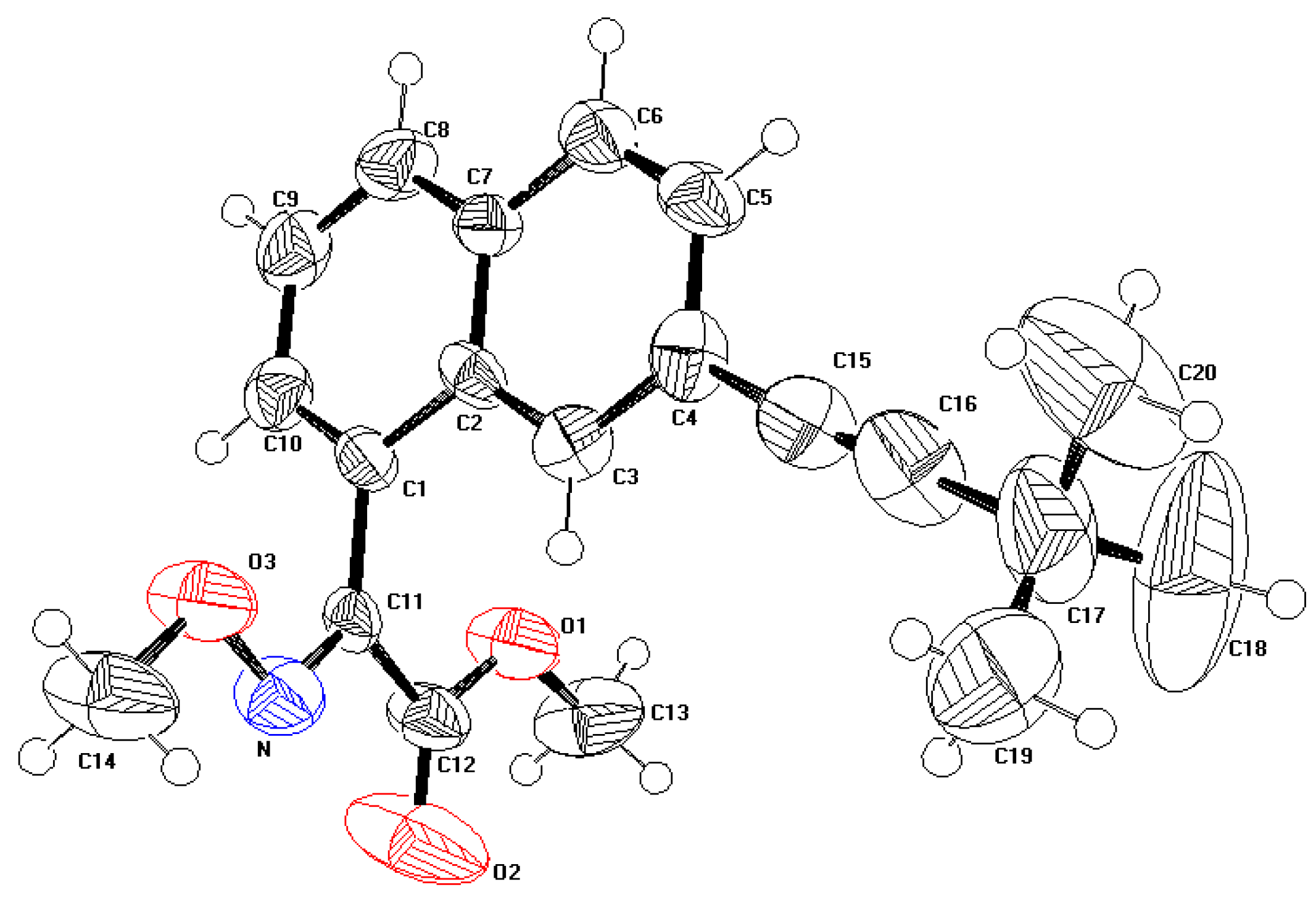
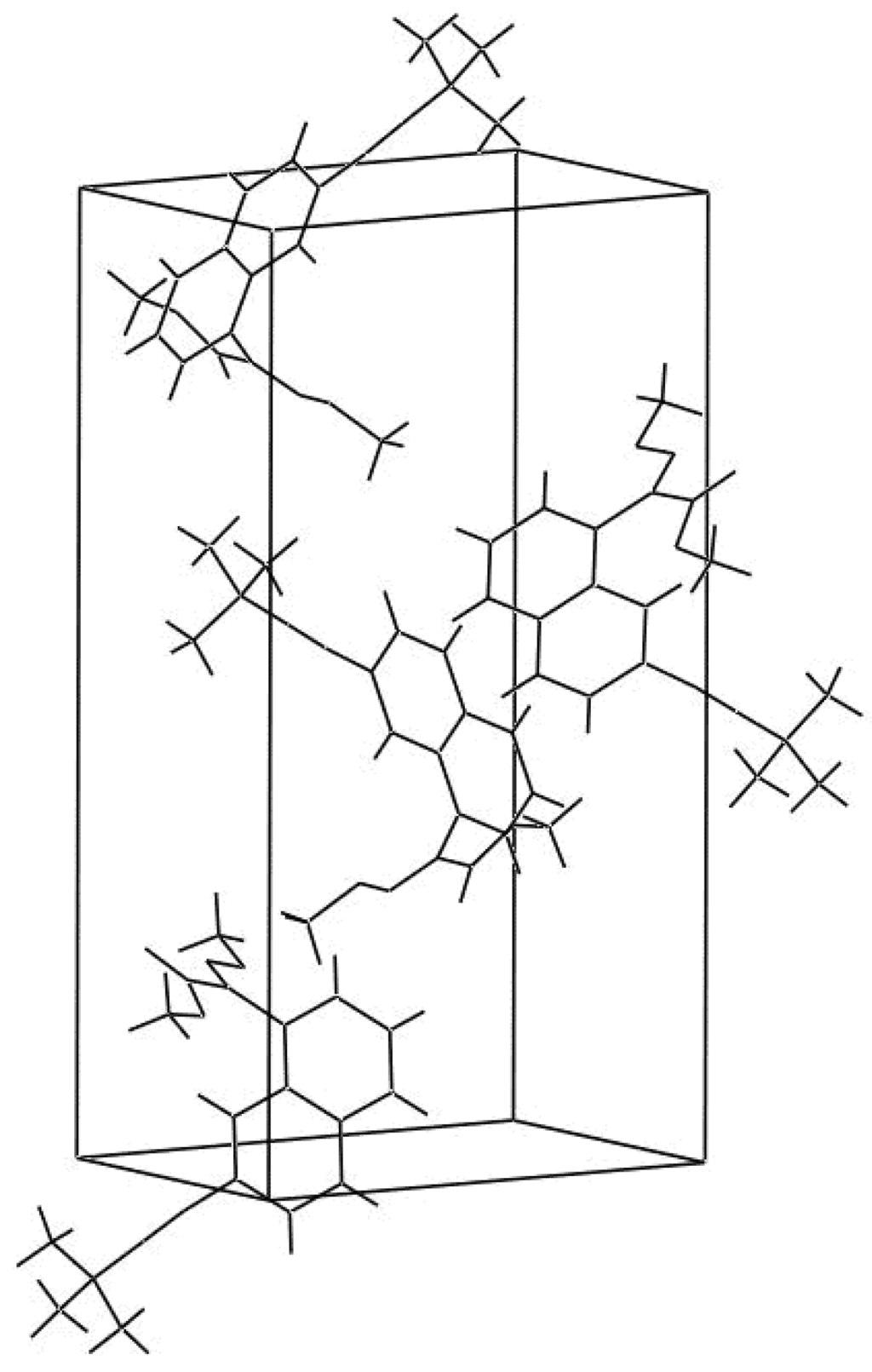
Figure 2.
A perspective view of the crystal packing in the unit cell.
Figure 2.
A perspective view of the crystal packing in the unit cell.
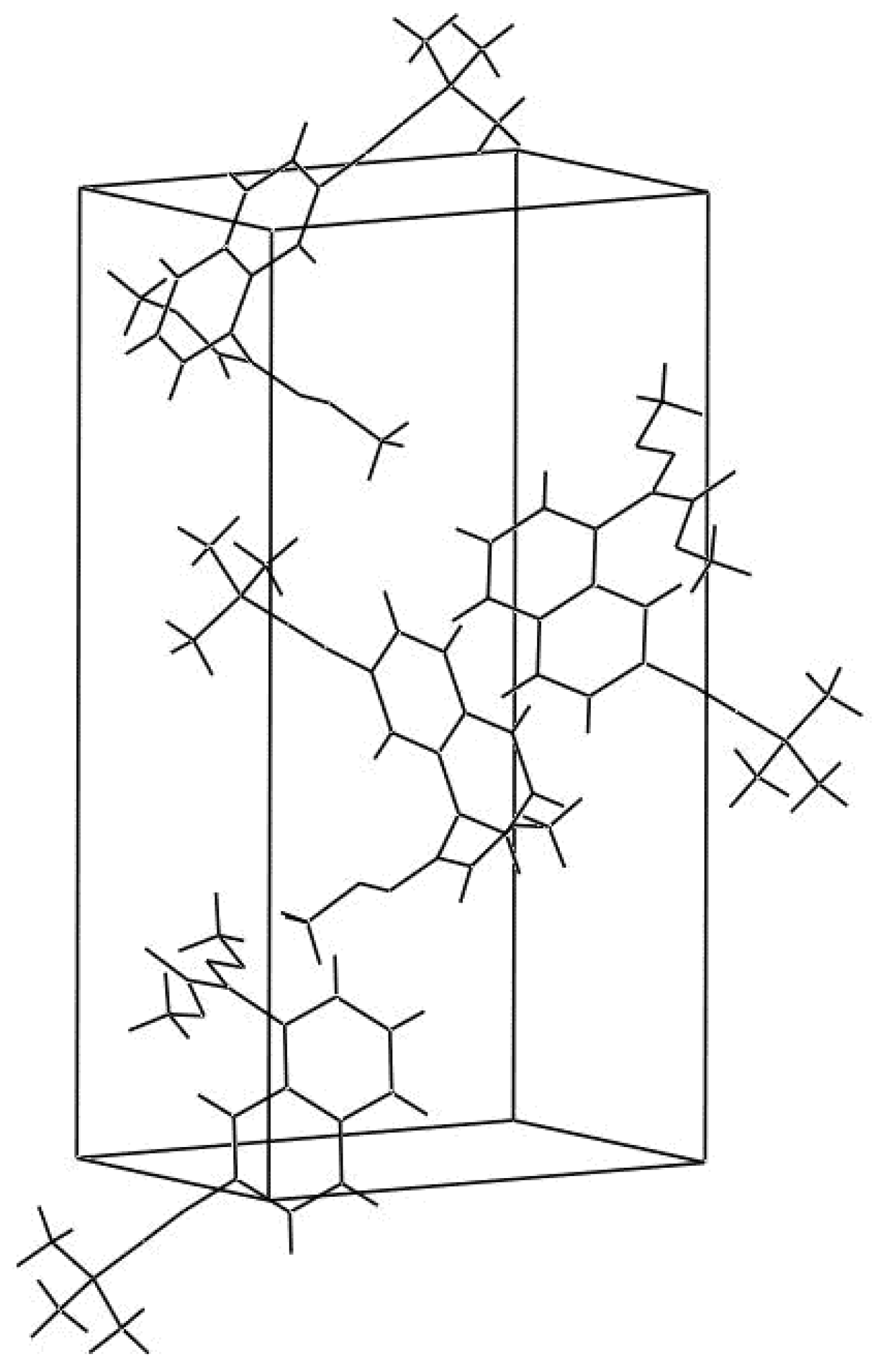

Table 1.
Selected bond distances (Å) by X-ray and theoretical calculations
| Atom 1 | Atom 2 | Distance (Å) | |
|---|---|---|---|
| X-ray | B3LYP/6-31G** | ||
