
Mobilome of Environmental Isolates of Clostridioides difficile



Abstract
1. Introduction
2. Results
2.1. Genetic Diversity of MGEs in Environmental C. difficile Isolates
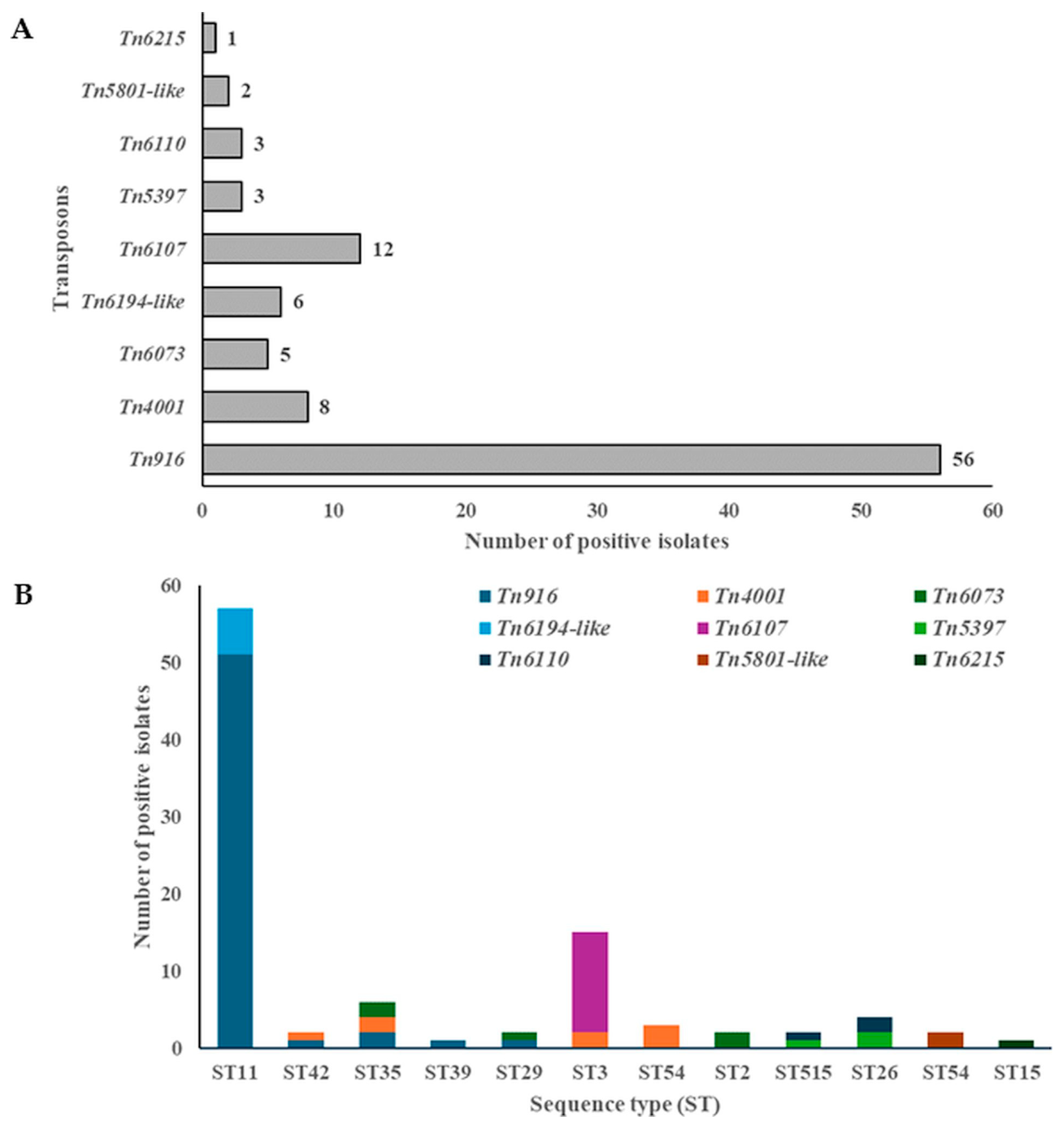
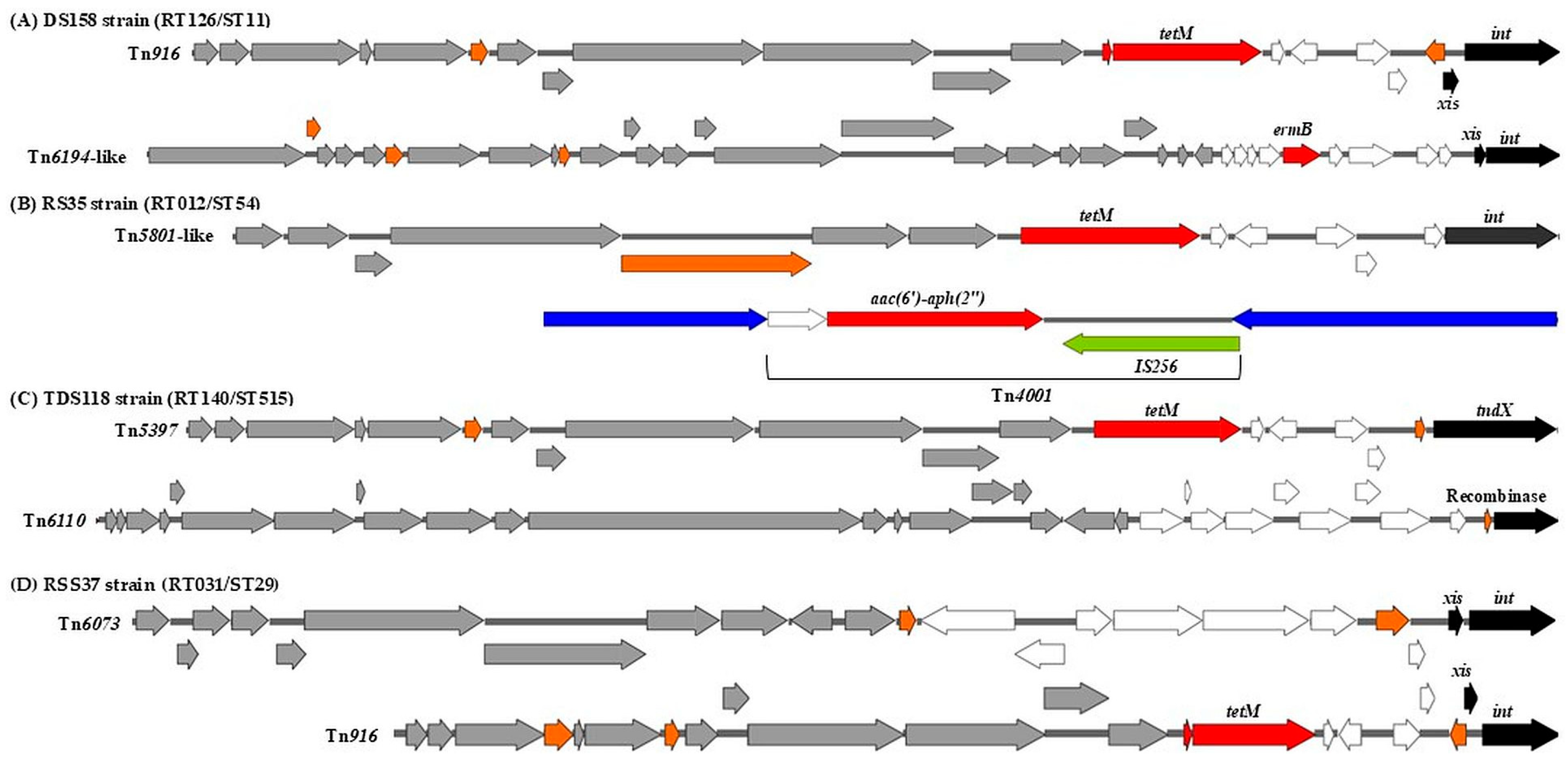
2.1.1. Mobilizable and Conjugative Transposons
2.1.2. Plasmids
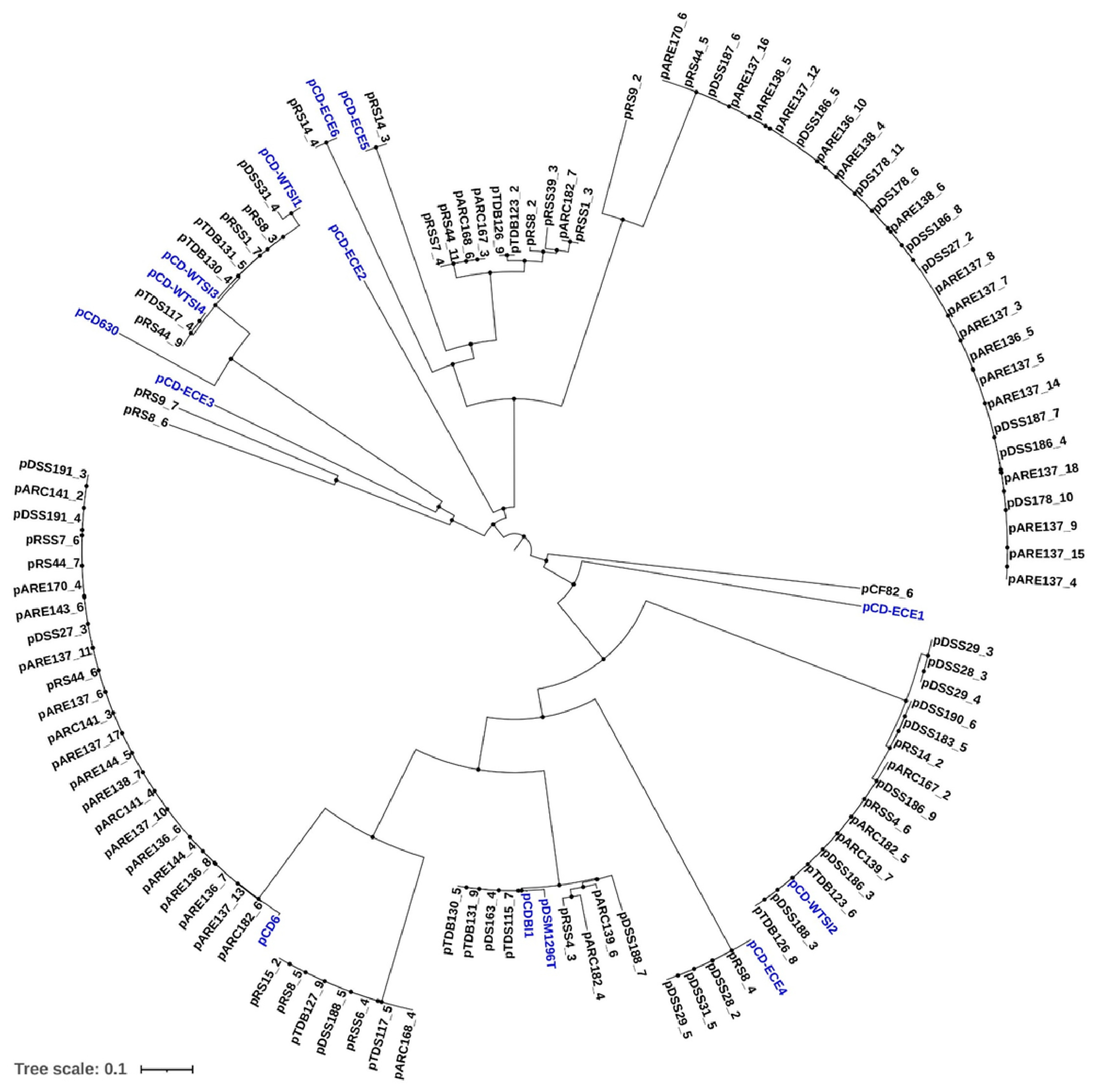
2.2. Phylogenetic Analysis of Identified Plasmids in Environmental C. difficile Genome Sequences
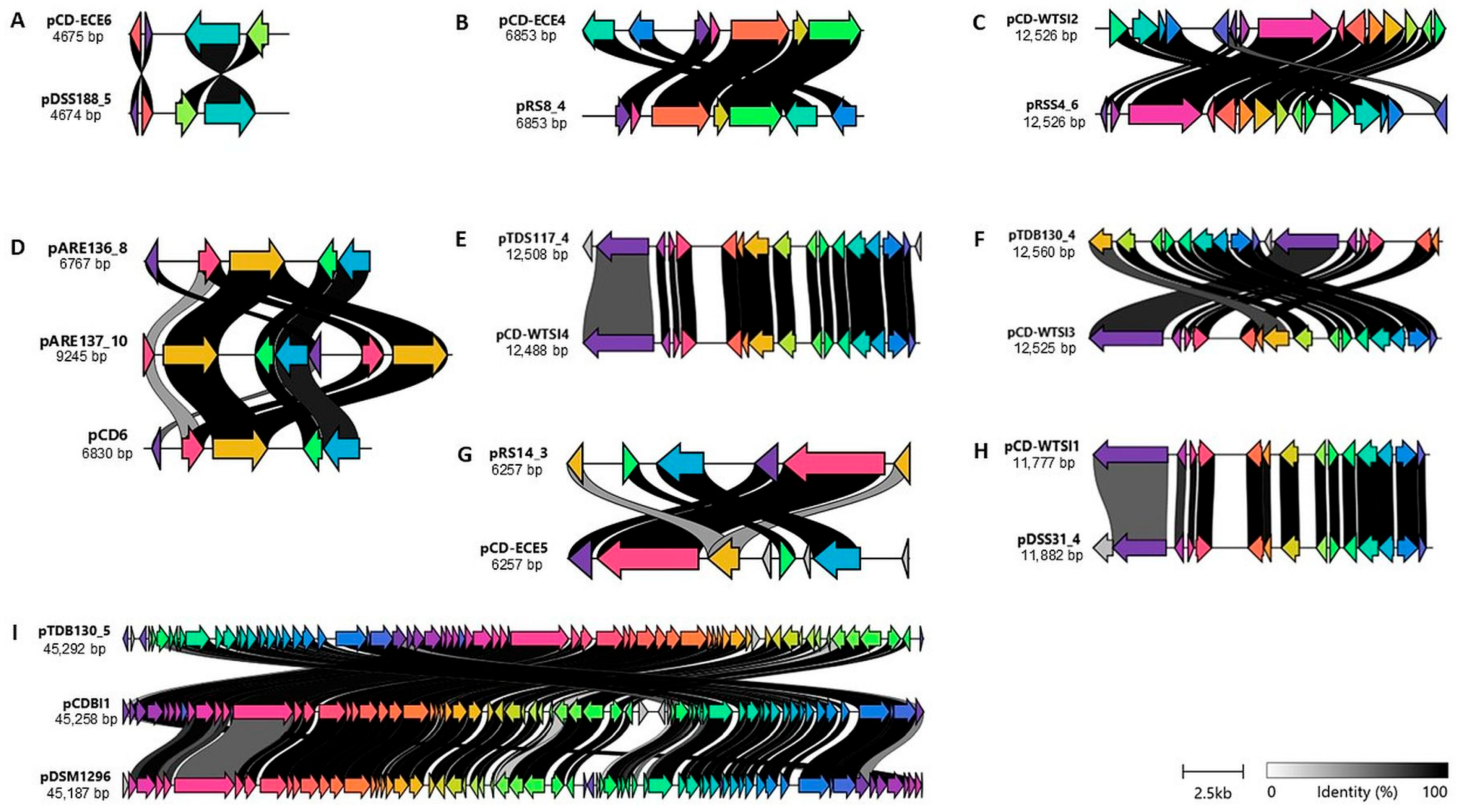
2.3. Comparison of the Identified Plasmids with Reference Plasmids
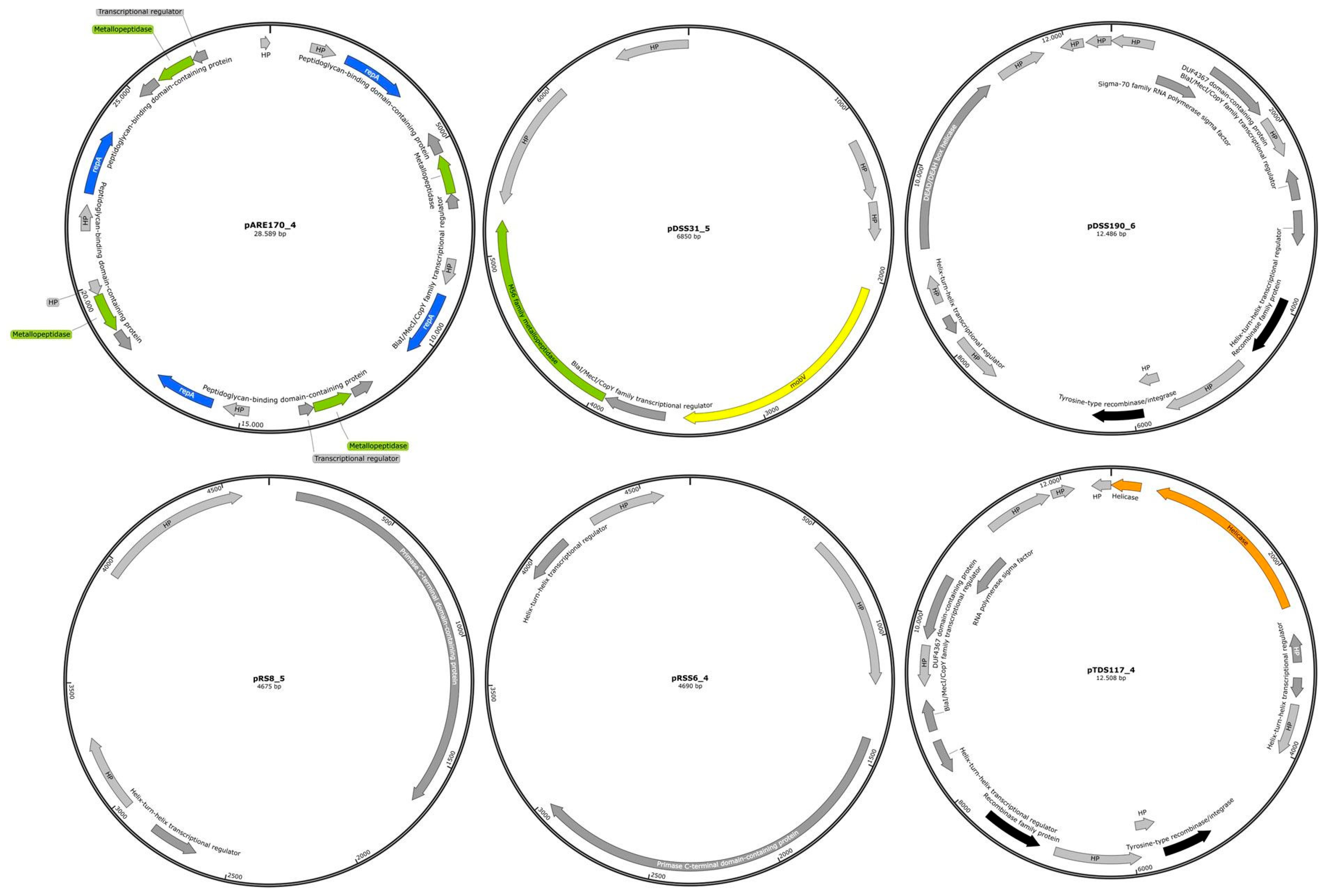
2.4. The Genome Organization of Identified Representative Cryptic Plasmids of C. difficile
3. Discussion
4. Materials and Methods
4.1. Isolation and Identification of C. difficile
4.2. Whole Genome Sequencing and Data Analysis
4.3. Analysis of MGEs
4.4. Phylogenetic Tree of Identified Plasmids in Environmental C. difficile Strains
4.5. Comparison of the Identified Plasmids with Reference Plasmids
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CDI | C. difficile infection |
| WGS | Whole genome sequencing |
| CTns | Conjugative transposons |
| MGEs | Mobile genetic elements |
| AMR | Antimicrobial resistance |
| MTns | Mobilizable transposons |
| MLST | Multilocus sequence typing |
| IS | Insertion sequence |
| ICEs | Integrative conjugative elements |
| HGT | Horizontal gene transfer |
| MLSB | Macrolide–lincosamide–streptogramin B |
| GI | Genomic island |
| ST | Sequence type |
| ORF | Open reading frame |
References
- Smits, W.K.; Lyras, D.; Lacy, D.B.; Wilcox, M.H.; Kuijper, E.J. Clostridium difficile infection. Nat. Rev. Dis. Prim. 2016, 2, 16020. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Schwan, C.; Jank, T. Clostridium difficile Toxin Biology. Annu. Rev. Microbiol. 2017, 71, 281–307. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.R.; Elliott, B.; Chang, B.J.; Perkins, T.T.; Riley, T.V. Diversity and Evolution in the Genome of Clostridium difficile. Clin. Microbiol. Rev. 2015, 28, 721–741. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, M.S.M.; Warburton, P.J.; Roberts, A.P.; Mullany, P.; Allan, E. Genetic organisation, mobility and predicted functions of genes on integrated, mobile genetic elements in sequenced strains of Clostridium difficile. PLoS ONE 2011, 6, e23014. [Google Scholar] [CrossRef]
- Sebaihia, M.; Wren, B.W.; Mullany, P.; Fairweather, N.F.; Minton, N.; Stabler, R.; Thomson, N.R.; Roberts, A.P.; Cerdeño-Tárraga, A.M.; Wang, H.; et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 2006, 38, 779–786. [Google Scholar] [CrossRef]
- Blau, K.; Gallert, C. Prophage Carriage and Genetic Diversity within Environmental Isolates of Clostridioides difficile. Int. J. Mol. Sci. 2023, 25, 2. [Google Scholar] [CrossRef]
- Peng, Z.; Jin, D.; Kim, H.B.; Stratton, C.W.; Wu, B.; Tang, Y.-W.; Sun, X. Update on Antimicrobial Resistance in Clostridium difficile: Resistance Mechanisms and Antimicrobial Susceptibility Testing. J. Clin. Microbiol. 2017, 55, 1998–2008. [Google Scholar] [CrossRef]
- Spigaglia, P. Recent advances in the understanding of antibiotic resistance in Clostridium difficile infection. Ther. Adv. Infect. Dis. 2016, 3, 23–42. [Google Scholar] [CrossRef]
- Mullany, P.; Williams, R.; Langridge, G.C.; Turner, D.J.; Whalan, R.; Clayton, C.; Lawley, T.; Hussain, H.; McCurrie, K.; Morden, N.; et al. Behavior and target site selection of conjugative transposon Tn916 in two different strains of toxigenic Clostridium difficile. Appl. Environ. Microbiol. 2012, 78, 2147–2153. [Google Scholar] [CrossRef]
- Roberts, A.P.; Pratten, J.; Wilson, M.; Mullany, P. Transfer of a conjugative transposon, Tn5397 in a model oral biofilm. FEMS Microbiol. Lett. 1999, 177, 63–66. [Google Scholar] [CrossRef]
