Abstract
Breast cancer is one of the most common cancers and the second leading cause of cancer death in the United States. Estrogen receptor (ER)-positive cancer is the most frequent subtype representing more than 70% of breast cancers. These tumors respond to endocrine therapy targeting the ER pathway including selective ER modulators (SERMs), selective ER downregulators (SERDs) and aromatase inhibitors (AIs). However, resistance to endocrine therapy associated with disease progression remains a significant therapeutic challenge. The precise mechanisms of endocrine resistance remain unclear. This is partly due to the complexity of the signaling pathways that influence the estrogen-mediated regulation in breast cancer. Mechanisms include ER modifications, alteration of coregulatory function and modification of growth factor signaling pathways. In this review, we provide an overview of epigenetic mechanisms of tamoxifen resistance in ER-positive luminal breast cancer. We highlight the effect of epigenetic changes on some of the key mechanisms involved in tamoxifen resistance, such as tumor-cell heterogeneity, ER signaling pathway and cancer stem cells (CSCs). It became increasingly recognized that CSCs are playing an important role in driving metastasis and tamoxifen resistance. Understanding the mechanism of tamoxifen resistance will provide insight into the design of novel strategies to overcome the resistance and make further improvements in breast cancer therapeutics.
1. Introduction
Breast cancer is one of the most common cancers in women worldwide and is a leading cause of cancer-related death in women in the United States [1]. Based on the expression of estrogen and progesterone receptors (ER/PR) and human epidermal growth factor receptor (HER2) status, breast cancer is classified into further subgroups that include: luminal A (ER+/PR+/HER2 negative), luminal B (ER+/PR+/HER2+), HER2 positive (ER-/PR-/HER2+), basal-like or triple negative (ER-/PR-/HER2−), claudin-low and normal-like [2]. These subgroups are associated with distinct pathological features and clinical outcomes [3]. Clinicians depend mainly on this immunopathological classification in the therapeutic decision-making process [4]. Recently, the American Society of Clinical Oncology/College of American Pathologists (ASCO/CAP) guidelines have defined the absence of ER/PR as less than 1% expression [5]. Approximately 70% of breast cancers express ER and/or PR, followed by triple negative breast cancers (TNBC 19%). The remaining are HER2-overexpressing breast cancer [1]. The presence of ER is considered a good prognostic marker and is commonly used to identify tumors that may respond to endocrine therapy targeting ER signaling pathways. HER2 subtype tumors can be treated by anti-HER2 monoclonal antibodies targeting HER2-dependnet signaling pathway. The TNBC subtype presents the worst prognosis subtype, since it is lacking targeted therapeutic options. Endocrine therapy is the main method of choice to treat luminal breast cancers. However, the development of endocrine resistance represents the main challenge to clinicians. In this review, we discuss recent efforts to understand epigenetic mechanism(s) of endocrine resistance and to resensitize resistant tumors to endocrine therapy.
2. Predisposition to Drug Resistance among Luminal Breast Cancer Patients
The luminal subtype tumors respond to ER-targeted therapies such as the mixed antiestrogen tamoxifen and the pure antiestrogen fulvestrant or to estrogen (E) deprivation therapies using aromatase inhibitors such as anastrozole, exemestane and letrozole [6,7]. However, for reasons that are unclear, over 30% of ER-positive (ER+) tumors are intrinsically hormone resistant (de novo resistance) at diagnosis, and among hormone-responsive ER+ tumors, the clinical behavior can be markedly heterogeneous even when they express similar ER levels [8]. Furthermore, approximately 40% of breast tumors that initially respond to hormone therapies eventually acquire resistance [8].
Luminal breast cancers are subdivided into two molecularly-distinct subtypes, luminal A and B, representing the majority of breast cancers. Genomic efforts have identified many genetic, epigenetic and transcriptional differences between these two luminal subtypes. They respond differently to endocrine therapy, with the luminal A subtype having a better prognosis and being more sensitive to endocrine therapy compared to the luminal B subtype [9]. Patients with luminal A breast cancers respond well to endocrine therapy, and the addition of chemotherapy provides minimal or no clinical benefit to patients with luminal A breast cancer [10]. Gene expression profiling demonstrates that the luminal A subtype is characterized by high ER and PR expression and low expression of proliferation (Ki67 < 14%) and growth factor genes, while the luminal B subtype expresses lower ER, lower or absent PR and high proliferation (Ki67 > 14%) and growth factor receptor gene expression. Likely to have poorer outcomes and to become metastatic [11,12,13,14,15]. In fact, the recurrence rate for luminal B tumors is similar to that of triple negative and HER2 positive tumors [3]. Several studies use different methods to distinguish between these two subtypes including the gene signature that predicts for a specific subtype such as Oncotype DX and MammaPrint [16,17,18]. The genomic signature and biomarkers help to identify groups of patients that benefit from endocrine therapy.
Hormone resistance of ER+ tumor cells appears to be due to a variety of factors (Figure 1). Examples include expression of mutant ERs or activation of E-independent growth factor signaling pathways. For the latter, activation of epidermal growth factor (EGFR/HER2) or insulin-like growth factor (IGFR) receptor pathways is the major culprit [19]. Unfortunately, these observations have not been translated into effective clinical treatments. Trials combining hormone therapies with EGFR inhibitors have shown little added benefit over hormone treatments alone [20,21], possibly because the appropriate subset of tumors likely to benefit have not been identified. Thus, tumors that relapse under continued hormone therapies remain an elusive problem. In order for long-term tumor suppression to be achieved, hormone responsiveness restoration remains an important clinical priority.
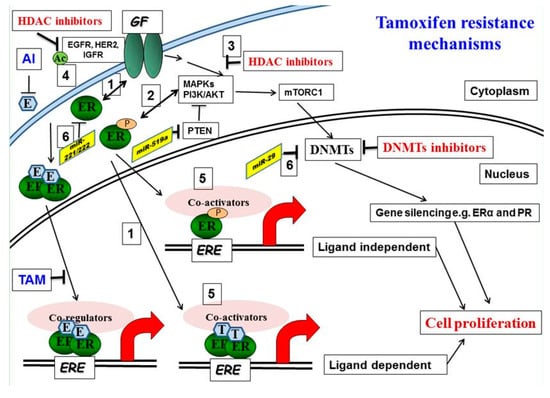
Figure 1.
Molecular mechanisms of tamoxifen resistance: (1) Increased bidirectional ER/growth factor (GF) receptor cross-talk converts tamoxifen into an agonist. (2) Activated downstream kinases, including ERK 1, 2 mitogen-activated protein kinase (MAPK) and AKT, phosphorylate both the ER and its accessory proteins. (3) HDAC inhibition reduces phosphorylation of MAPKs and AKT. (4, 5) Acetylation of EGFR promotes receptor tyrosine kinase (RTK) phosphorylation and activation. Corepressor complexes with NCoR Fare inactivated and dismissed from Tam-bound ER-promoter complexes, allowing instead the recruitment of the phosphorylated/activated coactivator complexes with AIB1. This results in an increase in the agonist versus the antagonist activity of tamoxifen on gene transcription. (6) microRNA involved in tamoxifen resistance, miRNA221/222, is upregulated and miR-29 is downregulated in tamoxifen-resistant cells.
The Molecular Mechanisms behind Luminal Tumor-Cell Heterogeneity
Cancer cells within solid tumors, especially in luminal breast disease, exhibit striking heterogeneity characterized by multiple phenotypic and genotypic cell subpopulations [22,23]. It is important to define these cell subpopulations to develop rational methods for targeting them and understand how each contributes to hormone resistance [24]. Non-genetic hypotheses speculate that some cell subpopulations are paradoxically destined for clonal expansion in response to stimuli like antiestrogens. Other hypotheses suggest that epigenetic, environmental or even dietary factors play a role in resistance. Tumor cell heterogeneity has been assessed with genetic changes such as copy number alteration and somatic mutation. However, recent studies have shown that epigenetic heterogeneity leads to cell-to-cell variation in response to therapy [25]. These epigenetic mechanisms include DNA methylation, histone post-translational modifications and chromatin remodeling (as discussed in next section). Epigenetic alterations are linked to genetic mutation of epigenome regulators, such as the writers, readers and erasers of epigenetic markers [25].
It has also been suggested that resistant cells emerge from a subpopulation of cancer-initiating/cancer stem-like cells, which gain growth advantages via epigenetic mechanisms. Thus, the origins of luminal breast cancer-cell heterogeneity remain unclear in regards to the roles of stem cells, somatic mutations, copy number changes and epigenetic alterations of genes involved in tumor growth, invasion, metastasis and resistance. Luminal tumor-cell heterogeneity is a continuing issue, for example the current clinical classifications that define luminal breast cancers as ones having at least 1% ER+ or PR+ cells [26]. This percentage of heterogeneity immediately raises practical questions: If so, what are the other 99% of cells? How do they contribute to tumor aggressiveness? How do they contribute to the development of hormone resistance? What treatment strategies could possibly be designed for such complex tumors?
3. Epigenetic Regulation of Gene Signaling in Breast Cancers
Epigenetic modifications are inheritable changes in gene expression without alterations in the DNA sequence. Compared to genetic changes, epigenetic modifications are often enzymatic and can be reversed by epigenetic inhibitors. In the nucleus, double-stranded DNA is compacted and organized into chromosomes. The DNA is wrapped around histone protein-complexes to form larger order nucleosomal structures, the basic structural units of chromosomes, allowing the selective accessibility of transcription machinery. The degree of DNA coiling determines whether chromatin is “open, euchromatin” and available for transcription or “closed, hetero-chromatin” and transcriptionally repressed. The balance between euchromatin and hetero-chromatin is determined by epigenetic regulation, allowing cells to regulate gene expression, resulting in significant changes in their biological functions. Epigenetic modifications include methylation of DNA, modification of histone proteins and alteration of miRNA expression; all of which influence gene expression patterns [27,28]. Since epigenetic modifications can be reversed, they appear to be desirable therapeutic targets for cancer patients. Below, we discuss each of these in detail.