| C1 | C2 | 1.395 (10) | 1.433 |
| C1 | C10 | 1.395 (11) | 1.381 |
| C1 | C11 | 1.474 (12) | 1.472 |
| C2 | C3 | 1.410 (09) | 1.416 |
| C2 | C7 | 1.395 (11) | 1.433 |
| C3 | C4 | 1.395 (13) | 1.388 |
| C4 | C5 | 1.395 (10) | 1.428 |
| C4 | C15 | 1.429 (10) | 1.428 |
| C5 | C6 | 1.395 (10) | 1.371 |
| C6 | C7 | 1.395 (09) | 1.422 |
| C7 | C8 | 1.395 (11) | 1.418 |
| C8 | C9 | 1.395 (11) | 1.375 |
| C9 | C10 | 1.395 (09) | 1.412 |
| C11 | N | 1.278 (13) | 1.290 |
| C11 | C12 | 1.514 (15) | 1.497 |
| C12 | O1 | 1.292 (10) | 1.351 |
| C12 | O2 | 1.152 (10) | 1.210 |
| C13 | O1 | 1.439 (12) | 1.436 |
| N | O3 | 1.376 (09) | 1.374 |
| C14 | O3 | 1.427 (11) | 1.467 |
| C15 | C16 | 1.163 (13) | 1.213 |
| C16 | C17 | 1.467 (12) | 1.459 |
| C17 | C18 | 1.439 (12) | 1.548 |
| C17 | C19 | 1.469 (11) | 1.541 |
| C17 | C20 | 1.531 (13) | 1.549 |
Table 2.
