
What Can De Novo Protein Design Bring to the Treatment of Hematological Disorders?

Abstract
Simple Summary
Abstract
1. Introduction
2. Development of De Novo Protein Design
3. Biomedical Applications of De Novo Protein Design
3.1. Novel Diagnostic and Therapeutic Drugs
3.2. Novel Vaccines
3.3. Novel Biological Materials
4. De Novo Protein Design in Treating Hematological Disorders
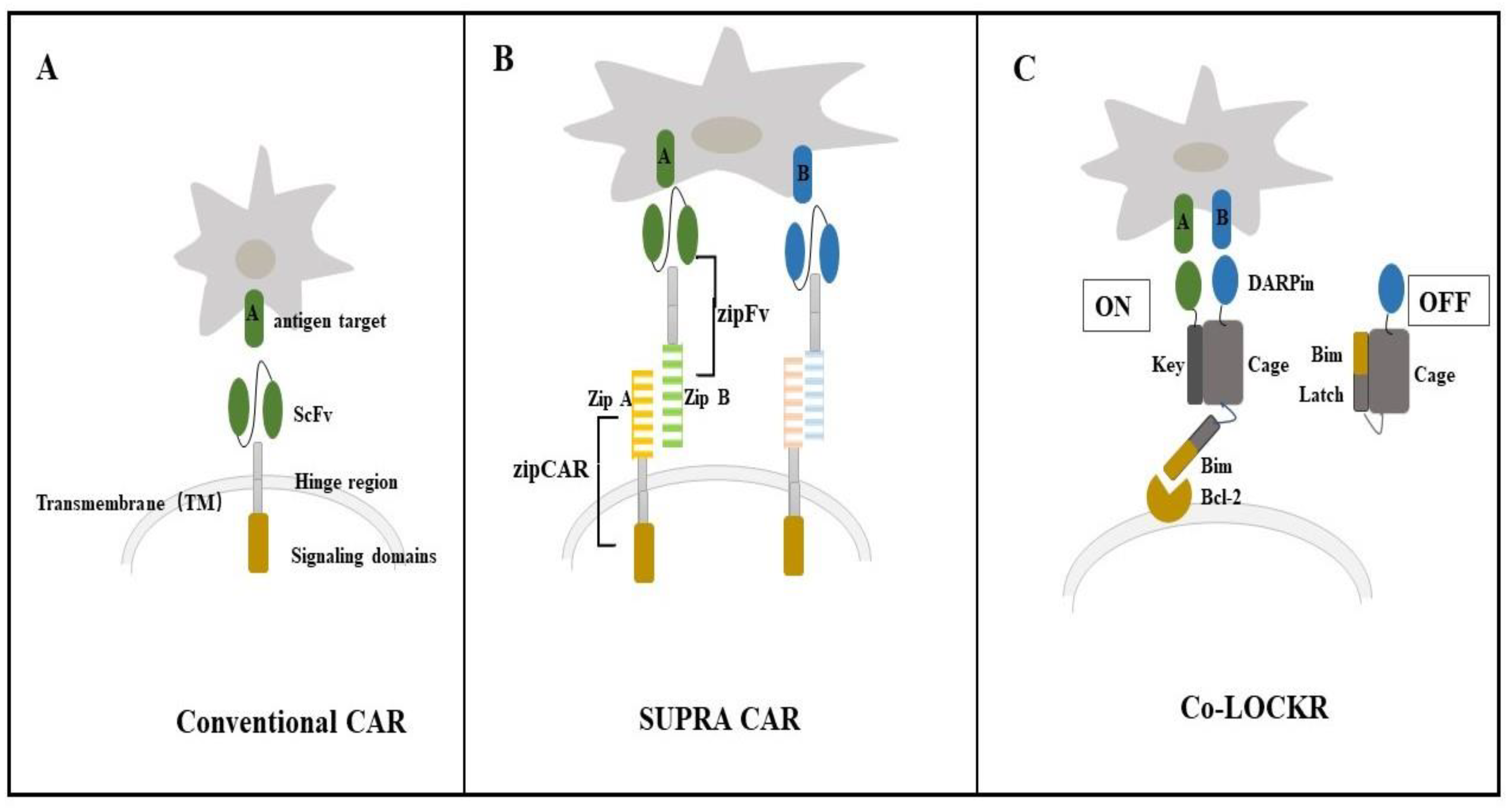
4.1. Colocalization-Dependent Protein Switches-Latching Orthogonal Cage-Key pRotein (Co-LOCKR) for CAR-T Cell Therapy
4.2. De Novo Designed Molecules
4.3. Neoleukin-2/15 (Neo-2/15)
5. Discussions, Success and Challenges
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pennisi, E. Bioinformatics. Gene counters struggle to get the right answer. Science 2003, 301, 1040–1041. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; Fitz Hugh, W.; et al. International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 412, 565. [Google Scholar]
- Gonzaga-Jauregui, C.; Lupski, J.R.; Gibbs, R.A. Human genome sequencing in health and disease. Annu. Rev. Med. 2012, 63, 35–61. [Google Scholar] [CrossRef] [PubMed]
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, C.F.; Simone, J.V.; Corrigan, J.J.; Seeler, R.A.; Edelstein, G.; Vanderheiden, J.; Schulman, I. Treatment of hemophilia with glycine-precipitated factor 8. N. Engl. J. Med. 1996, 275, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Roth, D.A.; Kessler, C.M.; Pasi, K.J.; Rup, B.; Courter, S.G.; Tubridy, K.L.; Recombinant Factor IX Study Group. Human recombinant factor IX: Safety and efficacy studies in hemophilia B patients previously treated with plasma-derived factor IX concentrates. Blood 2001, 98, 3600–3606. [Google Scholar] [CrossRef] [PubMed]
- Finfer, S.; Bellomo, R.; Boyce, N.; French, J.; Myburgh, J.; Norton, R.; SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N. Engl. J. Med. 2004, 350, 2247–2256. [Google Scholar]
- Corwin, H.L.; Gettinger, A.; Pearl, R.G.; Fink, M.P.; Levy, M.M.; Shapiro, M.J.; Corwin, M.J.; Colton, T.; EPO Critical Care Trials Group. Efficacy of recombinant human erythropoietin in critically ill patients: A randomized controlled trial. JAMA 2002, 288, 2827–2835. [Google Scholar] [CrossRef]
- Katzan, I.L.; Furlan, A.J.; Lloyd, L.E.; Frank, J.I.; Harper, D.L.; Hinchey, J.A.; Hammel, J.P.; Qu, A.; Sila, C.A. Use of tissue-type plasminogen activator for acute ischemic stroke: The Cleveland area experience. JAMA 2000, 283, 1151–1158. [Google Scholar] [CrossRef]