3.1. DNA Methylation
DNA methylation is the most important epigenetic modification in mammalian cells that is associated with gene expression. DNA methylation is associated with normal development and growth [29] and is dysregulated in tumors [30]. The covalent addition of methyl residues to cytosines residing in CpG dinucleotides is catalyzed by DNA methyltransferases (DNMTs) including DNMT1, DNMT3a and DNMT3b [29]. DNMT1 is required for maintenance of established DNA methylation patterns. Its deficiency may lead to global hypomethylation [31,32]. DNMT3a and DNMT3b are implicated in the generation of de novo methylation patterns [33]. Additionally, a family of nuclear methyl-CpG-binding protein domain (MBD) binds to methylated cytosines on DNA and regulates gene expression [34,35]. MBDs regulate gene transcription by recruiting histone modifying complexes such as the nucleosome remodeling deacetylase NuRD complex and histone methyltransferases (HMTases) [36]. In humans, ~70% of all CpG islands are hypermethylated and located in tightly-packed core regions of DNA. In contrast, CpG islands that remain hypomethylated are found in relaxed, open, frequent promoter regions of DNA, which enables gene expression [37].
It is estimated that 30% of breast cancer is linked to epigenetic modifications, particularly in DNA methylation [38]. Changes in DNA methylation patterns have been shown to be associated with breast cancer development, progression and metastasis [39]. Breast cancer has been associated with hypermethylation of tumor suppressor genes and with hypomethylation of oncogenes [38]. For example, the number of genes has been reported to be methylated and consequently silenced, including tumor suppressor genes, such as secreted frizzled-related protein (SFRP), RASSF1A, WNT inhibitory factory factor 1 (WIF1) inter-alpha-trypsin inhibitor heavy chain family member 5 (ITIH5), Dickkopf WNT signaling pathway inhibitor 3 (DKK3), ATM serine/threonine kinase (ATM), long interspersed nuclear element 1 (LINE1), cyclin-dependent kinase inhibitors (CDKN2 and CDKN1B), G1/S-specific cyclin-D2 (CCND2), breast cancer 1 ( BRCA1), mutL homology 1 (MLH1), glutathione S-transferase P (GSTP1), homeobox protein (HOXA5), cadherin-1 (CDH1), metalloproteinase inhibitor 3 (TIMP3), cAMP-responsive element-binding protein 3-like protein 1 (CREB3L1) and hormone receptors (ESR1 and PGR). These genes have been shown to be involved in cell cycle regulation, DNA repair, cell detoxification, apoptosis and cell adhesion and invasion [38,40,41,42,43,44,45,46,47]. The phospholipase A2 receptor (PLA2R1) is a transmembrane protein that plays a role in the clearance of phospholipase A2. The phospholipase A2 receptor (PLA2R1) acts as a tumor suppressor in certain tumors including breast cancer. PLA2R1 has been shown to be differentially expressed in normal and mammary cancer cells, and this expression is controlled by epigenetic mechanisms such as DNA methylation and histone modification [48].
DNA methylation signatures have been identified for the characterization and molecular subtyping of breast cancers [49,50,51,52,53,54]. Holm et al. report that Luminal B tumors has been shown to be more methylated than Basal-like or triple negative breast cancers and may contribute to tumor progression in this subtype [55,56]. In general, DNA methylation plays an important role in different subtypes of breast cancers, thereby providing valuable information on disease prognosis and response to treatment. Inhibition of DNMTs by cytosine nucleoside analogs such as 5-Aza-2′-deoxycytidine (AZA; decitabine) has been widely used to investigate the role of DNA methylation in breast cancer.
3.2. Histone Modifications
Post-translational modifications (PTMs) of histone tails such as phosphorylation, ubiquitination, SUMOylation, acetylation and methylation play an important role in modifying gene expression [57,58]. These modifications change the secondary structure of DNA and result in either induction or prevention of access by transcription factors to gene promoter regions.
Histone acetylation is a dynamic reaction catalyzed by histone acetyltransferases (HATs) or histone deacetylases (HDACs). HATs acetylate ε-amino groups of lysine residues in the N-terminal tails of core histones, relaxing chromatin and allowing transcription factor binding. The acetyl groups are removed from lysines by histone deacetylases (HDACs), which compact chromatin into tightly ordered nucleosomes and prevents access of transcription factors to DNA. In general, transcription activators recruit coactivators such as p300/CBP with HAT activity to DNA sites, whereas transcription repressors recruit corepressors with HDAC activity [59]. Histones can also be methylated, which turns genes “off", or demethylated, which turns them “on”, by loosening or either tightening histone tails, which allows or restricts transcription factor loading on DNA [58,60].
3.2.1. Histone Acetylation and HAT Inhibitors
HATs can stimulate or suppress tumor growth and progression. Depending on the target gene, hyper-acetylation of oncogenes leads to cancer progression. For example, increased histone acetylation was detected in hepatocellular carcinoma and is associated with prostate cancer recurrence [61]. The human HATs are classified into two types; type A HATs are nuclear enzymes responsible for acetylation of histones and non-histone proteins in the nucleolus, while type B HATs are cytoplasmic enzymes that modify free histones in the cytoplasm and then transport to the nucleus. Based on sequence homology, HATs are divided into three families: the GNAT (GCN5-related N-acetyltransferases) family consists of KAT2A and KAT2B; the MYST family (MOZ, Ybf2/Sas3, Sas2 and Tip60); and orphan HATs that include p300/CBP and steroid receptor coactivators (SRCs) [62,63,64]. The HAT enzymes have various substrate specificities for histone and non-histone proteins [61]. Because of their involvement in cancer development, HATs were proposed to be promising targets. Histone acetyltransferase MYST3 plays an important role in breast cancer development and activation of ER expression, and targeting MYST3 may serve as a novel strategy to block ER expression in MYST3-high ER+/HER2− breast tumors [65].
HAT modulators suppress the catalytic activity of the acetyl transferases. However, only a very limited amount of HAT modulators has been described or investigated [66], which are classified into bisulfate inhibitors, natural products, synthetic analogues and derivatives and small molecules (reviewed in [61,62,67]). These modulators have been limited to in vitro studies of growth inhibition of cancer cells [68]. Several small molecule HAT inhibitors have been derived from natural products, such as garcinol, curcumin and anacardic acid [61]. The lack of cellular permeability represents a major challenge of some of HAT inhibitors [69]. For example, anacardic acid, isolated from the shells of cashew nuts, is a potent in vitro inhibitor of both p300 and PCAF’s HAT activity [70]. Because cells are poorly permeable to anacardic acid, synthetic analogs are being analyzed for their HAT-inhibitory activity and effects on cancer cells [71].
3.2.2. Histone Deacetylation and HDAC Inhibitor
HDACs fall into two classes based on their structure: zinc-dependent classes I, II and IV; and NAD-dependent class III, also called sirtuins [72]. Class I consists of HDACs 1, 2, 3 and 8, whereas class IV has only one member HDAC11. Class II is divided into class IIa (HDAC 4, 5, 7, 9) and class IIb (HDAC 6 and 10) [73]. HDACs have been shown to have an important role in cancer development and progression. Previous reports indicate that HDAC levels are increased in certain types of cancer [74]. For example, HDAC1 is expressed in many cancers such as prostate, gastric, esophageal and breast cancers (reviewed in [74]). HDAC inhibitors increase cellular protein acetylation by inhibiting HDAC activity. There are four classes of HDAC inhibitors: hydroxamic acids (suberoylanilide hydroxamic acid (SAHA, vorinostat)), benzamides (MS-275), cyclic peptides (romidepsin) and short-chain fatty acids (valproic acid) [75]. HDAC inhibitors have been shown to inhibit tumor growth and promote apoptosis of cancer cells, while not affecting normal tissue [76,77]. Several clinical trials using HDAC inhibitors have been performed, and the results indicate that HDAC inhibitors have anticancer activity [74]. For example, HDAC inhibitors SAHA and romidepsin (FK228) were approved by the U.S. Food and Drug Administration (FDA) for the treatment of cutaneous T-cell lymphoma [78]. In addition, vorinostat, entinostat and panobinostat (LBH-589) have been demonstrated to exhibit potent activity when combined with cytotoxic drugs (paclitaxel), endocrine (tamoxifen) therapies; or with therapies targeted at HER2 (trastuzumab) or VEGF (bevacizumab) [41,45,74,79]. It has been shown recently that the combination of the HDAC inhibitor YCW1 with ionizing radiation induce cell death in triple-negative breast cancer cell lines in vivo and in mouse models [80].