Selected bond angles (°) by X-ray and theoretical calculations
| Atom 1 | Atom 2 | Atom 3 | Angle (°) | |
|---|---|---|---|---|
| X-ray | B3LYP/6-31G** | |||
| C2 | C1 | C10 | 120.0 (8) | 119.8 |
| C2 | C1 | C11 | 123.5 (5) | 121.1 |
| C10 | C1 | C11 | 116.2 (8) | 119.1 |
| C1 | C2 | C3 | 120.2 (6) | 122.6 |
| C1 | C2 | C7 | 120.0 (7) | 118.5 |
| C3 | C2 | C7 | 119.8 (7) | 118.7 |
| C2 | C3 | C4 | 120.0 (5) | 121.5 |
| C3 | C4 | C5 | 120.0 (5) | 119.2 |
| C3 | C4 | C15 | 122.1 (6) | 120.9 |
| C5 | C4 | C15 | 117.9 (8) | 119.9 |
| C4 | C5 | C6 | 120.0 (8) | 120.4 |
| C5 | C6 | C7 | 120.4 (7) | 121.2 |
| C2 | C7 | C6 | 119.8 (9) | 118.8 |
| C2 | C7 | C8 | 120.0 (5) | 119.6 |
| C6 | C7 | C8 | 120.2 (6) | 121.6 |
| C7 | C8 | C9 | 120.0 (5) | 120.6 |
| C8 | C9 | C10 | 120.0 (7) | 120.1 |
| C16 | C17 | C19 | 111.5 (9) | 109.4 |
| C16 | C17 | C20 | 107.7 (9) | 109.3 |
| C18 | C17 | C19 | 110.5 (8) | 109.7 |
| C18 | C17 | C20 | 107.3 (5) | 109.6 |
| C19 | C17 | C20 | 107.2 (4) | 109.5 |
| C1 | C10 | C9 | 120.0 (6) | 121.2 |
| C1 | C11 | N | 126.9 (8) | 125.7 |
| C1 | C11 | C12 | 121.6 (5) | 121.4 |
| N | C11 | C12 | 111.5 (4) | 112.9 |
| C12 | O1 | C13 | 117.0 (9) | 115.0 |
| N | O3 | C14 | 108.5 (5) | 109.2 |
| C11 | N | O3 | 110.8 (7) | 112.6 |
| C11 | C12 | O1 | 110.6 (7) | 110.5 |
| C11 | C12 | O2 | 126.0 (9) | 125.7 |
| O1 | C12 | O2 | 123.4 (5) | 123.8 |
| C4 | C15 | C16 | 174.8 (9) | 180.5 |
| C15 | C16 | C17 | 179.3 (6) | 180.1 |
| C16 | C17 | C18 | 112.6 (7) | 109.6 |
2.2. Optimized geometry
Calculated geometric parameters at B3LYP/6-311G** are equally displayed into Table 1 and Table 2. From the theoretical values, it is noteworthy that most of the optimized bond lengths have slightly larger values than the corresponding experimental ones, due to the fact that theoretical calculations imply isolated molecules in gaseous phase state while experimental results refer to molecules in the solid state. Comparing theoretical bond angles with those given in experimental data, the B3LYP calculated values correlate in a good agreement. In spite of the differences, calculated geometric parameters represent a good approximation and can provide a starting point to calculate other parameters, such as vibrational wavenumbers, as will be described below.
2.3. Vibrational wavenumbers
The theoretically derived DFT vibrational wavenumbers corresponding to the optimized geometry can be expressed in the more convenient internal coordinate space for a description of the potential energy distribution (PED) via the Redong Program [11]. This algorithm uses least square methods to fit the theoretical vibrational wavenumbers to the experimental ones by introducing scaling factors associated to the various types of force constants.
Table 3 displays the potential energy distribution among internal coordinates obtained for the scaled DFT normal modes analysis (scaling factor = 0.962). This last value is currently recommended to obtain correct vibrational wavenumbers and adequate assignments of the PED [12].
The νC=N stretching mode of the imine part is generally observed in the 1665-1675 cm-1 range by Raman spectroscopy. The presence of neigbouring aryl groups lowers this value by 10-20 cm-1. Using a general scaling factor of 0.962 leads to the 1594 cm-1 DFT theoretical wavenumber (1657 cm-1 without scaling). This assignment is in accordance with the Raman wavenumbers as reported by Dollish et al. [13]. The imine moiety participates also to Raman bands around 1274 cm-1, presently calculated at 1266 cm-1. Other vibrational modes of interest implying the imine group are predicted to occur at 314 cm-1 (δ(N-O-CH3), at 971.6 cm-1 (νN-O) and at 997 cm-1 for the (νO-CH3) stretching mode.
The alkyne νCΞC stretching mode displays intense Raman bands in the 2220-2240 cm-1 range. The DFT corresponding wavenumber is obtained presently at 2243 cm-1. Keeping the same 0.962 scaling factor, the δC-CΞC in plane bending mode is predicted to occur at 474.6 cm-1, this frequency being quite comparable to the experimental range centered at 484 cm-1[13].
The ester group gives a predicted DFT νC=O stretching mode located at 1749 cm-1 corresponding to current experimental bands obtained in the 1730-1750 cm-1 range. The associated νC-O stretching mode is predicted at 1215 cm-1, this value appearing quite consistent with experimental data (1200-1220 cm-1). The in plane δ(O-C=O) bending mode is observed in the 750-775 cm-1 range using Raman spectroscopy and the DFT derived corresponding wavenumber is obtained here at 752 cm-1. The δ(C-C=O) and δ(C-O-CH3) in plane bending motions are predicted to stand at 374 and 298 cm-1 respectively, these two wavenumbers being very close to the experimental data (300-340 cm-1).