- Smalley, R.V.; Andersen, J.W.; Hawkins, M.J.; Bhide, V.; O’Connell, M.J.; Oken, M.M.; Borden, E.C. Interferon alfa combined with cytotoxic chemotherapy for patients with non-Hodgkin’s lymphoma. N. Engl. J. Med. 1992, 327, 1336–1341. [Google Scholar] [CrossRef]
- Jazirehi, A.R.; Huerta-Yepez, S.; Cheng, G.; Bonavida, B. Rituximab (chimeric anti-CD20 monoclonal antibody) inhibits the constitutive nuclear factor-{kappa}B signaling pathway in non-Hodgkin’s lymphoma B-cell lines: Role in sensitization to chemotherapeutic drug-induced apoptosis. Cancer Res. 2005, 65, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Nashan, B.; Moore, R.; Amlot, P.; Schmidt, A.G.; Abeywickrama, K.; Soulillou, J.P. Randomised trial of basiliximab versus placebo for control of acute cellular rejection in renal allograft recipients. CHIB 201 International Study Group. Lancet 1997, 350, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Scott, L.J. Recombinant factor VIIa (Eptacog Alfa): A review of its use in congenital or acquired haemophilia and other congenital bleeding disorders. Drugs 2005, 65, 1161–1177. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, A.; Kumar, S.K. Chimeric antigen receptor T-cells, bispecific antibodies, and antibody-drug conjugates for multiple myeloma: An update. Am. J. Hematol. 2022, 97, 99–118. [Google Scholar] [CrossRef]
- Bock, A.M.; Nowakowski, G.S.; Wang, Y. Bispecific Antibodies for Non-Hodgkin Lymphoma Treatment. Curr. Treat. Options Oncol. 2022, 23, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Zhou, X.; Wang, X. Antibody-drug conjugates for the treatment of lymphoma: Clinical advances and latest progress. J. Hematol. Oncol. 2021, 14, 88. [Google Scholar] [CrossRef]
- Daher, M.; Basar, R.; Gokdemir, E.; Baran, N.; Uprety, N.; Nunez Cortes, A.K.; Mendt, M.; Kerbauy, L.N.; Banerjee, P.P.; Shanley, M.; et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood 2021, 137, 624–636. [Google Scholar] [CrossRef]
- Alabanza, L.M.; Xiong, Y.; Vu, B.; Webster, B.; Wu, D.; Hu, P.; Zhu, Z.; Dropulic, B.; Dash, P.; Schneider, D. Armored BCMA CAR T Cells Eliminate Multiple Myeloma and Are Resistant to the Suppressive Effects of TGF-β. Front. Immunol. 2022, 13, 832645. [Google Scholar] [CrossRef]
- The Nobel Prize in Chemistry 2018. Available online: https://www.nobelprize.org/prizes/chemistry/2018/summary/ (accessed on 3 October 2018).