3.2.3. Histone Methylation and Demethylation
Histone methylation occurs on the side chains of lysine (K) and arginine (R) residues; however, unlike acetylation, there is no change in the charge of the histone protein. Histone methylation is regulated by histone methyltransferases (HMTs) and histone demethylases (HDMs) [81]. Histone lysine methylation is associated with both transcriptional activation and repression. Methylation of histone 3 lysine 9, 20 or 27 (H3K9, H3K20 or H3K27) is associated with transcription silencing, whereas methylation of histone 3 lysine 4, 36 or 79 (H3K4, H3K36 or H3K79) is associated with transcription activation [57]. Beside gene transcription, histone methylation markers also recruit proteins associated with DNA repair [81]. Histone methylation regulates many cellular functions, including gene transcription, DNA replication and repair, developmental and differentiation processes, pluripotency and maintenance of genome integrity. It also affects the development of many diseases including malignancies [82]. Targeting histone methylation enzymes may restore normal methylation profile. Histone methylation is catalyzed by three families of enzymes, the set-domain containing protein family, the non-set domain protein family and the PRMT1 (protein arginine methyltransferases) family [83].
Enhancer of zeste homolog 2 (EZH2), a polycomb repressive complex 2 (PRC2) group protein, is a histone methyltransferase that methylates H3K27 and functions as a transcriptional repressor [84]. The overexpression of EZH2 is strongly associated with the development of breast cancer and breast cancer’s aggressiveness [85,86,87]. Indeed, it has been shown that EZH2 inhibits the expression of several tumor suppressor genes such as P16 INK4a, E-cadherin, BRCA1 and the adrenergic receptor β2 [82]. 3-Deazaneplanocin (DZNep), an inhibitor of the EZ2H, was widely used for experimental work. In spite of promising results, DZNep has a short plasma half-life, has nonspecific inhibition of histone methylation and is toxic in animal models [88]. DZNep induces antitumor activity and apoptosis in breast cancer cells, but not in normal cells [89]. Several EZH2 inhibitors have been developed in order to improve antitumor activity and reduce toxicity (reviewed in [88]). For example, tanshindiols are small molecule inhibitors of EZH2 that also possess anti-cancer activity in several tumor cell lines [90]. Lastly, inhibitory EZH2 peptides have been designed among which one termed SQ037 has been validated and shown to have considerable anti-EZH2 potency [91]. Some of these inhibitors have been moved to clinical trials, show early promising results and would be expected to have the desired efficacy with minimal side-effects [91].
The set and MYND domain containing protein 3 (SMYD3) is a novel histone lysine methyltransferase, and it specifically methylates H3K4 (reviewed in [92]). SMYD3 plays a significant role in the development and progression of human cancer via regulating gene transcription and promoting cells proliferation and migration. SMYD3 is overexpressed in several malignancies including esophageal squamous cell carcinoma, gastric cancer, hepatocellular carcinoma, prostate cancer, leukemia and breast cancers. Silencing using small interfering RNAs inhibits the growth of these cancer cells [93]. Similarly, inhibition of SMYD3 expression by Novobiocin inhibits the proliferation and migration of MDA-MB-231 breast cancer cells in a dose-dependent manner. The effect of Novobiocin is associated with the downregulation of SMYD3. Tranylcypromine is another potent H3K4 methylase. This small molecule demethylation inhibitor de-represses transcription of important target genes including the pluripotent stem cell marker Oct4 [94,95].
Lysine-specific demethylase 1 (LSD1) removes methyl groups from methylated proteins including histone H3 (H3K4) and non-histone proteins such as p53 and DNMT1, suggesting that it is involved in a wide variety of normal biological functions [96,97,98,99,100]. LSD1 is overexpressed in various types of solid tumors including basal-like breast cancer where it is a biomarker of poor prognosis and aggressiveness [101,102]. It has been shown recently that LSD1 is a potential target gene of miR-708. Overexpression of miR-708 inhibits breast cancer cell line MDA-MB-231 proliferation and invasion, whereas inhibition of miR-708 enhances these processes [103]. LSD1 together with ER remove methyl groups from H3K9 to activate gene expression [104]. There are several inhibitors of LSD1 that have been developed and tested for their effects on many forms of cancers. These include bizine, the tranylcypromine derivatives NCL1 and GSK2879552, biguanide and bisguanidine polyamine analogs [105]. GSK287 is an orally-bioavailable irreversible LSD1 inhibitor, currently under clinical evaluation for cancer treatment [106]. These LSD1 inhibitors alter promoter activity of multiple genes in breast cancer cells and are postulated to have considerable therapeutic potential [107,108,109]. It has been shown that LSD1 interacts with HDACs to control breast cancer cell growth. Combined treatment of triple-negative breast cancer cells with LSD1 inhibitor, pargyline, and HDAC inhibitor, SAHA, leads to growth inhibition [110,111].
G9a, a histone methyltransferase responsible for H3K9 methylation, has been reported to promote cancer aggressiveness, and its overexpression has been associated with poor prognosis [112,113,114,115,116]. Several small molecules inhibitors have been developed to inhibit the enzymatic activity of G9a [116]. Inhibition of G9a reduces the invasiveness and metastatic potential of human lung cancer cells [117] and inhibits the growth of prostate cancer cells [118]. BIX01294 (a diazepine-quinazoline-amine derivative) is one of the first molecules developed to reduce G9a-mediated H3K9 di-methylation [119]. BIX01294 treatment was shown to inhibit proliferation, motility and invasiveness of human neuroblastoma cells [120] and pancreatic cancer cells [121]. UNC0638 is another inhibitor characterized by high potency and specificity for G9a. In vitro, UNC0638 treatment has been shown to inhibit cell proliferation in various cell lines such as breast, squamous head and neck carcinoma, hepatocellular carcinoma, acute leukemia and cervical cancer [116].
3.3. microRNAs
Just as methylation modifies DNA and its ability to be transcribed, it modifies miRNAs and their ability to regulate protein expression post-transcriptionally. Epigenetic hypomethylation on miRNAs that regulate ER signaling is associated with deregulated ER function in breast cancers [122].
miRNAs are short, naturally-occurring noncoding RNAs (18–22 nucleotides in length) that regulate gene expression of target genes involved in different cellular functions including proliferation, differentiation and apoptosis [123,124]. Evidence indicates that alteration of miRNA expression is associated with cancer development and progression [125,126,127,128,129]. Several investigators have defined miRNA signatures that are differentially expressed in breast cancers compared to normal mammary tissues and are able to distinguish between different breast cancer subtypes [128,130,131,132,133]. At normal levels, miRNAs act as tumor suppressors or oncogenes. For example, miRNA-10b, miRNA-125b and miRNA-145, are downregulated, while miRNA-21 and miRNA-155 are upregulated in tumors compared to normal tissues [128]. The putative targets of miRNA-125b are the oncogenes YES, ETS1, TEL and AKT3; the growth factor FGFR2; and members of the MAPK pathway (MAP3K10, MAP3K11 and MAPK14) [128]. On the other hand, upregulated miRNA-21 targets the tumor suppressors PDCD4 and PTEN [127,134]. It has been shown that miRNA-30 expression is correlated with ER and PR levels and that miRNA let-7 isoforms regulate PR status (let-7c), lymph node metastasis (let-7f-1, let-7a-3, let-7a-2) and proliferation indices (let-7c, let-7d). miRNA let-7 also appears to be a tumor suppressor that is downregulated in breast cancer stem cells (CSC) [135,136]. Preclinical studies have shown that miRNAs play a functional role in different steps of the metastatic cascade (reviewed in [137]). Several miRNAs have been shown to act as EMT-negative regulators by targeting specific EMT-associated transcription factors (miR-1, miR-15b, miR-30c, miR-34a, miR-101, miR-124, miR-132, miR-137, miR-138, miR-150, 153, miR-200s, 203, miR-204, miR-205, miR-300, miR-335). Some miRNAs were found to regulate EZH2 (miR-15b, miR-138) or the NAD-dependent deacetylase sirtuin-1 (miR-200s, miR-204). In addition, miR-451 has been shown to play a role in tamoxifen resistance [82,83].
miRNAs have been shown to be easily extracted from different body fluids including blood, saliva and sputum. Several studies demonstrated that circulating miRNA can be used as biomarkers to discriminate between normal and diseased patients in many cancers, including breast cancer (reviewed in [138]). Overall, the ability of miRNA to very specifically target a desired miRNA has great promise as highly specific targeted therapies for cancer treatment.
4. Epigenetic Modulation of Tamoxifen Resistance
Estrogen Receptor
The interplay between epigenetics and ER signaling is believed to be one important factor that dictates breast cancer development and tumor response to conventional therapies. ER signaling plays a role in histone modifications including acetylation, phosphorylation and methylation through the interaction with histone modifying enzymes [139]. On the other hand, epigenetic pathways regulate ER signaling by controlling ER expression levels. Moreover, ER target genes are regulated by co-recruitment of ER and epigenetic cofactors, including HATs, HDACs, HMTs, DNMTs and polycomb proteins (reviewed in [140]). The overall balance among these coregulatory proteins controls ER functions including their responses to endocrine therapies. Downregulation of corepressors and/or overexpression of coactivators are very likely to contribute to endocrine resistance. Overexpression of ER co-regulators such as SRC1, TIF2, SRC3 and CBP contributes to tamoxifen resistance [141,142,143]. SRC1 overexpression is associated with tamoxifen resistance and disease recurrence only in the HER2− positive breast cancer subtype [144]. SRC3 overexpression is associated with shorter survival among tamoxifen-treated patients, suggesting that tamoxifen activity may be switched from antagonist to agonist. Cellular distribution of SRC3 has been shown to influence tamoxifen responsiveness. Nuclear SRC3 was associated with a favorable outcome in patients receiving endocrine therapy [145].
Tamoxifen inhibits cancer growth by inhibiting ER transcriptional activity. Its activity depends on the ability to stabilize the binding of ER to corepressors such as NCOR1 and smart (reviewed in [146]). Downregulation of these corepressors has been implicated in endocrine resistance. Girault et al. have shown that weak expression of NCOR1 is significantly associated with shorter relapse-free survival, suggesting that NCOR1 is required for full tamoxifen efficacy [143].