For the aromatic part of the molecule, we can observe a rough accordance between the DFT predicted wavenumbers (and corresponding vibrational assignments) and the experimental data obtained from vibrational analyses on benzene derivatives [14]. The pyrethoid molecule displays numerous and complex mixings of vibrational modes between the two rings and their substituents.
The ν8 degenerate (8a, 8b) mode (in Wilson’s notation) [14] implying νCA-CA ring stretching motions is generally observed around 1596 cm-1. DFT calculations predict contributions from these two modes at 1558 and in the1582-1594 cm-1 range.
νCA-CA and δCH modes related to the δ19 degenerate mode [14] give rise to experimental vibrational bands around 1485 cm-1. The DFT derived 1484.6 cm-1 wavenumber corresponds correctly to such an assignment.
The mode 4 of benzene generally gives rise to weak Raman and strong IR bands. This out of plane mode (γCH) is generally observed to occur around 693 cm-1. The DFT wavenumber obtained presently at 738 cm-1 could correspond to this mode.
The intense νCA-CA (ν1 in Wilson’s notation) ring stretching mode appears in Raman spectra at 992 cm-1. This mode has been identified as being slightly dependent of δCH bending motions. Due to large mixings of vibrational motions taking place between the two rings, an attempt in the assignment of this mode from DFT calculation is given here at 971.6 cm-1.
From DFT vibrational analyses, out of plane γCH motions display in and out of phase motions depending on the ring. These modes are predicted at 819 and 825 cm-1. Other contributions to the out of plane motions have been also obtained theoretically at 887 and 933 cm-1. An attempt to assign these wavenumbers to the ν5, 10a and 10b normal modes as observed for benzene derivatives [13,14,15] remains however difficult.
Table 3.
Normal modes analysis of RU60358 from DFT (RB3LYP/6-31G (d, p).
| ν (cm-1)* | Assignments | ν (cm-1)* | Assignments |
|---|---|---|---|
| 10.6 | τC17CT, τC1C11, δC4,C15,C16 | 702.1 | νCACA, νC1C11, δCACACA, δCT,O,N |
| 13.8 | τC1C11 | 723.4 | νsCT(CH3)3 (tBu) |
| 24.7 | τC11C12 | 738.1 | γCAH, γCACA, τCACA |
| 30.9 | δCA,C4,C15 | 749.6 | γC12=O2, γC11N, δC12=O2 |
| 35.4 | τC4C15, τC1C2, δC15,C16,C17 | 752.2 | γC12=O2, δC12=O2, δC12O1CT, νC12O1 |
| 61.9 | τC1C11, τC1CA, τC4C15 | 778.7 | γCAH, γC1C11, γCACA |
| 67.2 | τNO, γC11N | 819.0 | γCAH#, γCACA |
| 98.1 | τC14O, τNO | 825.3 | δCACACA(ring 1), γCAH##, νCACA |
| 102.4 | δC15,C16,C17, τC6C7 | 832.4 | δCACACA, νC16C17, νC17CT |
| 108.4 | δC12,C11,N, τO1C13, δC1,C11,C12 | 873.0 | γC3H, τ CACA |
| 112.8 | τO1C13 | 887.1 | γCAH(ring 1) |
| 129.4 | δC4,C15,C16, τO1C12 | 897.4 | νO3N, νO3CT, νO1C12, νO1CT,νC17CT |
| 158.9 | τC2C7, τC2C3, τC7C8 | 897.8 | νC17CT, ρ(tBu) |
| 163.6 | τO1C12, τO3C14, τON | 899.7 | νC17CT |
| 173.9 | τON, δC4,C15,C16, τO3C14 | 926.0 | νC17CT, νNO3, νC4CA, δCACACA |
| 185.2 | τC6C7, τC1C2, τC4C15 | 933.0 | γCAH( all rings) |
| 218.6 | νC4C15, νC16C17, δC3,C4,C5 | 933.8 | ρ(tBu) |
| 221.9 | τC17CH3 (tBu) | 942.9 | γCAH(all rings) |
| 246.0 | δC12,O1,C13, δO1,C12,C11, τC4C15 | 971.6 | νO3N, νCACA, δCACACA, δCAH, δC11,N,O3 |