- Taverna, D.M.; Goldstein, R.A. Why are proteins marginally stable? Proteins 2002, 46, 105–109. [Google Scholar] [CrossRef]
- Pan, X.; Kortemme, T. Recent advances in de novo protein design: Principles, methods, and applications. J. Biol. Chem. 2021, 296, 100558. [Google Scholar] [CrossRef]
- Huang, P.S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Coventry, B.; Goreshnik, I.; Huang, B.; Sheffler, W.; Park, J.S.; Jude, K.M.; Marković, I.; Kadam, R.U.; Verschueren, K.H.G.; et al. Design of protein-binding proteins from the target structure alone. Nature 2022, 605, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Fleming, P.J.; Rose, G.D. Do all backbone polar groups in proteins form hydrogen bonds? Protein Sci. 2005, 14, 1911–1917. [Google Scholar] [CrossRef] [PubMed]
- Korendovych, I.V.; DeGrado, W.F. De novo protein design, a retrospective. Q. Rev. Biophys. 2020, 53, e3. [Google Scholar] [CrossRef]
- Woolfson, D.N. A Brief History of De Novo Protein Design: Minimal, Rational, and Computational. J. Mol. Biol. 2021, 433, 167160. [Google Scholar] [CrossRef]
- Hill, R.B.; Raleigh, D.P.; Lombardi, A.; DeGrado, W.F. De novo design of helical bundles as models for understanding protein folding and function. Acc. Chem. Res. 2000, 33, 745–754. [Google Scholar] [CrossRef]
- Schafmeister, C.E.; LaPorte, S.L.; Miercke, L.J.; Stroud, R.M. A designed four helix bundle protein with native-like structure. Nat. Struct. Biol. 1997, 4, 1039–1046. [Google Scholar] [CrossRef]
- Harbury, P.B.; Plecs, J.J.; Tidor, B.; Alber, T.; Kim, P.S. High-resolution protein design with backbone freedom. Science 1998, 282, 1462–1467. [Google Scholar] [CrossRef]
- Krantz, B.A.; Sosnick, T.R. Engineered metal binding sites map the heterogeneous folding landscape of a coiled coil. Nat. Struct. Biol. 2001, 8, 1042–1047. [Google Scholar] [CrossRef]
- Das, R.; Baker, D. Macromolecular modeling with rosetta. Annu. Rev. Biochem. 2008, 77, 363–382. [Google Scholar] [CrossRef]
- O’Meara, M.J.; Leaver-Fay, A.; Tyka, M.D.; Stein, A.; Houlihan, K.; DiMaio, F.; Bradley, P.; Kortemme, T.; Baker, D.; Snoeyink, J.; et al. Combined covalent-electrostatic model of hydrogen bonding improves structure prediction with Rosetta. J. Chem. Theory Comput. 2015, 11, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Xu, Y.; Hu, X.; Liu, Y.; Liao, S.; Zhang, J.; Huang, C.; Hong, J.; Chen, Q.; Liu, H. A backbone-centred energy function of neural networks for protein design. Nature 2022, 602, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Koga, N.; Tatsumi-Koga, R.; Liu, G.; Xiao, R.; Acton, T.B.; Montelione, G.T.; Baker, D. Principles for designing ideal protein structures. Nature 2012, 491, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Correia, B.E.; Bates, J.T.; Loomis, R.J.; Baneyx, G.; Carrico, C.; Jardine, J.G.; Rupert, P.; Correnti, C.; Kalyuzhniy, O.; Vittal, V.; et al. Proof of principle for epitope-focused vaccine design. Nature 2014, 507, 201–206. [Google Scholar] [CrossRef]
- Jacobs, T.M.; Williams, B.; Williams, T.; Xu, X.; Eletsky, A.; Federizon, J.F.; Szyperski, T.; Kuhlman, B. Design of structurally distinct proteins using strategies inspired by evolution. Science 2016, 352, 689–690. [Google Scholar] [CrossRef]
- Lombardi, A.; Pirro, F.; Maglio, O.; Chino, M.; DeGrado, W.F. De Novo Design of Four-Helix Bundle Metalloproteins: One Scaffold, Diverse Reactivities. Acc. Chem. Res. 2019, 52, 1148–1159. [Google Scholar] [CrossRef]
- Watkins, D.W.; Jenkins, J.M.X.; Grayson, K.J.; Wood, N.; Steventon, J.W.; Le Vay, K.K.; Goodwin, M.I.; Mullen, A.S.; Bailey, H.J.; Crump, M.P.; et al. Construction and in vivo assembly of a catalytically proficient and hyperthermostable de novo enzyme. Nat. Commun. 2017, 8, 358. [Google Scholar] [CrossRef]
- Joh, N.H.; Wang, T.; Bhate, M.P.; Acharya, R.; Wu, Y.; Grabe, M.; Hong, M.; Grigoryan, G.; DeGrado, W.F. De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science 2014, 346, 1520–1524. [Google Scholar] [CrossRef]
- Joh, N.H.; Grigoryan, G.; Wu, Y.; DeGrado, W.F. Design of self-assembling transmembrane helical bundles to elucidate principles required for membrane protein folding and ion transport. Philos. Trans. R. Soc. B 2017, 372, 20160214. [Google Scholar] [CrossRef]
- Baker, D. What has de novo protein design taught us about protein folding and biophysics? Protein Sci. 2019, 28, 678–683. [Google Scholar] [CrossRef]