Recruitment of HDACs to an ER-corepressor complex enhances the actions of tamoxifen, while the absence of HDAC recruitment to corepressors at a tamoxifen-bound ER complex results in drug resistance [147]. Inactivation of the ER corepressor could predispose cancer cells to the anti-tumorigenic effect of HDACi, while genomic alterations in ER corepressors or coactivators are candidate biomarkers that could predict response to HDACi in tamoxifen-resistant breast cancers [147]. Along those lines, it has been reported that depletion of the nuclear receptor coactivator SRC3 enhances the sensitivity of breast cancer cells to the HDACi vorinostat (SAHA). In contrast, overexpression of SRC3 decreases SAHA-induced cancer cell apoptosis [148].
5. Epigenetic Regulation of Breast Cancer Stem Cells
Breast CSCs are theoretically a rare, immortal cell subset within the heterogeneous population of solid-tumor cancer cells. They can both self-renew and give rise to all other cell subpopulations present in that tumor. Such CSCs would represent a significant clinical challenge as they would not only be resistant to therapies, but would also play essential roles in tumor recurrence and metastasis [149].
Cellular heterogeneity within breast cancers and the putative existence of breast CSCs are also possible reasons for therapeutic failures [150]. Assuming they exist, breast CSCs are predicted to be a minor (possibly <1%) non-proliferative precursor cell subpopulation within a tumor, able to give rise to and maintain more differentiated downstream cell types within a tumor [151,152,153], but are resistant to endocrine, radiation and chemotherapies that target the more differentiated cells [154,155,156,157,158,159,160,161,162,163,164,165]. If so, therapeutic approaches that kill CSCs may be the best hope for curing cancers. However, with regard to human breast CSCs, much uncertainty remains about their identity, markers that identify them and whether there is a single definable breast CSC or if there are multiple CSCs that differ among breast cancer subtypes.
Conventional treatments only kill differentiated cancer cells. Targeting putative CSCs is a promising therapeutic approach, although, especially in luminal breast cancers, identification of these cells remains elusive. One approach would be to target their self-renewal capacity by inducing their differentiation; a switch that would presumably reduce their resistance to drugs. This is not outside the realm of possibility; it has been reported that histone modifier (Bmi-1 and EZH2) and non-coding RNA (let7, miR-93, miR-100 and HOTAIR) are involved in the regulation of CSC phenotypes. For instance, exposure to vorinostat reduces mammosphere formation capacity, an index of CSC function [149]. Witt et al. [166] have shown that when compared to non-stem-tumor-cells, the deacetylases HDAC1 and HDAC7 are overexpressed in CSCs. They demonstrated that currently available HDACi such as trichostatin A, a pan HDACi, suppress HDAC1 and HDAC, and may therefore modify the epigenetic marks that characterize CSCs.
Epigenetic modifications are involved in the formation, maintenance and function of breast CSCs. However, there are not many data on DNA methylation, and its interplay with breast CSCs. DNA methylation has been shown to regulate hematopoietic stem cells where it targets pluripotency factors [152,167,168,169,170,171] and targets CD133 in colon, ovarian, blood, prostate and brain CSCs [172,173,174,175]. The association between DNA methylation and CSCs [176] suggests that hypomethylating agents have the potential to induce CSC differentiation generating cells that would be sensitive to therapeutic agents. A methyltransferase inhibitor, 3-deazaneplanocin, can disrupt the polycomb 2 complex and reduce CSCs in acute myeloid leukemia [177], hepatocellular carcinoma [178], glioblastoma [179] and prostate cancer [180].
In addition to targeting regulatory genes in differentiated cells, histone modifications also play a role in CSC biology. The polycomb repressive complex (PRC), which represses gene expression through histone modification and chromatin compaction [181], regulates CSCs of the breast [182], prostate [180], ovary [183] and glioblastomas [184]. Paranjape et al. have shown that overexpression of Bmil, a polycomb protein, increased self-renewal and stemness in mammary epithelial cells [185]. Inhibitor of LSD1, the enzyme responsible for demethylating H3K4, acts specifically on embryonal carcinoma stem cells of pluripotent cancers such as teratocarcinomas, seminomas and embryonic carcinomas [186], suggesting that H3K4 demethylation is involved in the formation of such tumors.
Duru and coworkers [187] have discussed the role of epigenetics in the regulation of miRNA with relation to CSCs. They have shown that miR-140 can be activated by epigenetic therapy or dietary compounds targeting stem cells in ductal carcinoma in situ, thereby preventing relapse or progression to invasive disease.
Histone acetylation also plays an important role in the regulation of CSC miRNAs. For example, miR-34a is downregulated in pancreatic CSCs. HDAC inhibitor SAHA (vorinostat) treatment restores miR-34a levels and decreases CSCs viability. In breast cancers, miR-34a suppresses HDAC1 and HDAC7 expression and its level is inversely correlated with HDAC1 and HDAC7 activity, as well as tumor characteristics such as grade and stage [188]. Therapy-resistant and aggressive breast cancers are associated with low miR-34a expression and high HDAC1 and HDAC7 expression, which deacetylate HSP70K 246 [189].
Previous studies have identified unique miRNA expression profiles for breast CSCs compared to non-tumorigenic cells, and the dysregulation of miRNA plays an important role in breast CSC biology [135,190,191,192,193]. These miRNAs may function as oncomirs or tumor suppressor genes to regulate self-renewal, invasiveness and drug resistance of CSCs [191]. miRNA-200 isoforms a, b and c are downregulated in breast CSCs whose targets include stem cell self-renewal factor Bmi, and the transcriptional repressors of E-cadherin, ZEB1/ZEB2 [192,194]. miRNA let-7, miRNA-200c and miRNA-107 have been shown to be downregulated by Lin28, an RNA-binding protein that induces specific miRNA uridylation and blocks miRNA processing by Dicer [195]. miRNA-103/107 overexpression induces epithelial-mesenchymal transition (EMT) and increases the risk of metastases in breast cancer patients [196]. EMT has been directly linked to the generation of cells with CSC-like properties [197]. Other miRNAs reportedly downregulated in breast CSCs include miRNA-30, miRNA-128, miRNA-34c, miRNA-34a and miRNA-16; while upregulated miRNAs include miRNA-181 and miRNA-495 [190].
6. Summary and Future Perspectives
In summary, resistance to hormone therapies in breast cancers can arise from a variety of mechanisms among which epigenetic changes are likely to be important. DNA methylation for instance alters mRNA expression of genes important for estrogen-dependent growth. It is not surprising then that hormone resistance is likely due to a combination of factors that include both a selection of pre-existing resistant cells harboring irreversible genetic defects as well as cells carrying reversible epigenetic errors. The latter is targetable. For this reason, understanding specifically how epigenetic changes contribute to the broad phenotype of hormone resistance could uncover treatment modalities and pathways for which drugs are already available. These could be used either to prevent development of resistance or to restore drug sensitivity to previously resistant cells. However, it is diagnostically important to identify the tumor and cell types that would respond to such treatments. For example, luminal breast cancers respond differently to epigenetic drugs than do basal and receptor-negative breast cancers [198]. Additionally, it is critically important to evaluate the combinatorial effects of hormone therapies together with epigenetic therapies in order to answer even the most rudimentary questions: i.e., should such treatments be combined or sequenced? Despite the large number of studies dealing with epigenetics and breast cancers published in the past few years, much remains to be learned at the basic research level before translational applications can be rationally deployed.