- Mullany, P.; Wilks, M.; Lamb, I.; Clayton, C.; Wren, B.; Tabaqchali, S. Genetic analysis of a tetracycline resistance element from Clostridium difficile and its conjugal transfer to and from Bacillus subtilis. J. Gen. Microbiol. 1990, 136, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Jasni, A.S.; Mullany, P.; Hussain, H.; Roberts, A.P. Demonstration of conjugative transposon (Tn5397)-mediated horizontal gene transfer between Clostridium difficile and Enterococcus faecalis. Antimicrob. Agents Chemother. 2010, 54, 4924–4926. [Google Scholar] [CrossRef]
- Mullany, P.; Allan, E.; Roberts, A.P. Mobile genetic elements in Clostridium difficile and their role in genome function. Res. Microbiol. 2015, 166, 361–367. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Sebaihia, M.; Lawley, T.D.; Stabler, R.A.; Dawson, L.F.; Martin, M.J.; Holt, K.E.; Seth-Smith, H.M.B.; Quail, M.A.; Rance, R.; et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc. Natl. Acad. Sci. USA 2010, 107, 7527–7532. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Miyajima, F.; Roberts, P.; Ellison, L.; Pickard, D.J.; Martin, M.J.; Connor, T.R.; Harris, S.R.; Fairley, D.; Bamford, K.B.; et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat. Genet. 2013, 45, 109–113. [Google Scholar] [CrossRef]
- Wasels, F.; Monot, M.; Spigaglia, P.; Barbanti, F.; Ma, L.; Bouchier, C.; Dupuy, B.; Mastrantonio, P. Inter- and intraspecies transfer of a Clostridium difficile conjugative transposon conferring resistance to MLSB. Microb. Drug Resist. 2014, 20, 555–560. [Google Scholar] [CrossRef]
- Amy, J.; Bulach, D.; Knight, D.; Riley, T.; Johanesen, P.; Lyras, D. Identification of large cryptic plasmids in Clostridioides (Clostridium) difficile. Plasmid 2018, 96–97, 25–38. [Google Scholar] [CrossRef]
- Hornung, B.V.H.; Kuijper, E.J.; Smits, W.K. An in silico survey of Clostridioides difficile extrachromosomal elements. Microb. Genom. 2019, 5, e000296. [Google Scholar] [CrossRef]
- Ramírez-Vargas, G.; López-Ureña, D.; Badilla, A.; Orozco-Aguilar, J.; Murillo, T.; Rojas, P.; Riedel, T.; Overmann, J.; González, G.; Chaves-Olarte, E.; et al. Novel Clade C-I Clostridium difficile strains escape diagnostic tests, differ in pathogenicity potential and carry toxins on extrachromosomal elements. Sci. Rep. 2018, 8, 13951. [Google Scholar] [CrossRef]