- Woolfson, D.N.; Bartlett, G.J.; Burton, A.J.; Heal, J.W.; Niitsu, A.; Thomson, A.R.; Wood, C.W. De novo protein design: How do we expand into the universe of possible protein structures? Curr. Opin. Struct. Biol. 2015, 33, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Smadbeck, J.; Kieslich, C.A.; Floudas, C.A. Protein folding and de novo protein design for biotechnological applications. Trends Biotechnol. 2014, 2, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Kuhlman, B.; Bradley, P.; Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef]
- Dawson, W.M.; Rhys, G.G.; Woolfson, D.N. Towards functional de novo designed proteins. Curr. Opin. Chem. Biol. 2019, 52, 102–111. [Google Scholar] [CrossRef]
- D’Amone, L.; Matzeu, G.; Quijano-Rubio, A.; Callahan, G.P.; Napier, B.; Baker, D.; Omenetto, F.G. Reshaping de Novo Protein Switches into Bioresponsive Materials for Biomarker, Toxin, and Viral Detection. Adv. Mater. 2022, e2208556. [Google Scholar] [CrossRef]
- Leong, S.W.; Lim, T.S.; Ismail, A.; Choong, Y.S. Integration of molecular dynamics simulation and hotspot residues grafting for de novo scFv design against Salmonella Typhi TolC protein. J. Mol. Recognit. 2018, 31, e2695. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Serohijos, A.W.; Newton, G.; Tassone, G.; Wang, Z.; Sgroi, D.C.; Dokholyan, N.V.; Basilion, J.P. Identification and rational redesign of peptide ligands to CRIP1, a novel biomarker for cancers. PLoS Comput. Biol. 2008, 4, e1000138. [Google Scholar] [CrossRef]
- Lin, S.; Wade, J.D.; Liu, S. De Novo Design of Flavonoid-Based Mimetics of Cationic Antimicrobial Peptides: Discovery, Development, and Applications. Acc. Chem. Res. 2021, 54, 104–119. [Google Scholar] [CrossRef]
- Chen, C.H.; Starr, C.G.; Troendle, E.; Wiedman, G.; Wimley, W.C.; Ulmschneider, J.P.; Ulmschneider, M.B. Simulation-Guided Rational de Novo Design of a Small Pore-Forming Antimicrobial Peptide. J. Am. Chem. Soc. 2019, 141, 4839–4848. [Google Scholar] [CrossRef]
- Khara, J.S.; Obuobi, S.; Wang, Y.; Hamilton, M.S.; Robertson, B.D.; Newton, S.M.; Yang, Y.Y.; Langford, P.R.; Ee, P.L.R. Disruption of drug-resistant biofilms using de novo designed short α-helical antimicrobial peptides with idealized facial amphiphilicity. Acta Biomater. 2017, 57, 103–114. [Google Scholar] [CrossRef]
- Park, H.; Lee, S.; Hong, S. Structure-based de novo design and synthesis of aminothiazole-based p38 MAP kinase inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 3784–3787. [Google Scholar] [CrossRef] [PubMed]
- Bellows, M.L.; Taylor, M.S.; Cole, P.A.; Shen, L.; Siliciano, R.F.; Fung, H.K.; Floudas, C.A. Discovery of entry inhibitors for HIV-1 via a new de novo protein design framework. Biophys. J. 2010, 99, 3445–3453. [Google Scholar] [CrossRef] [PubMed]
- Correia, B.E.; Ban, Y.E.; Holmes, M.A.; Xu, H.; Ellingson, K.; Kraft, Z.; Carrico, C.; Boni, E.; Sather, D.N.; Zenobia, C.; et al. Computational design of epitope-scaffolds allows induction of antibodies specific for a poorly immunogenic HIV vaccine epitope. Structure 2010, 18, 1116–1126. [Google Scholar] [CrossRef]
- Fleishman, S.J.; Whitehead, T.A.; Ekiert, D.C.; Dreyfus, C.; Corn, J.E.; Strauch, E.M.; Wilson, I.A.; Baker, D. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 2011, 332, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Leal, E.S.; Adler, N.S.; Fernández, G.A.; Gebhard, L.G.; Battini, L.; Aucar, M.G.; Videla, M.; Monge, M.E.; Hernández de Los Ríos, A.; Acosta Dávila, J.A.; et al. De novo design approaches targeting an envelope protein pocket to identify small molecules against dengue virus. Eur. J. Med. Chem. 2019, 182, 111628. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Pearce, R.; Zhang, Y. De novo design of protein peptides to block association of the SARS-CoV-2 spike protein with human ACE2. Aging 2020, 12, 11263–11276. [Google Scholar] [CrossRef]
- Li, S.; Wang, L.; Meng, J.; Zhao, Q.; Zhang, L.; Liu, H. De Novo design of potential inhibitors against SARS-CoV-2 Mpro. Comput. Biol. Med. 2022, 147, 105728. [Google Scholar] [CrossRef]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef]
- Allen, W.J.; Fochtman, B.C.; Balius, T.E.; Rizzo, R.C. Customizable de novo design strategies for DOCK: Application to HIVgp41 and other therapeutic targets. J. Comput. Chem. 2017, 38, 2641–2663. [Google Scholar] [CrossRef]
- Linsky, T.W.; Vergara, R.; Codina, N.; Nelson, J.W.; Walker, M.J.; Su, W.; Hsiang, T.Y.; Esser-Nobis, K.; Yu, K.; Hou, Y.J.; et al. De novo design of ACE2 protein decoys to neutralize SARS-CoV-2. bioRxiv 2020. [CrossRef]