Acknowledgments
I would like to acknowledge Kate Horwitz, Mohamed Abdel-Hafiz and Tammy Trudeau for their support and valuable advice.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Mendes, T.F.; Kluskens, L.D.; Rodrigues, L.R. Triple Negative Breast Cancer: Nanosolutions for a Big Challenge. Adv. Sci. 2015, 2, 1500053. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.N.; Wronski, A.; Castano, Z.; Dake, B.; Malone, C.; De Raedt, T.; Enos, M.; DeRose, Y.S.; Zhou, W.; Guerra, S.; et al. Loss of RasGAP Tumor Suppressors Underlie the Aggressive Nature of Luminal B Breast Cancers. Cancer Discov. 2016, 7, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Tran, B; Bedard, P.L. Luminal-B breast cancer and novel therapeutic targets. Breast Cancer Res. 2011, 13, 221. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Hammond, M.E.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch. Pathol. Lab. Med. 2010, 134, e48–e72. [Google Scholar] [PubMed]
- Allred, D.C.; Brown, P.; Medina, D. The origins of estrogen receptor alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast Cancer Res. 2004, 6, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K. Steroid hormone receptors in breast cancer management. Breast Cancer Res. Treat. 1998, 51, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Badia, E.; Oliva, J.; Balaguer, P.; Cavailles, V. Tamoxifen resistance and epigenetic modifications in breast cancer cell lines. Curr. Med. Chem. 2007, 14, 3035–3045. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.D.; Sanna, G.; Siclari, O.; Biganzoli, L.; Di Leo, A. Defining optimal duration and predicting benefit from chemotherapy in patients with luminal-like subtypes. Breast 2015, 24 (Suppl. 2), S136–S142. [Google Scholar] [CrossRef] [PubMed]
- Diessner, J.; Wischnewsky, M.; Blettner, M.; Hausler, S.; Janni, W.; Kreienberg, R.; Stein, R.; Stuber, T.; Schwentner, L.; Bartmann, C.; et al. Do Patients with Luminal A Breast Cancer Profit from Adjuvant Systemic Therapy? A Retrospective Multicenter Study. PLoS ONE 2016, 11, e0168730. [Google Scholar] [CrossRef] [PubMed]
- Cheang, M.C.; Chia, S.K.; Voduc, D.; Gao, D.; Leung, S.; Snider, J.; Watson, M.; Davies, S.; Bernard, P.S.; Parker, J.S.; et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst. 2009, 101, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Prat, A; Cheang, M.C.; Martin, M.; Parker, J.S.; Carrasco, E.; Caballero, R.; Tyldesley, S.; Gelmon, K.; Bernard, P.S.; Nielsen, T.O.; et al. Prognostic significance of progesterone receptor-positive tumor cells within immunohistochemically defined luminal A breast cancer. J. Clin. Oncol. 2013, 31, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Cancello, G.; Maisonneuve, P.; Rotmensz, N.; Viale, G.; Mastropasqua, M.G.; Pruneri, G.; Montagna, E.; Iorfida, M.; Mazza, M.; Balduzzi, A.; et al. Progesterone receptor loss identifies Luminal B breast cancer subgroups at higher risk of relapse. Ann. Oncol. 2013, 24, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, C.; Neo, S.Y.; McShane, L.M.; Korn, E.L.; Long, P.M.; Jazaeri, A.; Martiat, P.; Fox, S.B.; Harris, A.L.; Liu, E.T. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc. Natl. Acad. Sci. USA 2003, 100, 10393–10398. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Mackay, A.; A’Hern, R.; Natrajan, R.; Tan, D.S.; Dowsett, M.; Ashworth, A.; Reis-Filho, J.S. Breast cancer molecular profiling with single sample predictors: A retrospective analysis. Lancet Oncol. 2010, 11, 339–349. [Google Scholar] [CrossRef]
- Dowsett, M.; Sestak, I.; Lopez-Knowles, E.; Sidhu, K.; Dunbier, A.K.; Cowens, J.W.; Ferree, S.; Storhoff, J.; Schaper, C.; Cuzick, J. Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J. Clin. Oncol. 2013, 31, 2783–2790. [Google Scholar] [CrossRef] [PubMed]
- Wirapati, P.; Sotiriou, C.; Kunkel, S.; Farmer, P.; Pradervand, S.; Haibe-Kains, B.; Desmedt, C.; Ignatiadis, M.; Sengstag, T.; Schutz, F.; et al. Meta-analysis of gene expression profiles in breast cancer: Toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 2008, 10, R65. [Google Scholar] [CrossRef] [PubMed]
- Ali, S; Coombes, R.C. Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer 2002, 2, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.E.; Walsh, G.; Skene, A.; Llombart, A.; Mayordomo, J.I.; Detre, S.; Salter, J.; Clark, E.; Magill, P.; Dowsett, M. A phase II placebo-controlled trial of neoadjuvant anastrozole alone or with gefitinib in early breast cancer. J. Clin. Oncol. 2007, 25, 3816–3822. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R.; Martin, L.A.; Leary, A.; Head, J.; Dowsett, M. Clinical strategies for rationale combinations of aromatase inhibitors with novel therapies for breast cancer. J. Steroid Biochem. Mol. Biol. 2007, 106, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.L.; Polyak, K. Breast tumor heterogeneity: Cancer stem cells or clonal evolution? Cell Cycle 2007, 6, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- Marjanovic, N.D.; Weinberg, R.A.; Chaffer, C.L. Cell plasticity and heterogeneity in cancer. Clin. Chem. 2013, 59, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Pankov, A.; Song, J.S.; Costello, J.F. Intratumoral Heterogeneity of the Epigenome. Cancer Cell 2016, 29, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.E.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Choudhury, S.; Kowalczyk, A.; Bessarabova, M.; Beresford-Smith, B.; Conway, T.; Kaspi, A.; Wu, Z.; Nikolskaya, T.; Merino, V.F.; et al. Epigenetic regulation of cell type-specific expression patterns in the human mammary epithelium. PLoS Genet. 2011, 7, e1001369. [Google Scholar] [CrossRef] [PubMed]
- Lustberg, M.B.; Ramaswamy, B. Epigenetic targeting in breast cancer: Therapeutic impact and future direction. Drug News Perspect. 2009, 22, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, P.; Shaw, P. Loss of imprinting and cancer. J. Pathol. 2007, 211, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. DNA methylation and cancer. Oncogene 2002, 21, 5358–5360. [Google Scholar] [CrossRef] [PubMed]
- Colacino, J.A.; Arthur, A.E.; Dolinoy, D.C.; Sartor, M.A.; Duffy, S.A.; Chepeha, D.B.; Bradford, C.R.; Walline, H.M.; McHugh, J.B.; D’Silva, N.; et al. Pretreatment dietary intake is associated with tumor suppressor DNA methylation in head and neck squamous cell carcinomas. Epigenetics 2012, 7, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Duthie, S.J. Epigenetic modifications and human pathologies: Cancer and CVD. Proc. Nutr. Soc. 2011, 70, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Duthie, S.J. Folate and cancer: How DNA damage, repair and methylation impact on colon carcinogenesis. J. Inherit. Metab. Dis. 2011, 34, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Hua, S.; He, X.; Zhang, Y. DNA methyltransferases and methyl-binding proteins of mammals. Acta Biochim. Biophys. Sin. (Shanghai) 2010, 42, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. DNA methylation signatures for breast cancer classification and prognosis. Genome Med. 2012, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Lubecka, K.; Kurzava, L.; Flower, K.; Buvala, H.; Zhang, H.; Teegarden, D.; Camarillo, I.; Suderman, M.; Kuang, S.; Andrisani, O.; et al. Stilbenoids remodel the DNA methylation patterns in breast cancer cells and inhibit oncogenic NOTCH signaling through epigenetic regulation of MAML2 transcriptional activity. Carcinogenesis 2016, 37, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.I.; Aumsuwan, P.; Khan, I.A.; Walker, L.A.; Dasmahapatra, A.K. Epigenetic events associated with breast cancer and their prevention by dietary components targeting the epigenome. Chem. Res. Toxicol. 2012, 25, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Sunami, E.; Shinozaki, M.; Sim, M.S.; Nguyen, S.L.; Vu, A.T.; Giuliano, A.E.; Hoon, D.S. Estrogen receptor and HER2/neu status affect epigenetic differences of tumor-related genes in primary breast tumors. Breast Cancer Res. 2008, 10, R46. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Nayak, S.; Jankowitz, R.; Davidson, N.E. Epigenetics in breast cancer: What’s new? Breast Cancer Res. 2011, 13, 225. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.H.; Esteller, M. Chromatin remodeling in mammary gland differentiation and breast tumorigenesis. Cold Spring Harb. Perspect. Biol. 2010, 2, a004515. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, J.; Ronneberg, J.A.; Tost, J.; Kristensen, V. The epigenetics of breast cancer. Mol. Oncol. 2010, 4, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.K.; Sukumar, S. Epigenomics and breast cancer. Pharmacogenomics 2008, 9, 1879–1902. [Google Scholar] [CrossRef] [PubMed]
- Pathiraja, T.N.; Stearns, V.; Oesterreich, S. Epigenetic regulation in estrogen receptor positive breast cancer--role in treatment response. J. Mammary Gland Biol. Neoplasia 2010, 15, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, M.; Hoon, D.S.; Giuliano, A.E.; Hansen, N.M.; Wang, H.J.; Turner, R.; Taback, B. Distinct hypermethylation profile of primary breast cancer is associated with sentinel lymph node metastasis. Clin. Cancer Res. 2005, 11, 2156–2162. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.K.; Mellor, P.; Smith, S.E.; Kendall, S.; Just, N.A.; Vizeacoumar, F.S.; Sarker, S.; Phillips, Z.; Alvi, R.; Saxena, A.; et al. Epigenetic silencing of CREB3L1 by DNA methylation is associated with high-grade metastatic breast cancers with poor prognosis and is prevalent in triple negative breast cancers. Breast Cancer Res. 2016, 18, 12. [Google Scholar] [CrossRef] [PubMed]