- Ramírez-Vargas, G.; Rodríguez, C. Putative Conjugative Plasmids with tcdB and cdtAB Genes in Clostridioides difficile. Emerg. Infect. Dis. 2020, 26, 2287–2290. [Google Scholar] [CrossRef]
- Boekhoud, I.M.; Hornung, B.V.H.; Sevilla, E.; Harmanus, C.; Bos-Sanders, I.M.J.G.; Terveer, E.M.; Bolea, R.; Corver, J.; Kuijper, E.J.; Smits, W.K. Plasmid-mediated metronidazole resistance in Clostridioides difficile. Nat. Commun. 2020, 11, 598. [Google Scholar] [CrossRef] [PubMed]
- Blau, K.; Gallert, C. Prevalence, Antimicrobial Resistance and Toxin-Encoding Genes of Clostridioides difficile from Environmental Sources Contaminated by Feces. Antibiotics 2023, 12, 162. [Google Scholar] [CrossRef] [PubMed]
- Blau, K.; Berger, F.K.; Mellmann, A.; Gallert, C. Clostridioides difficile from Fecally Contaminated Environmental Sources: Resistance and Genetic Relatedness from a Molecular Epidemiological Perspective. Microorganisms 2023, 11, 2497. [Google Scholar] [CrossRef]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A free online platform for data visualization and graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.L.M.; Chooi, Y.-H. clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Kartalidis, P.; Skoulakis, A.; Tsilipounidaki, K.; Florou, Z.; Petinaki, E.; Fthenakis, G.C. Clostridioides difficile as a Dynamic Vehicle for the Dissemination of Antimicrobial-Resistance Determinants: Review and In Silico Analysis. Microorganisms 2021, 9, 1383. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, W.; Xiao, T.; Chen, Y.; Lv, T.; Wang, Y.; Zhang, S.; Cai, H.; Chi, X.; Kong, X.; et al. Comparative genomic and transmission analysis of Clostridioides difficile between environmental, animal, and clinical sources in China. Emerg. Microbes Infect. 2021, 10, 2244–2255. [Google Scholar] [CrossRef]