- Smadbeck, J.; Peterson, M.B.; Zee, B.M.; Garapaty, S.; Mago, A.; Lee, C.; Giannis, A.; Trojer, P.; Garcia, B.A.; Floudas, C.A. De novo peptide design and experimental validation of histone methyltransferase inhibitors. PLoS ONE 2014, 9, e95535. [Google Scholar] [CrossRef] [PubMed]
- Arya, H.; Coumar, M.S. Design of novel ROCK inhibitors using fragment-based de novo drug design approach. J. Mol. Model. 2020, 26, 249. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.S.; Jin, W.Y.; Zhou, L.; Lu, X.H.; Li, W.Y.; Ma, Y.; Wang, R.L. Structure based design of selective SHP2 inhibitors by De novo design, synthesis and biological evaluation. J. Comput. Aided Mol. Des. 2019, 33, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Zhang, H.; Duan, Y.Q.; Dong, W.L.; Cheng, X.C.; Wang, S.Q.; Wang, R.L. Design novel inhibitors for treating cancer by targeting Cdc25B catalytic domain with de novo design. Comb. Chem. High Throughput Screen. 2014, 17, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Jeong, Y.; Hong, S. Structure-based de novo design and biochemical evaluation of novel BRAF kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Istivan, T.S.; Pirogova, E.; Gan, E.; Almansour, N.M.; Coloe, P.J.; Cosic, I. Biological effects of a de novo designed myxoma virus peptide analogue: Evaluation of cytotoxicity on tumor cells. PLoS ONE 2011, 6, e24809. [Google Scholar] [CrossRef]
- Wang, Q.; Liang, G.; Zhang, M.; Zhao, J.; Patel, K.; Yu, X.; Zhao, C.; Ding, B.; Zhang, G.; Zhou, F.; et al. De novo design of self-assembled hexapeptides as β-amyloid (Aβ) peptide inhibitors. ACS Chem. Neurosci. 2014, 5, 972–981. [Google Scholar] [CrossRef]
- Sievers, S.A.; Karanicolas, J.; Chang, H.W.; Zhao, A.; Jiang, L.; Zirafi, O.; Stevens, J.T.; Münch, J.; Baker, D.; Eisenberg, D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 2011, 475, 96–100. [Google Scholar] [CrossRef]
- Rajadas, J.; Liu, C.W.; Novick, P.; Kelley, N.W.; Inayathullah, M.; Lemieux, M.C.; Pande, V.S. Rationally designed turn promoting mutation in the amyloid-β peptide sequence stabilizes oligomers in solution. PLoS ONE 2011, 6, e21776. [Google Scholar] [CrossRef]
- Wu, J.; Ma, Y.; Zhou, H.; Zhou, L.; Du, S.; Sun, Y.; Li, W.; Dong, W.; Wang, R. Identification of protein tyrosine phosphatase 1B (PTP1B) inhibitors through De Novo Evolution, synthesis, biological evaluation and molecular dynamics simulation. Biochem. Biophys. Res. Commun. 2020, 526, 273–280. [Google Scholar] [CrossRef]
- Walls, A.C.; Fiala, B.; Schäfer, A.; Wrenn, S.; Pham, M.N.; Murphy, M.; Tse, L.V.; Shehata, L.; O’Connor, M.A.; Chen, C.; et al. Elicitation of Potent Neutralizing Antibody Responses by Designed Protein Nanoparticle Vaccines for SARS-CoV-2. Cell 2020, 183, 1367–1382. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, P.S.; Walls, A.C.; Golden, N.; Atyeo, C.; Fischinger, S.; Li, C.; Aye, P.; Navarro, M.J.; Lai, L.; Edara, V.V.; et al. Adjuvanting a subunit COVID-19 vaccine to induce protective immunity. Nature 2021, 594, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Marcandalli, J.; Fiala, B.; Ols, S.; Perotti, M.; de van der Schueren, W.; Snijder, J.; Hodge, E.; Benhaim, M.; Ravichandran, R.; Carter, L.; et al. Induction of Potent Neutralizing Antibody Responses by a Designed Protein Nanoparticle Vaccine for Respiratory Syncytial Virus. Cell 2019, 176, 1420–1431. [Google Scholar] [CrossRef] [PubMed]
- Sesterhenn, F.; Yang, C.; Bonet, J.; Cramer, J.T.; Wen, X.; Wang, Y.; Chiang, C.I.; Abriata, L.A.; Kucharska, I.; Castoro, G.; et al. De novo protein design enables the precise induction of RSV-neutralizing antibodies. Science 2020, 368, eaay5051. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, P.J.M.; Antanasijevic, A.; Berndsen, Z.; Yasmeen, A.; Fiala, B.; Bijl, T.P.L.; Bontjer, I.; Bale, J.B.; Sheffler, W.; Allen, J.D.; et al. Enhancing and shaping the immunogenicity of native-like HIV-1 envelope trimers with a two-component protein nanoparticle. Nat. Commun. 2019, 10, 4272. [Google Scholar] [CrossRef]
- Boyoglu-Barnum, S.; Ellis, D.; Gillespie, R.A.; Hutchinson, G.B.; Park, Y.-J.; Moin, S.M.; Acton, O.; Ravichandran, R.; Murphy, M.; Pettie, D. Elicitation of broadly protective immunity to influenza by multivalent hemagglutinin nanoparticle vaccines. bioRxiv 2020. [CrossRef]
- Bruun, T.U.J.; Andersson, A.C.; Draper, S.J.; Howarth, M. Engineering a Rugged Nanoscaffold To Enhance Plug-and-Display Vaccination. ACS Nano 2018, 12, 8855–8866. [Google Scholar] [CrossRef]