- Menschikowski, M.; Hagelgans, A.; Nacke, B.; Jandeck, C.; Sukocheva, O.; Siegert, G. Epigenetic control of phospholipase A2 receptor expression in mammary cancer cells. BMC Cancer 2015, 15, 971. [Google Scholar] [CrossRef] [PubMed]
- Ordway, J.M.; Budiman, M.A.; Korshunova, Y.; Maloney, R.K.; Bedell, J.A.; Citek, R.W.; Bacher, B.; Peterson, S.; Rohlfing, T.; Hall, J.; et al. Identification of novel high-frequency DNA methylation changes in breast cancer. PLoS ONE 2007, 2, e1314. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Turcan, S.; Rimner, A.; Kaufman, A.; Giri, D.; Morris, L.G.; Shen, R.; Seshan, V.; Mo, Q.; Heguy, A.; et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci. Transl. Med. 2011, 3, 75ra25. [Google Scholar] [CrossRef] [PubMed]
- Bediaga, N.G.; Acha-Sagredo, A.; Guerra, I.; Viguri, A.; Albaina, C.; Ruiz Diaz, I.; Rezola, R.; Alberdi, M.J.; Dopazo, J.; Montaner, D.; et al. DNA methylation epigenotypes in breast cancer molecular subtypes. Breast Cancer Res. 2010, 12, R77. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Shen, L.; Wen, S.; Rosen, D.G.; Jelinek, J.; Hu, X.; Huan, S.; Huang, M.; Liu, J.; Sahin, A.A.; et al. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res. 2007, 9, R57. [Google Scholar] [CrossRef] [PubMed]
- Bertolo, C.; Guerrero, D.; Vicente, F.; Cordoba, A.; Esteller, M.; Ropero, S.; Guillen-Grima, F.; Martinez-Penuela, J.M.; Lera, J.M. Differences and molecular immunohistochemical parameters in the subtypes of infiltrating ductal breast cancer. Am. J. Clin. Pathol. 2008, 130, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Fiegl, H.; Millinger, S.; Goebel, G.; Muller-Holzner, E.; Marth, C.; Laird, P.W. Breast cancer DNA methylation profiles in cancer cells and tumor stroma: association with HER-2/neu status in primary breast cancer. Cancer Res. 2006, 66, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Holm, K.; Hegardt, C.; Staaf, J.; Vallon-Christersson, J.; Jonsson, G.; Olsson, H.; Borg, A.; Ringner, M. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010, 12, R36. [Google Scholar] [CrossRef] [PubMed]
- Holm, K.; Staaf, J.; Lauss, M.; Aine, M.; Lindgren, D.; Bendahl, P.O.; Vallon-Christersson, J.; Barkardottir, R.B.; Hoglund, M.; Borg, A.; et al. An integrated genomics analysis of epigenetic subtypes in human breast tumors links DNA methylation patterns to chromatin states in normal mammary cells. Breast Cancer Res. 2016, 18, 27. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Sawan, C.; Herceg, Z. Histone modifications and cancer. Adv. Genet. 2010, 70, 57–85. [Google Scholar] [PubMed]
- Zhao, Q.Y.; Lei, P.J.; Zhang, X.; Zheng, J.Y.; Wang, H.Y.; Zhao, J.; Li, Y.M.; Ye, M.; Li, L.; Wei, G.; et al. Global histone modification profiling reveals the epigenomic dynamics during malignant transformation in a four-stage breast cancer model. Clin. Epigenetics 2016, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, H. Silencing of Wnt5a during colon cancer metastasis involves histone modifications. Epigenetics 2012, 7, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Manzo, F.; Tambaro, F.P.; Mai, A.; Altucci, L. Histone acetyltransferase inhibitors and preclinical studies. Expert Opin. Ther. Pat. 2009, 19, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Abmayr, S.M.; Workman, J.L. Regulation of KAT6 Acetyltransferases and Their Roles in Cell Cycle Progression, Stem Cell Maintenance, and Human Disease. Mol. Cell Biol. 2016, 36, 1900–1907. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Liang, Y.; Cao, X.; Wang, X.; Gao, H.; Lin, S.Y.; Schiff, R.; Wang, X.S.; Li, K. Identification of MYST3 as a novel epigenetic activator of ERalpha frequently amplified in breast cancer. Oncogene 2016, 36, 2910–2918. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Bourke, E.; Scobie, M.; Famme, M.A.; Koolmeister, T.; Helleday, T.; Eriksson, L.A.; Lowndes, N.F.; Brown, J.A. Rational design and validation of a Tip60 histone acetyltransferase inhibitor. Sci. Rep. 2014, 4, 5372. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.P.; Robaa, D.; Alhalabi, Z.; Sippl, W.; Jung, M. KATching-Up on Small Molecule Modulators of Lysine Acetyltransferases. J. Med. Chem. 2016, 59, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Stimson, L.; Rowlands, M.G.; Newbatt, Y.M.; Smith, N.F.; Raynaud, F.I.; Rogers, P.; Bavetsias, V.; Gorsuch, S.; Jarman, M.; Bannister, A.; et al. Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Mol. Cancer Ther. 2005, 4, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Gajer, J.M.; Furdas, S.D.; Grunder, A.; Gothwal, M.; Heinicke, U.; Keller, K.; Colland, F.; Fulda, S.; Pahl, H.L.; Fichtner, I.; et al. Histone acetyltransferase inhibitors block neuroblastoma cell growth in vivo. Oncogenesis 2015, 4, e137. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanyam, K.; Swaminathan, V.; Ranganathan, A.; Kundu, T.K. Small molecule modulators of histone acetyltransferase p300. J. Biol. Chem. 2003, 278, 19134–19140. [Google Scholar] [CrossRef] [PubMed]
- Eliseeva, E.D.; Valkov, V.; Jung, M.; Jung, M.O. Characterization of novel inhibitors of histone acetyltransferases. Mol. Cancer Ther. 2007, 6, 2391–2398. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Olzscha, H.; Sheikh, S.; La Thangue, N.B. Deacetylation of chromatin and gene expression regulation: A new target for epigenetic therapy. Crit. Rev. Oncog. 2015, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; Davakis, S.; et al. Histone Deacetylase Inhibitors: An Attractive Therapeutic Strategy Against Breast Cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell Biochem. 2005, 96, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Marson, C.M. Histone deacetylase inhibitors: Design, structure-activity relationships and therapeutic implications for cancer. Anticancer Agents Med. Chem. 2009, 9, 661–692. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.; La Thangue, N.B. Drug Insight: Histone deacetylase inhibitor-based therapies for cutaneous T-cell lymphomas. Nat. Clin. Pract. Oncol. 2008, 5, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Atadja, P.; Davidson, N.E. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer Biol. Ther. 2007, 6, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Stearns, V.; Zhou, Q.; Davidson, N.E. Epigenetic regulation as a new target for breast cancer therapy. Cancer Invest. 2007, 25, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.W.; Yeh, Y.L.; Wang, Y.C.; Huang, W.J.; Ho, S.Y.; Lin, P.; Wang, Y.J. Combination of the novel histone deacetylase inhibitor YCW1 and radiation induces autophagic cell death through the downregulation of BNIP3 in triple-negative breast cancer cells in vitro and in an orthotopic mouse model. Mol. Cancer 2016, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Lin, C.; Zhong, L.L.; Zhao, L.; Zhang, G.; Lu, A.; Wu, J.; Bian, Z. Targeting histone methylation for colorectal cancer. Ther. Adv. Gastroenterol. 2017, 10, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Poreba, E.; Kamieniarz, K.; Schneider, R. Histone modifiers in cancer: Friends or foes? Genes Cancer 2011, 2, 631–647. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetics in cancer: Fundamentals and Beyond. Pharmacol. Ther. 2017, 173, 118–134. [Google Scholar] [CrossRef] [PubMed]
- van der Vlag, J; Otte, A.P. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat. Genet. 1999, 23, 474–478. [Google Scholar] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Collett, K.; Eide, G.E.; Arnes, J.; Stefansson, I.M.; Eide, J.; Braaten, A.; Aas, T.; Otte, A.P.; Akslen, L.A. Expression of enhancer of zeste homologue 2 is significantly associated with increased tumor cell proliferation and is a marker of aggressive breast cancer. Clin. Cancer Res. 2006, 12, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.; Tan, P.B.; Liu, E.T.; Yu, Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007, 21, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Kim, H.Y.; Byun, B.J.; Chae, C.H.; Lee, J.Y.; Ryu, S.Y.; Park, W.K.; Cho, H.; Choi, G. Biological evaluation of tanshindiols as EZH2 histone methyltransferase inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2486–2492. [Google Scholar] [CrossRef] [PubMed]
- Smadbeck, J.; Peterson, M.B.; Zee, B.M.; Garapaty, S.; Mago, A.; Lee, C.; Giannis, A.; Trojer, P.; Garcia, B.A.; Floudas, C.A. De novo peptide design and experimental validation of histone methyltransferase inhibitors. PLoS ONE 2014, 9, e95535. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, A.M. SET and MYND domain containing protein 3 in cancer. Am. J. Transl. Res. 2017, 9, 1–14. [Google Scholar] [PubMed]