- Imwattana, K.; Kiratisin, P.; Riley, T.V.; Knight, D.R. Genomic basis of antimicrobial resistance in non-toxigenic Clostridium difficile in Southeast Asia. Anaerobe 2020, 66, 102290. [Google Scholar] [CrossRef]
- Dingle, K.E.; Didelot, X.; Quan, T.P.; Eyre, D.W.; Stoesser, N.; Marwick, C.A.; Coia, J.; Brown, D.; Buchanan, S.; Ijaz, U.Z.; et al. A Role for Tetracycline Selection in Recent Evolution of Agriculture-Associated Clostridium difficile PCR Ribotype 078. mBio 2019, 10, e02790-18. [Google Scholar] [CrossRef]
- O’Grady, K.; Knight, D.R.; Riley, T.V. Antimicrobial resistance in Clostridioides difficile. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 2459–2478. [Google Scholar] [CrossRef]
- de Vries, L.E.; Hasman, H.; Jurado Rabadán, S.; Agersø, Y. Sequence-Based Characterization of Tn5801-Like Genomic Islands in Tetracycline-Resistant Staphylococcus pseudintermedius and Other Gram-positive Bacteria from Humans and Animals. Front. Microbiol. 2016, 7, 576. [Google Scholar] [CrossRef]
- Imwattana, K.; Putsathit, P.; Collins, D.A.; Leepattarakit, T.; Kiratisin, P.; Riley, T.V.; Knight, D.R. Global evolutionary dynamics and resistome analysis of Clostridioides difficile ribotype 017. Microb. Genom. 2022, 8, 000792. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Vargas, G.; Quesada-Gómez, C.; Acuña-Amador, L.; López-Ureña, D.; Murillo, T.; Del Mar Gamboa-Coronado, M.; Chaves-Olarte, E.; Thomson, N.; Rodríguez-Cavallini, E.; Rodríguez, C. A Clostridium difficile Lineage Endemic to Costa Rican Hospitals Is Multidrug Resistant by Acquisition of Chromosomal Mutations and Novel Mobile Genetic Elements. Antimicrob. Agents Chemother. 2017, 61, e02054-16. [Google Scholar] [CrossRef] [PubMed]
- Lyon, B.R.; May, J.W.; Skurray, R.A. Tn4001: A gentamicin and kanamycin resistance transposon in Staphylococcus aureus. Mol. Gen. Genet. 1984, 193, 554–556. [Google Scholar] [CrossRef] [PubMed]
- Amy, J.; Johanesen, P.; Lyras, D. Extrachromosomal and integrated genetic elements in Clostridium difficile. Plasmid 2015, 80, 97–110. [Google Scholar] [CrossRef]