- Sundaram, R.; Lynch, M.P.; Rawale, S.V.; Sun, Y.; Kazanji, M.; Kaumaya, P.T. De novo design of peptide immunogens that mimic the coiled coil region of human T-cell leukemia virus type-1 glycoprotein 21 transmembrane subunit for induction of native protein reactive neutralizing antibodies. J. Biol. Chem. 2004, 279, 24141–24151. [Google Scholar] [CrossRef]
- Descalzi-Montoya, D.; Montel, R.A.; Smith, K.; Dziopa, E.; Darwich, A.; Yang, Z.; Bitsaktsis, C.; Korngold, R.; Sabatino, D. Synthetic Antibody Mimics Based on Cancer-Targeting Immunostimulatory Peptides. ChemBioChem 2021, 22, 1589–1596. [Google Scholar] [CrossRef]
- Céspedes, M.V.; Unzueta, U.; Tatkiewicz, W.; Sánchez-Chardi, A.; Conchillo-Solé, O.; Álamo, P.; Xu, Z.; Casanova, I.; Corchero, J.L.; Pesarrodona, M.; et al. In vivo architectonic stability of fully de novo designed protein-only nanoparticles. ACS Nano 2014, 8, 4166–4176. [Google Scholar] [CrossRef]
- Karch, C.P.; Doll, T.A.P.F.; Paulillo, S.M.; Nebie, I.; Lanar, D.E.; Corradin, G.; Burkhard, P. The use of a P. falciparum specific coiled-coil domain to construct a self-assembling protein nanoparticle vaccine to prevent malaria. J. Nanobiotechnol. 2017, 15, 62. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Gu, X.; Zhang, Q.; Xu, H.; Zhong, Z.; Deng, C. Cancer Nanomedicines Based on Synthetic Polypeptides. Biomacromolecules 2019, 20, 4299–4311. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Lee, N.K.; Kim, I.S. Bioengineered protein-based nanocage for drug delivery. Adv Drug Deliv Rev. 2016, 106, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.E.; Colovos, C.; Yeates, T.O. Nanohedra: Using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 2217–2221. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.T.; Reading, E.; Hura, G.L.; Tsai, K.L.; Laganowsky, A.; Asturias, F.J.; Tainer, J.A.; Robinson, C.V.; Yeates, T.O. Structure of a designed protein cage that self-assembles into a highly porous cube. Nat. Chem. 2014, 6, 1065–1071. [Google Scholar] [CrossRef]
- Quijano-Rubio, A.; Yeh, H.W.; Park, J.; Lee, H.; Langan, R.A.; Boyken, S.E.; Lajoie, M.J.; Cao, L.; Chow, C.M.; Miranda, M.C.; et al. De novo design of modular and tunable protein biosensors. Nature 2021, 591, 482–487. [Google Scholar] [CrossRef]
- Courbet, A.; Hansen, J.; Hsia, Y.; Bethel, N.; Park, Y.J.; Xu, C.; Moyer, A.; Boyken, S.E.; Ueda, G.; Nattermann, U.; et al. Computational design of mechanically coupled axle-rotor protein assemblies. Science 2022, 376, 383–390. [Google Scholar] [CrossRef]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef]
- Rodríguez-Carmona, E.; Villaverde, A. Nanostructured bacterial materials for innovative medicines. Trends Microbiol. 2010, 9, 423–430. [Google Scholar] [CrossRef]
- Sharifi, S.; Behzadi, S.; Laurent, S.; Forrest, M.L.; Stroeve, P.; Mahmoudi, M. Toxicity of Nanomaterials. Chem. Soc. Rev. 2012, 41, 2323–2343333. [Google Scholar] [CrossRef]
- Pieters, B.J.; van Eldijk, M.B.; Nolte, R.J.; Mecinović, J. Natural supramolecular protein assemblies. Chem. Soc. Rev. 2016, 45, 24–39. [Google Scholar] [CrossRef]
- Herrera Estrada, L.P.; Champion, J.A. Protein nanoparticles for therapeutic protein delivery. Biomater. Sci. 2015, 3, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Munro, H.N.; Linder, M.C. Ferritin: Structure, biosynthesis, and role in iron metabolism. Physiol. Rev. 1978, 58, 317–396. [Google Scholar] [CrossRef] [PubMed]
- Kramer, R.M.; Li, C.; Carter, D.C.; Stone, M.O.; Naik, R.R. Engineered protein cages for nanomaterial synthesis. J. Am. Chem. Soc. 2004, 126, 13282–13286. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, E.M.; Scorsato, V.; Dos Santos, M.L.; Júnior, A.T.; Tada, S.F.; Dos Santos, C.A.; de Toledo, M.A.; de Souza, A.P.; Polikarpov, I.; Aparicio, R. Crystal structure of a small heat-shock protein from Xylella fastidiosa reveals a distinct high-order structure. Acta Crystallogr. Sect. F Struct. Biol. 2017, 73, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Kickhoefer, V.A.; Han, M.; Raval-Fernandes, S.; Poderycki, M.J.; Moniz, R.J.; Vaccari, D.; Silvestry, M.; Stewart, P.L.; Kelly, K.A.; Rome, L.H. Targeting vault nanoparticles to specific cell surface receptors. ACS Nano 2009, 3, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Stein, V.; Alexandrov, K. Synthetic protein switches: Design principles and applications. Trends Biotechnol. 2015, 33, 101–110. [Google Scholar] [CrossRef]