- Hamamoto, R.; Silva, F.P.; Tsuge, M.; Nishidate, T.; Katagiri, T.; Nakamura, Y.; Furukawa, Y. Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci. 2006, 97, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.G.; Zou, J.N.; Wang, S.Z.; Zhang, T.C.; Xi, T. Novobiocin decreases SMYD3 expression and inhibits the migration of MDA-MB-231 human breast cancer cells. IUBMB Life 2010, 62, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Wynder, C.; Schmidt, D.M.; McCafferty, D.G.; Shiekhattar, R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem. Biol. 2006, 13, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Wynder, C.; Cooch, N.; Shiekhattar, R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005, 437, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hevi, S.; Kurash, J.K.; Lei, H.; Gay, F.; Bajko, J.; Su, H.; Sun, W.; Chang, H.; Xu, G.; et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009, 41, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Hino, S.; Kohrogi, K.; Nakao, M. Histone demethylase LSD1 controls the phenotypic plasticity of cancer cells. Cancer Sci. 2016, 107, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schule, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, S.; Sedukhina, A.S.; Nakagawa, Y.; Maeda, I.; Kubota, M.; Ohnuma, S.; Tsugawa, K.; Ohta, T.; Roche-Molina, M.; Bernal, J.A.; et al. LSD1 overexpression is associated with poor prognosis in basal-like breast cancer, and sensitivity to PARP inhibition. PLoS ONE 2015, 10, e0118002. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ma, S.; Zhao, G.; Yang, L.; Zhang, P.; Yi, Q.; Cheng, S. miR-708/LSD1 axis regulates the proliferation and invasion of breast cancer cells. Cancer Med. 2016, 5, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Hayward, D.; Cole, P.A. LSD1 Histone Demethylase Assays and Inhibition. Methods Enzymol. 2016, 573, 261–278. [Google Scholar] [PubMed]
- Morera, L.; Lubbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics 2016, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Greene, E.; Murray Stewart, T.; Goodwin, A.C.; Baylin, S.B.; Woster, P.M.; Casero, R.A., Jr. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc. Natl. Acad. Sci. USA 2007, 104, 8023–8028. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Stewart, T.M.; Wu, Y.; Baylin, S.B.; Marton, L.J.; Perkins, B.; Jones, R.J.; Woster, P.M.; Casero, R.A., Jr. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin. Cancer Res. 2009, 15, 7217–7228. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Huang, Y.; Marton, L.J.; Woster, P.M.; Davidson, N.E.; Casero, R.A., Jr. Polyamine analogs modulate gene expression by inhibiting lysine-specific demethylase 1 (LSD1) and altering chromatin structure in human breast cancer cells. Amino Acids 2011, 42, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Vasilatos, S.N.; Katz, T.A.; Oesterreich, S.; Wan, Y.; Davidson, N.E.; Huang, Y. Crosstalk between lysine-specific demethylase 1 (LSD1) and histone deacetylases mediates antineoplastic efficacy of HDAC inhibitors in human breast cancer cells. Carcinogenesis 2013, 34, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Vasilatos, S.N.; Bhargava, R.; Fine, J.L.; Oesterreich, S.; Davidson, N.E.; Huang, Y. Functional interaction of histone deacetylase 5 (HDAC5) and lysine-specific demethylase 1 (LSD1) promotes breast cancer progression. Oncogene 2017, 36, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dorsey, J.; Chuikov, S.; Perez-Burgos, L.; Zhang, X.; Jenuwein, T.; Reinberg, D.; Berger, S.L. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 2010, 285, 9636–9641. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Shen, L.; Suzuki, S.; Kurokawa, T.; Masuko, K.; Tanaka, Y.; Kato, H.; Mizuno, Y.; Yokoe, M.; Sugauchi, F.; et al. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol. Res. 2007, 37, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.T.; Weisenberger, D.J.; Velicescu, M.; Gonzales, F.A.; Lin, J.C.; Liang, G.; Jones, P.A. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer Res. 2002, 62, 6456–6461. [Google Scholar] [PubMed]
- Casciello, F.; Windloch, K.; Gannon, F.; Lee, J.S. Functional Role of G9a Histone Methyltransferase in Cancer. Front. Immunol. 2015, 6, 487. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.W.; Hua, K.T.; Kao, H.J.; Chi, C.C.; Wei, L.H.; Johansson, G.; Shiah, S.G.; Chen, P.S.; Jeng, Y.M.; Cheng, T.Y.; et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010, 70, 7830–7840. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Shen, L.; Ahmed, S.; Boumber, Y.; Sekido, Y.; Haddad, B.R.; Issa, J.P. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS ONE 2008, 3, e2037. [Google Scholar] [CrossRef] [PubMed]
- Kubicek, S.; O’Sullivan, R.J.; August, E.M.; Hickey, E.R.; Zhang, Q.; Teodoro, M.L.; Rea, S.; Mechtler, K.; Kowalski, J.A.; Homon, C.A.; et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 2007, 25, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Tian, Y.; Salwen, H.R.; Chlenski, A.; Godley, L.A.; Raj, J.U.; Yang, Q. Histone-lysine methyltransferase EHMT2 is involved in proliferation, apoptosis, cell invasion, and DNA methylation of human neuroblastoma cells. Anticancer Drugs 2013, 24, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, Q.; Paulk, J.; Kubicek, S.; Kemp, M.M.; Adams, D.J.; Shamji, A.F.; Wagner, B.K.; Schreiber, S.L. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem. Biol. 2012, 7, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- De Souza Rocha Simonini, P.; Breiling, A.; Gupta, N.; Malekpour, M.; Youns, M.; Omranipour, R.; Malekpour, F.; Volinia, S.; Croce, C.M.; Najmabadi, H.; et al. Epigenetically deregulated microRNA-375 is involved in a positive feedback loop with estrogen receptor alpha in breast cancer cells. Cancer Res. 2010, 70, 9175–9184. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Zamore, P.D. microPrimer: The biogenesis and function of microRNA. Development 2005, 132, 4645–4652. [Google Scholar] [CrossRef] [PubMed]
- Davis-Dusenbery, B.N.; Hata, A. MicroRNA in Cancer: The Involvement of Aberrant MicroRNA Biogenesis Regulatory Pathways. Genes Cancer 2011, 1, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Mulrane, L.; McGee, S.F.; Gallagher, W.M.; O’Connor, D.P. miRNA Dysregulation in Breast Cancer. Cancer Res. 2013, 73, 6554–6562. [Google Scholar] [CrossRef] [PubMed]
- Davoren, P.A.; McNeill, R.E.; Lowery, A.J.; Kerin, M.J.; Miller, N. Identification of suitable endogenous control genes for microRNA gene expression analysis in human breast cancer. BMC Mol. Biol. 2008, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.B.; Christoffersen, N.R.; Jacobsen, A.; Lindow, M.; Krogh, A.; Lund, A.H. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J. Biol. Chem. 2008, 283, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Si, M.L.; Zhu, S.; Wu, H.; Lu, Z.; Wu, F.; Mo, Y.Y. miR-21-mediated tumor growth. Oncogene 2007, 26, 2799–2803. [Google Scholar] [CrossRef] [PubMed]
- Lowery, A.J.; Miller, N.; Devaney, A.; McNeill, R.E.; Davoren, P.A.; Lemetre, C.; Benes, V.; Schmidt, S.; Blake, J.; Ball, G.; et al. MicroRNA signatures predict oestrogen receptor, progesterone receptor and HER2/neu receptor status in breast cancer. Breast Cancer Res. 2009, 11, R27. [Google Scholar] [CrossRef] [PubMed]
- Mattie, M.D.; Benz, C.C.; Bowers, J.; Sensinger, K.; Wong, L.; Scott, G.K.; Fedele, V.; Ginzinger, D.; Getts, R.; Haqq, C. Optimized high-throughput microRNA expression profiling provides novel biomarker assessment of clinical prostate and breast cancer biopsies. Mol. Cancer 2006, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bracken, C.P.; Bert, A.G.; Goodall, G.J. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 2008, 7, 3112–3118. [Google Scholar] [CrossRef] [PubMed]
- Sempere, L.F.; Christensen, M.; Silahtaroglu, A.; Bak, M.; Heath, C.V.; Schwartz, G.; Wells, W.; Kauppinen, S.; Cole, C.N. Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res. 2007, 67, 11612–11620. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Polytarchou, C.; Hatziapostolou, M.; Kottakis, F.; Maroulakou, I.G.; Struhl, K.; Tsichlis, P.N. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci. Signal 2009, 2, ra62. [Google Scholar] [CrossRef] [PubMed]
- Gandellini, P.; Doldi, V.; Zaffaroni, N. microRNAs as players and signals in the metastatic cascade: Implications for the development of novel anti-metastatic therapies. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cava, C.; Bertoli, G.; Castiglioni, I. Integrating genetics and epigenetics in breast cancer: Biological insights, experimental, computational methods and therapeutic potential. BMC Syst. Biol. 2015, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Cortez, V.; Vadlamudi, R.K. Epigenetics of estrogen receptor signaling: Role in hormonal cancer progression and therapy. Cancers (Basel) 2011, 3, 1691–1707. [Google Scholar] [CrossRef] [PubMed]
- Hervouet, E.; Cartron, P.F.; Jouvenot, M.; Delage-Mourroux, R. Epigenetic regulation of estrogen signaling in breast cancer. Epigenetics 2013, 8, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Green, K.A.; Carroll, J.S. Oestrogen-receptor-mediated transcription and the influence of co-factors and chromatin state. Nat. Rev. Cancer 2007, 7, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Girault, I.; Bieche, I.; Lidereau, R. Role of estrogen receptor alpha transcriptional coregulators in tamoxifen resistance in breast cancer. Maturitas 2006, 54, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Fleming, F.J.; Myers, E.; Kelly, G.; Crotty, T.B.; McDermott, E.W.; O’Higgins, N.J.; Hill, A.D.; Young, L.S. Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. J. Clin. Pathol. 2004, 57, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Iwase, H.; Omoto, Y.; Toyama, T.; Yamashita, H.; Hara, Y.; Sugiura, H.; Zhang, Z. Clinical significance of AIB1 expression in human breast cancer. Breast Cancer Res. Treat. 2003, 80, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.M.; Guo, C.; Zhang, J. Nuclear receptor corepressor complexes in cancer: Mechanism, function and regulation. Am. J. Clin. Exp. Urol. 2014, 2, 169–187. [Google Scholar] [PubMed]
- Legare, S; Basik, M. Minireview: The Link Between ERalpha Corepressors and Histone Deacetylases in Tamoxifen Resistance in Breast Cancer. Mol. Endocrinol. 2016, 30, 965–976. [Google Scholar]