- Dost, I.; Abdel-Glil, M.; Persson, S.; Conza, K.L.; Oleastro, M.; Alves, F.; Maurischat, S.; Scholtzek, A.; Mazuet, C.; Diancourt, L.; et al. Genomic study of European Clostridioides difficile ribotype 002/sequence type 8. Microb. Genom. 2024, 10, 001270. [Google Scholar] [CrossRef]
- Smits, W.K.; Roseboom, A.M.; Corver, J. Plasmids of Clostridioides difficile. Curr. Opin. Microbiol. 2022, 65, 87–94. [Google Scholar] [CrossRef]
- Roseboom, A.M.; Ducarmon, Q.R.; Hornung, B.V.H.; Harmanus, C.; Crobach, M.J.T.; Kuijper, E.J.; Vossen, R.H.A.M.; Kloet, S.L.; Smits, W.K. Carriage of three plasmids in a single human clinical isolate of Clostridioides difficile. Plasmid 2023, 125, 102669. [Google Scholar] [CrossRef]
- Purdy, D.; O’KEeffe, T.A.T.; Elmore, M.; Herbert, M.; McLeod, A.; Bokori-Brown, M.; Ostrowski, A.; Minton, N.P. Conjugative transfer of clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Mol. Microbiol. 2002, 46, 439–452. [Google Scholar] [CrossRef]
- Ransom, E.M.; Ellermeier, C.D.; Weiss, D.S. Use of mCherry Red fluorescent protein for studies of protein localization and gene expression in Clostridium difficile. Appl. Environ. Microbiol. 2015, 81, 1652–1660. [Google Scholar] [CrossRef]
- Garnier, T.; Cole, S.T. Identification and molecular genetic analysis of replication functions of the bacteriocinogenic plasmid pIP404 from Clostridium perfringens. Plasmid 1988, 19, 151–160. [Google Scholar] [CrossRef]
- Larcombe, S.; Williams, G.C.; Amy, J.; Lim, S.C.; Riley, T.V.; Muleta, A.; Barugahare, A.A.; Powell, D.R.; Johanesen, P.A.; Cheng, A.C.; et al. A genomic survey of Clostridioides difficile isolates from hospitalized patients in Melbourne, Australia. Microbiol. Spectr. 2023, 11, e01352-23. [Google Scholar] [CrossRef]
- Lemee, L.; Dhalluin, A.; Testelin, S.; Mattrat, M.-A.; Maillard, K.; Lemeland, J.-F.; Pons, J.-L. Multiplex PCR Targeting tpi (Triose Phosphate Isomerase), tcdA (Toxin A), and tcdB (Toxin B) Genes for Toxigenic Culture of Clostridium difficile. J. Clin. Microbiol. 2004, 42, 5710–5714. [Google Scholar] [CrossRef] [PubMed]