- Langan, R.A.; Boyken, S.E.; Ng, A.H.; Samson, J.A.; Dods, G.; Westbrook, A.M.; Nguyen, T.H.; Lajoie, M.J.; Chen, Z.; Berger, S.; et al. De novo design of bioactive protein switches. Nature 2019, 572, 205–210. [Google Scholar] [CrossRef]
- Junge, W.; Nelson, N. ATP synthase. Annu. Rev. Biochem. 2015, 84, 631–657. [Google Scholar] [CrossRef]
- Okuno, D.; Iino, R.; Noji, H. Rotation and structure of FoF1-ATP synthase. J. Biochem. 2011, 149, 655–664. [Google Scholar] [CrossRef]
- Deme, J.C.; Johnson, S.; Vickery, O.; Aron, A.; Monkhouse, H.; Griffiths, T.; James, R.H.; Berks, B.C.; Coulton, J.W.; Stansfeld, P.J.; et al. Structures of the stator complex that drives rotation of the bacterial flagellum. Nat. Microbiol. 2020, 5, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Mueller, K.T.; Maude, S.L.; Porter, D.L.; Frey, N.; Wood, P.; Han, X.; Waldron, E.; Chakraborty, A.; Awasthi, R.; Levine, B.L.; et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 2017, 130, 2317–2325. [Google Scholar] [CrossRef] [PubMed]
- Landoni, E.; Savoldo, B. Treating hematological malignancies with cell therapy: Where are we now? Expert Opin. Biol. Ther. 2018, 18, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The application of CAR-T cell therapy in hematological malignancies: Advantages and challenges. Acta Pharm. Sin. B 2018, 8, 539–551. [Google Scholar] [CrossRef]
- Roschewski, M.; Longo, D.L.; Wilson, W.H. CAR T-Cell Therapy for Large B-Cell Lymphoma—Who, When, and How? N. Engl. J. Med. 2022, 386, 692–696. [Google Scholar] [CrossRef]
- FDA. Anna Kwilas, PhD, Chair of the Review Committee, OTAT/DCGT. Summary Basis for Regulatory Action—Abecma. 2021. Available online: https://www.fda.gov/vaccines-blood-biologics/abecma-idecabtagene-vicleucel (accessed on 21 April 2021).
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438. [Google Scholar] [CrossRef]
- Delgado-Soler, L.; Pinto, M.; Tanaka-Gil, K.; Rubio-Martinez, J. Molecular determinants of Bim(BH3) peptide binding to pro-survival proteins. J. Chem. Inf. Model 2012, 52, 2107–2118. [Google Scholar] [CrossRef]
- Lajoie, M.J.; Boyken, S.E.; Salter, A.I.; Bruffey, J.; Rajan, A.; Langan, R.A.; Olshefsky, A.; Muhunthan, V.; Bick, M.J.; Gewe, M.; et al. Designed protein logic to target cells with precise combinations of surface antigens. Science 2020, 369, 1637–1643. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Cohen, P. Protein kinases--the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, H.; Hu, C.; Wang, H.; Liu, J.; Wang, W.; Liu, Q. An overview of kinase downregulators and recent advances in discovery approaches. Signal Transduct. Target. Ther. 2021, 6, 423. [Google Scholar] [CrossRef] [PubMed]
- Pereira, W.; Camps, I. De Novo Design of New Inhibitor of Mutated Tyrosine-Kinase for the Myeloid Leukemia Treatment. Curr. Pharm. Des. 2016, 22, 5057–5064. [Google Scholar] [CrossRef]
- Akdis, M.; Burgler, S.; Crameri, R.; Eiwegger, T.; Fujita, H.; Gomez, E.; Klunker, S.; Meyer, N.; O’Mahony, L.; Palomares, O.; et al. Interleukins, from 1 to 37, and interferon-γ: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2011, 127, 701–721. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Cretney, E.; Kershaw, M.H.; Hayakawa, Y. Cytokines in cancer immunity and immunotherapy. Immunol. Rev. 2004, 202, 275–293. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Lombardi, R.; Loetsch, C.; Zinkl, D.; Jackson, J.; Schofield, P.; Deenick, E.K.; King, C.; Phan, T.G.; Webster, K.E.; Sprent, J.; et al. Potent antitumour activity of interleukin-2-Fc fusion proteins requires Fc-mediated depletion of regulatory T-cells. Nat. Commun. 2017, 8, 15373. [Google Scholar] [CrossRef]
- Levin, A.M.; Bates, D.L.; Ring, A.M.; Krieg, C.; Lin, J.T.; Su, L.; Moraga, I.; Raeber, M.E.; Bowman, G.R.; Novick, P.; et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature 2012, 484, 529–533. [Google Scholar] [CrossRef]
- Spangler, J.B.; Moraga, I.; Mendoza, J.L.; Garcia, K.C. Insights into cytokine-receptor interactions from cytokine engineering. Annu. Rev. Immunol. 2015, 33, 139–167. [Google Scholar] [CrossRef]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef]
- Siegel, J.P.; Puri, R.K. Interleukin-2 toxicity. J. Clin. Oncol. 1991, 9, 694–704. [Google Scholar] [CrossRef]
- Krieg, C.; Létourneau, S.; Pantaleo, G.; Boyman, O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11906–11911. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.A.; Yu, S.; Ulge, U.Y.; Spangler, J.B.; Jude, K.M.; Labão-Almeida, C.; Ali, L.R.; Quijano-Rubio, A.; Ruterbusch, M.; Leung, I.; et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature 2019, 565, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.P.; Chari, A.; Ciferri, C.; Liu, W.T.; Rémigy, H.W.; Stark, H.; Wiesmann, C. Cryo-EM in drug discovery: Achievements, limitations and prospects. Nat. Rev. Drug. Discov. 2018, 17, 471–492. [Google Scholar] [CrossRef]