| 269.5 | τC17CH3(tBu) | 996.9 | νCTO1, νO3CT |
| 276.5 | τC17CH3(tBu) | 1013.9 | ρ(tBu) |
| 279.4 | δC12,O1,C13, νC1C11, δC1,C11,C12 | 1014.1 | ρ(tBu) |
| 297.8 | τC11N, δC12,O1,C13, τO1C12 | 1045.1 | νCACA, νO3CT, νO3N |
| 314.3 | δC14,O,N, νC1C11, τC11N, δCACACA | 1060.2 | νO3CT, νCACA, νO3N |
| 332.4 | δCT,C17,CT(tBu) | 1095.2 | νC12O1, νCTO1, δC11,N,O3, νO3CT |
| 336.7 | δCT,C17,CT(tBu) | 1130.9 | ρC14H3 |
| 357.3 | δCT,C17,CT(tBu), δC1,C2,C3, δC6,C7,C8 | 1133.8 | ρC13H3 |
| 367.4 | νC11C12, δC11,N,O, δC12,C11,N | 1134.5 | δCAH, νCACA(ring 2) |
| 373.6 | δC11,C12,O2, δC12,O1,C13 | 1148.6 | δCAH, νCACA(ring 1) |
| 387.9 | τC4C15, τC4CA, γC6C7 | 1163.1 | ρC13H3 |
| 424.2 | δCT,C17,CT(tBu) | 1172.1 | ρC14H3 |
| 425.2 | τCACA, γCAH | 1181.6 | ρ(tBu) |
| 474.6 | δC4,C15,C16, δC16,C17,CT, γC-N | 1183.4 | ρ(tBu) |
| 501.5 | δC16,C17,CT, δCA,C4,C15, δCACACA | 1184.8 | νO1C12, ρC14H3, νC4C15, ρC13H3, ρ(tBu) |
| 512.7 | δC16,C17,CT, δC15,C16,C17, τCACA | 1196.4 | νCACA, δCAH |
| 528.1 | γCACA, τCACA, γC1C11, δC14,O3,N | 1215.3 | δCAH, νO1C12 |
| 545.3 | τC4C15, δC15,C16,C17, δC16,C17,CT,τCACA, γC4C15 | 1241.2 | δCAH(ring 2) |
| 550.4 | δC4,C15,C16, δC16,C17,CT | 1266.0 | νC11C12, νC1C11, νO1C12, δCAH(ring 1) |
| 583.3 | τC11N, νC16C17, δCA,C1,C11, γC11N, γC12=O2 | 1270.9 | νC16C17, νC4C15, δCAH(ring 1) |
| 590.2 | γC11N, τC11C12, γC12=O2, g C4C15 | 1303.8 | νCACA, δCAH |
| 616.1 | δCACACA, δCT,O,N, νC17CT | 1352.3 | νCACA |
| 652.7 | γC4C15, γC1C11, τCACA, γCAH | 1353.7 | dsCH3(tBu) |
| 1354.4 | dsCH3(tBu) | 2243.4 | νC15C16 |
| 1364.7 | νCACA, δCAH | 2925.2 | νsCH3 (tBu) |
| 1384.7 | δsCH3(tBu) | 2925.8 | νsCH3(tBu) |
| 1416.2 | δsC14H3 | 2931.6 | νsC14H3 |
| 1421.4 | δsC13H3 | 2931.8 | νsC14H3, nsCH3(tBu) |
| 1423.3 | δsC13H3 | 2947.1 | νsC13H3 |
| 1433.6 | δaCH3(tBu) | 2996.8 | νaCH3(tBu) |
| 1434.7 | δaC14H3 | 2997.4 | νaCH3(tBu) |
| 1436.2 | νCACA, δCAH | 3002.5 | νaCH3 (tBu) |
| 1437.8 | δaC13H3 | 3006.2 | νaC14H3 |
| 1443.9 | δaCH3 (tBu) | 3006.8 | νaCH3(tBu) |
| 1444.4 | δaCH3(tBu) | 3010.2 | νaCH3(tBu) |
| 1451.5 | δaC13H3 | 3010.9 | νaCH3(tBu) |
| 1456.3 | δaCH3(tBu) | 3020.7 | νaC13H3 |
| 1456.8 | δaCH3(tBu) | 3041.2 | νaC14H3 |
| 1457.4 | δaC14H3 | 3054.8 | νaC13H3 |
| 1474.6 | δaCH3(tBu) | 3056.3 | νCAH(all rings) |
| 1484.6 | νCACA, δCAH | 3059.4 | νCAH(all rings) |
| 1557.7 | νCACA, δCAH | 3068.8 | νCAH(ring 1) |
| 1582.1 | νC11N, νCACA | 3083.6 | νCAH(ring 1) |
| 1593.9 | νC11N, νCACA | 3087.9 | νCAH(ring 2) |
| 1609.1 | νCACA | 3096.9 | νCAH(ring 2) |
| 1749.0 | νC12=O2 |
(tBu) : Tertiobutyl group. ring 1 is defined by atoms C1, C2, C7, C8, C9 and C10 . ring 2 is defined by atoms C2, C3, C4, C5, C6 and C7# CA-H out of plane bending (in phase in each ring but out of phase between rings)## CA-H out of plane bending in phase for both rings. ν : stretching modes (νs : symmetric, νa : antisymmetric)δ : in plane valence angle bending, (δs : symmetric, δa : antisymmetric). τ, γ, ρ : torsion, out of plane wagging and rocking deformations respectively(*scaled vibrational frequencies originate from application of a general scaling factor (0.962) applied to all types of internal force constants).