- van Almsick, V.; Schuler, F.; Mellmann, A.; Schwierzeck, V. The Use of Long-Read Sequencing Technologies in Infection Control: Horizontal Transfer of a blaCTX-M-27 Containing lncFII Plasmid in a Patient Screening Sample. Microorganisms 2022, 10, 491. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Liang, Q.; Deng, M.; He, J.; Wang, M.; Hong, W.; Wu, J.; Lu, B.; Leptihn, S.; Yu, Y.; et al. BacAnt: A Combination Annotation Server for Bacterial DNA Sequences to Identify Antibiotic Resistance Genes, Integrons, and Transposable Elements. Front. Microbiol. 2021, 12, 649969. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.-M. ARG-ANNOT, a New Bioinformatic Tool To Discover Antibiotic Resistance Genes in Bacterial Genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef]
- Wang, M.; Goh, Y.-X.; Tai, C.; Wang, H.; Deng, Z.; Ou, H.-Y. VRprofile2: Detection of antibiotic resistance-associated mobilome in bacterial pathogens. Nucleic Acids Res. 2022, 50, W768–W773. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, C.; Li, W.-G.; Xu, J.-L.; Zhang, W.-Z.; Dai, Y.-F.; Lu, J.-X. Independent Microevolution Mediated by Mobile Genetic Elements of Individual Clostridium difficile Isolates from Clade 4 Revealed by Whole-Genome Sequencing. mSystems 2019, 4, e00252-18. [Google Scholar] [CrossRef]
- Wishart, D.S.; Han, S.; Saha, S.; Oler, E.; Peters, H.; Grant, J.R.; Stothard, P.; Gautam, V. PHASTEST: Faster than PHASTER, better than PHAST. Nucleic Acids Res. 2023, 51, W443–W450. [Google Scholar] [CrossRef]
- Lemoine, F.; Correia, D.; Lefort, V.; Doppelt-Azeroual, O.; Mareuil, F.; Cohen-Boulakia, S.; Gascuel, O. NGPhylogeny.fr: New generation phylogenetic services for non-specialists. Nucleic Acids Res. 2019, 47, W260–W265. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate No. | RT/ST | Tn/IS | AMR Genes |
|---|---|---|---|