- Zou, Z.; Luo, X.; Nie, P.; Wu, B.; Zhang, T.; Wei, Y.; Wang, W.; Geng, G.; Jiang, J.; Mi, Y. Inhibition of SRC-3 enhances sensitivity of human cancer cells to histone deacetylase inhibitors. Biochem. Biophys. Res. Commun. 2016, 478, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Phan, N.L.; Trinh, N.V.; Pham, P.V. Low concentrations of 5-aza-2′-deoxycytidine induce breast cancer stem cell differentiation by triggering tumor suppressor gene expression. Onco Targets Ther. 2016, 9, 49–59. [Google Scholar] [PubMed]
- Lorico, A.; Rappa, G. Phenotypic heterogeneity of breast cancer stem cells. J. Oncol. 2011, 2011, 135039. [Google Scholar]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef] [PubMed]
- van Vlerken, L.E.; Hurt, E.M.; Hollingsworth, R.E. The role of epigenetic regulation in stem cell and cancer biology. J. Mol. Med. (Berl.) 2012, 90, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Fuller, M. Stem cells and cancer: Two faces of eve. Cell 2006, 124, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Pajonk, F.; Vlashi, E.; McBride, W.H. Radiation resistance of cancer stem cells: The 4 R’s of radiobiology revisited. Stem Cells 2010, 28, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Moncharmont, C.; Levy, A.; Gilormini, M.; Bertrand, G.; Chargari, C.; Alphonse, G.; Ardail, D.; Rodriguez-Lafrasse, C.; Magne, N. Targeting a cornerstone of radiation resistance: Cancer stem cell. Cancer Lett. 2012, 322, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Kim, Y.S.; Kang, M.J.; Farrar, W.L.; Hurt, E.M. Long-term recovery of irradiated prostate cancer increases cancer stem cells. Prostate 2012, 72, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Krause, M.; Baumann, M. Cancer stem cells at the crossroads of current cancer therapy failures--radiation oncology perspective. Semin. Cancer Biol. 2010, 20, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Glass, J. The phenotypic radiation resistance of CD44+/CD24(-or low) breast cancer cells is mediated through the enhanced activation of ATM signaling. PLoS ONE 2011, 6, e24080. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Sacca, M.; Vigneri, P.; De Maria, R. Cancer stem cells and chemosensitivity. Clin. Cancer Res. 2011, 17, 4942–4947. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Zeppernick, F.; Ahmadi, R.; Campos, B.; Dictus, C.; Helmke, B.M.; Becker, N.; Lichter, P.; Unterberg, A.; Radlwimmer, B.; Herold-Mende, C.C. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin. Cancer Res. 2008, 14, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Balic, M.; Lin, H.; Young, L.; Hawes, D.; Giuliano, A.; McNamara, G.; Datar, R.H.; Cote, R.J. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin. Cancer Res. 2006, 12, 5615–5621. [Google Scholar] [CrossRef] [PubMed]
- Witt, A.E.; Lee, C.W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 2016, 36, 1707–1720. [Google Scholar] [CrossRef] [PubMed]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Leis, O.; Eguiara, A.; Lopez-Arribillaga, E.; Alberdi, M.J.; Hernandez-Garcia, S.; Elorriaga, K.; Pandiella, A.; Rezola, R.; Martin, A.G. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 2011, 31, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Liu, S.; Wang, P.; Zhao, S.; Wang, F.; Bing, L.; Zhang, Y.; Ling, E.A.; Gao, J.; Hao, A. Expression profile of embryonic stem cell-associated genes Oct4, Sox2 and Nanog in human gliomas. Histopathology 2011, 59, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Jeter, C.R.; Liu, B.; Liu, X.; Chen, X.; Liu, C.; Calhoun-Davis, T.; Repass, J.; Zaehres, H.; Shen, J.J.; Tang, D.G. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene 2011, 30, 3833–3845. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.L.; Fiscus, R.R.; Tung, J.W.; Tin, V.P.; Cheng, L.C.; Sihoe, A.D.; Fink, L.M.; Ma, Y.; Wong, M.P. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS ONE 2010, 5, e14062. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Convery, P.A.; Matsumura, N.; Whitaker, R.S.; Kondoh, E.; Perry, T.; Huang, Z.; Bentley, R.C.; Mori, S.; Fujii, S.; et al. Epigenetic regulation of CD133 and tumorigenicity of CD133 + ovarian cancer cells. Oncogene 2009, 28, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Tabu, K.; Sasai, K.; Kimura, T.; Wang, L.; Aoyanagi, E.; Kohsaka, S.; Tanino, M.; Nishihara, H.; Tanaka, S. Promoter hypomethylation regulates CD133 expression in human gliomas. Cell Res. 2008, 18, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.M.; Tsai, H.C.; Glockner, S.C.; Lin, S.; Ohm, J.E.; Easwaran, H.; James, C.D.; Costello, J.F.; Riggins, G.; Eberhart, C.G.; et al. Abnormal DNA methylation of CD133 in colorectal and glioblastoma tumors. Cancer Res. 2008, 68, 8094–8103. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Ding, W.; Rountree, C.B. Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-beta. Hepatology 2010, 51, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Fouse, S.D.; Shen, Y.; Pellegrini, M.; Cole, S.; Meissner, A.; Van Neste, L.; Jaenisch, R.; Fan, G. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell 2008, 2, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bi, C.; Cheong, L.L.; Mahara, S.; Liu, S.C.; Tay, K.G.; Koh, T.L.; Yu, Q.; Chng, W.J. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood 2011, 118, 2830–2839. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Suzuki, E.; Negishi, M.; Saraya, A.; Miyagi, S.; Konuma, T.; Tanaka, S.; Tada, M.; Kanai, F.; Imazeki, F.; et al. 3-Deazaneplanocin A is a promising therapeutic agent for the eradication of tumor-initiating hepatocellular carcinoma cells. Int. J. Cancer 2011, 130, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Suva, M.L.; Riggi, N.; Janiszewska, M.; Radovanovic, I.; Provero, P.; Stehle, J.C.; Baumer, K.; Le Bitoux, M.A.; Marino, D.; Cironi, L.; et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009, 69, 9211–9218. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Hurt, E.M.; Mathews, L.A.; Cabarcas, S.M.; Sun, L.; Marquez, V.E.; Danesi, R.; Farrar, W.L. Pharmacologic disruption of Polycomb Repressive Complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol. Cancer 2011, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Pietersen, A.M.; van Lohuizen, M. Stem cell regulation by polycomb repressors: Postponing commitment. Curr. Opin. Cell Biol. 2008, 20, 201–207. [Google Scholar] [CrossRef] [PubMed]
- van Vlerken, L.E.; Kiefer, C.M.; Morehouse, C.; Li, Y.; Groves, C.; Wilson, S.D.; Yao, Y.; Hollingsworth, R.E.; Hurt, E.M. EZH2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl. Med. 2013, 2, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, S.; Hersey, J.M.; Mellor, P.; Dai, W.; Santos-Silva, A.; Liber, D.; Luk, L.; Titley, I.; Carden, C.P.; Box, G.; et al. Ovarian cancer stem cell-like side populations are enriched following chemotherapy and overexpress EZH2. Mol. Cancer Ther. 2011, 10, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Hurt, E.M.; Farrar, W.L. Clinical significance of Polycomb gene expression in brain tumors. Mol. Cancer 2010, 9, 265. [Google Scholar] [CrossRef] [PubMed]
- Paranjape, A.N.; Balaji, S.A.; Mandal, T.; Krushik, E.V.; Nagaraj, P.; Mukherjee, G.; Rangarajan, A. Bmi1 regulates self-renewal and epithelial to mesenchymal transition in breast cancer cells through Nanog. BMC Cancer 2014, 14, 785. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, F.; Ren, Q.; Sun, H.; Xu, Z.; Lan, R.; Liu, Y.; Ward, D.; Quan, J.; Ye, T.; et al. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 2011, 71, 7238–7249. [Google Scholar] [CrossRef] [PubMed]
- Duru, N.; Gernapudi, R.; Eades, G.; Eckert, R.; Zhou, Q. Epigenetic Regulation of miRNAs and Breast Cancer Stem Cells. Curr. Pharmacol. Rep. 2015, 1, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Nalls, D.; Tang, S.N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Fu, J.; Xiao, X.; Wu, J.; Wu, R.C. MiR-34a regulates therapy resistance by targeting HDAC1 and HDAC7 in breast cancer. Cancer Lett. 2014, 354, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbacher, D.; Balic, M.; Pichler, M. The role of microRNAs in breast cancer stem cells. Int. J. Mol. Sci. 2013, 14, 14712–14723. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Tan, W.; Chen, K.; You, N.; Zhu, S.; Liang, G.; Xie, X.; Li, Q.; Zeng, Y.; Ouyang, N.; et al. Prognostic Value of a BCSC-associated MicroRNA Signature in Hormone Receptor-Positive HER2-Negative Breast Cancer. EBioMedicine 2016, 11, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Valdez, K.E.; Carletti, M.Z.; Behbod, F. Targeting the perpetrator: Breast cancer stem cell therapeutics. Curr. Drug Targets 2010, 11, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Heo, I.; Joo, C.; Kim, Y.K.; Ha, M.; Yoon, M.J.; Cho, J.; Yeom, K.H.; Han, J.; Kim, V.N. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 2009, 138, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Martello, G.; Rosato, A.; Ferrari, F.; Manfrin, A.; Cordenonsi, M.; Dupont, S.; Enzo, E.; Guzzardo, V.; Rondina, M.; Spruce, T.; et al. A MicroRNA targeting dicer for metastasis control. Cell 2010, 141, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Dagdemir, A.; Judes, G.; Lebert, A.; Echegut, M.; Karsli-Ceppioglu, S.; Rifai, K.; Daures, M.; Ngollo, M.; Dubois, L.; Penault-Llorca, F.; et al. Epigenetic Modifications with DZNep, NaBu and SAHA in Luminal and Mesenchymal-like Breast Cancer Subtype Cells. Cancer Genom. Proteom. 2016, 13, 291–303. [Google Scholar]

© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).