3. Experimental Section
3.1 X-ray structure determination
A needle-shaped crystal of dimensions 0.32 ´ 0.27 ´ 0.10 mm was mounted on a Philips Enraf Nonius four-circle diffractometer. Data were collected with MoKa radiation. Intensities for 3082 reflections were measured by the w/2q scan method, scan width = 1.4°, scan speed 0.02°s-1 and 0 < h < 10, 0 < l < 15, 0 < k < 30. Lattice parameters from 25 reflections (2 < q < 30). Three standard reflections monitored every two hours, no significant variation was observed. The data were corrected for Lorentz and Polarization effects, but not for absorption or extinction. The structure was determined by considering 1583 reflections with I ≥ 3σ(I).
The structure was solved by direct methods implemented in Shelxs [16]. A Fourier synthesis revealed the complete structure, which was refined by full-matrix least squares. All non-H atoms refined anisotropically. The H atoms were located from a difference Fourier map and included in the refinement with the isotropic temperature factor of the carrier atom. Fig. 1 shows the atomic numbering of the molecule.
The final least-squares cycle using Shelxl [17] gave R = 0.068 for all reflections with I ≥ 3σ(I), wR = 6.8%, S = 1.25, w =1. (Δρ)min= -0.271 eÅ-3 , (Δρ)max = 0.380 eÅ-3.
Atomic scattering factors for heavy atoms were taken from International Tables for X-ray Crystallography [18] while the factors for H were those of Stewart, Davidson & Simpson [19]. A summary of the key crystallographic information is given in Table 4.
Crystallographic data has been deposited with the Cambridge Crystallographic Data Centre with the deposition number CCDC 272 478. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
3.2. Computational methods
Molecular modeling, geometry optimizations, harmonic wavenumbers for the normal modes of vibration and force field in cartesian coordinates were calculated on RU60358 using the Density Functional Theory and the B3LYP [20,21] (Becke’s three parameter Hybrid Functional using the LYP correlation Functional) functional together with the 6-31G(d,p) basis set. Such combination is being used with good results for organic molecules [22] and hydrogen-bonded systems [23,24,25], and represents a good compromise between economy of computational resources, accuracy and applicability to many-atoms molecules. All the calculations were made with the Gaussian 03 set of programs [26]. Ab initio geometry optimization on RU60358 was performed starting from the geometry found in the X-ray refinement.
Table 4.
Crystallographic data and experimental details for C20H21NO3
| Crystal data | |
|---|---|
| Formula | C20H21NO3 |
| Molecular weight | 323.34 |
| Crystal system | Orthorhombic |
| Space group | P212121 |
| Unit cell determination | Least-squares fit from 25 reflections (2° < q < 30°) |
| a (Å) | 7.7575 |
| b (Å) | 11.3182 |
| c (Å) | 21.3529 |
| V (Å3) | 1874.80 |
| Z | 4 |
| dcalc(g. cm-3) | 1.16 |
| mu (mm-1) | 0.077 |
| Crystal colour | Colourless |
| Crystal size | 0.32 ´ 0.27 ´ 0.10 ( mm) |
| Experimental data | |
| Technique | Four circles diffractometer, CAD4 Enraf Nonius kappa geometry |
| Graphite oriented monochromator : Moka | |
| λ = 0.71070 Å, ω/2q scan | |
| Scanning range for θ | 2.04 ¾ 29.96 |
| Number of reflections measured | 3082 |
| Number of reflections observed | 1583 (I ≥ 3σ(I)criterion) |
| Limiting indices | h 0 ® 10 |
| k 0 ® 15 | |
| l 0 ® 30 | |
| T (K) | 293 |
| Refinement data | |
| Refinement method | Full-matrix least-squares on F |
| Final R indices | R=0.068, wR=0.068 |
| S | 1.249 |
| H atoms | constrained refinement |
| Parameters | 224 |
| (Δ/σ)max | 0.380 |
| (Δρ)min | -0.271 eÅ-3 |
| (Δρ)max | 0.380 eÅ-3 |
| Extinction | No extinction correction applied |
References
- Baert, F.; Guelzim, A. X-ray Structure of the Pyrethroid Insecticide { 1R-[1 α(S*),2α] }-2-(2,2-Dichlorovinyl)-3,3-dimethylcyclopropanecarboxylic Acid Cyano(3-phenoxyphenyl)methyl Ester (RU 24501). Acta Cryst. C 1991, 47, 606–608. [Google Scholar] [CrossRef]
- Baert, F.; Guelzim, A.; Germain, G. Structure of Two Pyrethroid Insecticides: Acrynathryn (RU 38702) and a Derivative (RU 38181). Acta Cryst. C 1991, 47, 768–771. [Google Scholar] [CrossRef]
- Hamzaoui, F.; Lamiot, J.; Baert, F. X-ray Structure of a New Pyrethroid RU 52259. Acta Cryst. C 1993, 49, 818–820. [Google Scholar] [CrossRef]
- Hamzaoui, F.; Baert, F. A New Pyrethroid Insecticide RU41414. Acta Cryst. C 1996, 52, 689–690. [Google Scholar] [CrossRef]
- Tessier, J. Recent Advances in the Chemistry of Insect Control; Janes, N.F., Ed.; The Royal Society of Chemistry: London, 1985; pp. 26–52. [Google Scholar]
- Tessier, J.; Teche, A.; Demoute, J. P. Pesticide Chemistry: Human Welfare and the Environment; Miyamoto, J., Kearney, P.C., Eds.; Pergamon Press: Oxford, New York, 1983; Vol. 1, pp. 95–100. [Google Scholar]
- Tessier, J.; Teche, A.; Demoute, J. P. Proceedings of the 5th IUPAC International Congress of Pesticide Chemistry; Miyamoto, J., Kearney, P.C., Eds.; Pergamon Press: Oxford, New York, 1983; pp. 197–202.