| BP201, BP199, BP198, BP197, CF196, CF194, CF193, CF192, CF132, CF129, RSS62, RSS39, RSS64, RSS63, RSS61, RSS66, RSS68, RSS67, CF70, CF72, RSS65, BP71, CF88, CF109, CF90, TDS128, CF89, CF113, CF92 | RT127/ST11 | Tn916 | tetM |
| DSS189 | RT106/ST42 | Tn916 | tetM |
| Tn4001/IS256 | aac(6′)-aph(2″) | ||
| DSS185, DSS184 | RT328/ST35 | Tn916 | tetM |
| Tn4001/IS256 | aac(6′)-aph(2″) | ||
| Tn6073 | |||
| CF107, CF79, ASS23, ASS24, ASS25, ASS26, CF80, CF69, CF75, CF74, CF73, CF77, CF76, CF83, CF81, CF78 | RT126/ST11 | Tn916 | tetM |
| DS181, DS159, DS158 | Tn916 | tetM | |
| Tn6194-like | ermB | ||
| DS180, DS179, DS171 | RT078/ST11 | Tn916 | tetM |
| DS177, DS155, DS156 | Tn6194-like | ermB | |
| ARE146 | RT085/ST39 | Tn916 | tetM |
| RSS37 | RT031/ST29 | Tn916 | tetM |
| Tn6073 | |||
| DS161, DS157 | RT001/ST3 | Tn4001/IS256 | aac(6′)-aph(2″) |
| Tn6107 | |||
| DS163, RS154, RS153, RS148, TDB131, TDB130, RS17, TDB126, TDS115, TDS123 | Tn6107 | ||
| RS35, ASS20 | RT012/ST54 | Tn4001/IS256 | aac(6′)-aph(2″) |
| Tn5801-like | tetM | ||
| RSS10 | Tn4001/IS256 | aac(6′)-aph(2″) | |
| S45 | RT014/ST2 | Tn6073 | |
| RSS5 | RT020/ST2 | ||
| TDS118 | RT140/ST515 | Tn5397 | tetM |
| Tn6110 | |||
| RSS12, RSS52 | RT140/ST26 | Tn5397 | tetM |
| Tn6110 | |||
| RSS11 | RT010/ST15 | Tn6215 | ermB |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blau, K.; Gallert, C. Mobilome of Environmental Isolates of Clostridioides difficile. Antibiotics 2025, 14, 678. https://doi.org/10.3390/antibiotics14070678
Blau K, Gallert C. Mobilome of Environmental Isolates of Clostridioides difficile. Antibiotics. 2025; 14(7):678. https://doi.org/10.3390/antibiotics14070678
Chicago/Turabian StyleBlau, Khald, and Claudia Gallert. 2025. "Mobilome of Environmental Isolates of Clostridioides difficile" Antibiotics 14, no. 7: 678. https://doi.org/10.3390/antibiotics14070678
APA StyleBlau, K., & Gallert, C. (2025). Mobilome of Environmental Isolates of Clostridioides difficile. Antibiotics, 14(7), 678. https://doi.org/10.3390/antibiotics14070678