- Longchamp, J.N.; Rauschenbach, S.; Abb, S.; Escher, C.; Latychevskaia, T.; Kern, K.; Fink, H.W. Imaging proteins at the single-molecule level. Proc. Natl. Acad. Sci. USA 2017, 114, 1474–1479. [Google Scholar] [CrossRef]
- Sauerborn, M.; Brinks, V.; Jiskoot, W.; Schellekens, H. Immunological mechanism underlying the immune response to recombinant human protein therapeutics. Trends Pharmacol. Sci. 2010, 31, 53–59. [Google Scholar] [CrossRef]
- Hermeling, S.; Crommelin, D.J.; Schellekens, H.; Jiskoot, W. Structure-immunogenicity relationships of therapeutic proteins. Pharm. Res. 2004, 21, 897–903. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type | Application | Summary | Reference |
|---|---|---|---|
| Novel diagnostic drugs | infectious diseases | De novo designed protein could sensitively detect diverse targets, such as salmonella typhi TolC protein, anti-hepatitis B antibodies, botulinum neurotoxin B, and SARS COV-2. | [46,47] |
| neoplastic diseases | A high-affinity peptide (A1M) has been designed by computational protein design techniques, which may help the early diagnosis of breast cancer. | [48] | |
| Novel therapeutic drugs | infectious diseases | Bacterial infectious diseases: various de novo antimicrobial peptides have been designed for the treatment of bacteria-induced infections. Viral infectious diseases: various de novo designed inhibitors have been used for the treatment of viral infectious diseases, such as HIV, hepatitis C, H1N1, dengue fever, and COVID-19. | [49,50,51,52,53,54,55,56,57,58,59,60,61] |
| neoplastic diseases | Various de novo designed inhibitors, such as histone methyltransferase inhibitors, ROCK inhibitors, SHP2 inhibitors, BRAF kinase inhibitors, and Cdc25B inhibitors, could be considered as anticancer agents for further investigation. | [62,63,64,65,66,67] | |
| other diseases | De novo peptides, such as all-D-amino acid inhibitors and β-amyloid peptide inhibitors have been designed for the treatment of AD. De novo protein tyrosine phosphatase 1B has been designed for the treatment of type-2 diabetes mellitus. | [68,69,70,71] | |
| Novel vaccines | protein nanoparticle vaccines | Designed protein nanoparticles have been used as vaccines for SARS COV-2, perfusion respiratory syncytial virus, HIV-1 envelope, influenza hemagglutinin, and Plasmodium falciparum cysteine–rich protective antigen. | [72,73,74,75,76,77,78] |
| cancer-targeting peptide vaccines | Designed peptides could be used as vaccines for adult T-cell leukemia/lymphoma. A designed trimeric peptide could target and activate NK cell immunotoxicity directly toward tumors. | [79,80] | |
| Novel biological materials | designed protein-based nanoparticles and vehicles | Designed protein-based nanoparticles and vehicles could be used for imaging, drug delivery, and gene therapy. | [81,82,83] |
| designed protein-based nanocages | Designed protein-based nanocages could be used for vaccine, gene, and small molecule delivery. | [84,85] | |
| designed protein-based biosensors | Designed biosensors could be used to detect the anti-apoptosis protein Bcl-2, the IgG1 Fc domain, the Her2 receptor, botulinum neurotoxin B, cardiac Troponin I, and RBD of SARS-COV-2. | [59,86,87] | |
| designed protein-based machinery | Designed mechanical systems provide opportunities for genetically encodable nanomachines. | [88] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, H.; Cheng, Z.; Hu, Y.; Tang, L.V. What Can De Novo Protein Design Bring to the Treatment of Hematological Disorders? Biology 2023, 12, 166. https://doi.org/10.3390/biology12020166
Lu H, Cheng Z, Hu Y, Tang LV. What Can De Novo Protein Design Bring to the Treatment of Hematological Disorders? Biology. 2023; 12(2):166. https://doi.org/10.3390/biology12020166
Chicago/Turabian StyleLu, Hui, Zhipeng Cheng, Yu Hu, and Liang V. Tang. 2023. "What Can De Novo Protein Design Bring to the Treatment of Hematological Disorders?" Biology 12, no. 2: 166. https://doi.org/10.3390/biology12020166
APA StyleLu, H., Cheng, Z., Hu, Y., & Tang, L. V. (2023). What Can De Novo Protein Design Bring to the Treatment of Hematological Disorders? Biology, 12(2), 166. https://doi.org/10.3390/biology12020166