- Babin, D.; Demassey, J.; Demoute, J. P.; Dutheil, P.; Terrie, I.; Tessier, J. A New Way toward Zα,β Unsaturated Esters: A Pyrethroid Application. J. Org. Chem. 1992, 57, 584–589. [Google Scholar] [CrossRef]
- Elliott, M. The Relationship between the Structure and the Activity of Pyrethroids. Bull. Wld Hlth Org. 1970, 44, 315–324. [Google Scholar]
- Elliott, M.; Farnham, A. W.; Janes, N. F.; Needham, P. H.; Pulman, D. A. Insecticidal Activity of the Pyrethrins and Related Compounds. Pestic. Sci. 1975, 6, 537–542. [Google Scholar] [CrossRef]
- Allouche, A.; Pourcin, J. Ab initio calculation of vibrational force fields: Determination of non-redundant symmetry coordinates by least-square component analysis. Spectrochim. Acta, Part A 1993, 49, 571–580. [Google Scholar] [CrossRef]
- Pulay, P. Possibilities and limitations of ab initio calculation of vibrational spectra. J. Mol. Struct. 1995, 347, 293–308. [Google Scholar] [CrossRef]
- Dollish, F. R.; Fateley, W. G.; Bentley, F. F. Characteristic Raman Frequencies of Organic Compounds; John Wiley& Sons: New York, 1974. [Google Scholar]
- Varsányi, G.; Szöke, S. Vibrational Spectra of Benzene Derivatives; Academic Press: New York, 1969. [Google Scholar]
- Wilson, E. B., Jr. The Normal Modes and Frequencies of Vibration of the Regular Plane Hexagon Model of the Benzene Molecule. Phys. Rev. 1934, 45, 706–714. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS97: Program for crystal structure determination; University of Göttingen: Germany, 1997. [Google Scholar]
- Sheldrick, G. M. SHELXL97: Program for crystal structure determination; University of Göttingen: Germany, 1997. [Google Scholar]
- Brown, P. J.; Fox, A. G.; Maslen, E. N.; O'Keefe, M. A.; Willis, B. T. M. International Tables for X-ray Crystallography; Wilson, A. J.C., Prince, E., Eds.; Kluwer Academic Publishers: Dordrecht, 1999; Vol. C, pp. 548–589. [Google Scholar]
- Stewart, R. F.; Davidson, E. R.; Simpson, W. T. Coherent X-Ray Scattering for the Hydrogen Atom in the Hydrogen Molecule. J. Chem. Phys. 1965, 42, 3175–3187. [Google Scholar] [CrossRef]
- Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Rauhut, G.; Pulay, P. Transferable scaling factors for density functional derived vibrational force fields. J. Phys. Chem. 1995, 99, 3093–3100. [Google Scholar] [CrossRef]
- Gómez Marigliano, A. C.; Varetti, E. L. Self-Association of Formamide in Carbon Tetrachloride Solutions: An Experimental and Quantum Chemistry Vibrational and Thermodynamic Study. J. Phys. Chem. A 2002, 106, 1100–1106. [Google Scholar] [CrossRef]
- Gómez Marigliano, A.C.; Varetti, E.L. Unpublished results.
- Sim, F.; St.-Amant, A.; Papai, I.; Salahub, D. R. Gaussian density functional calculations on hydrogen-bonded systems. J. Am. Chem. Soc. 1992, 114, 4391–4400. [Google Scholar] [CrossRef]
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03, Revision A.1. Gaussian, Inc.: Pittsburgh PA, 2003. [Google Scholar]
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.