
Associating Biological Activity and Predicted Structure of Antimicrobial Peptides from Amphibians and Insects
 , , , , , , , , ,
, , , , , , , , ,
Abstract
1. Introduction
2. Results
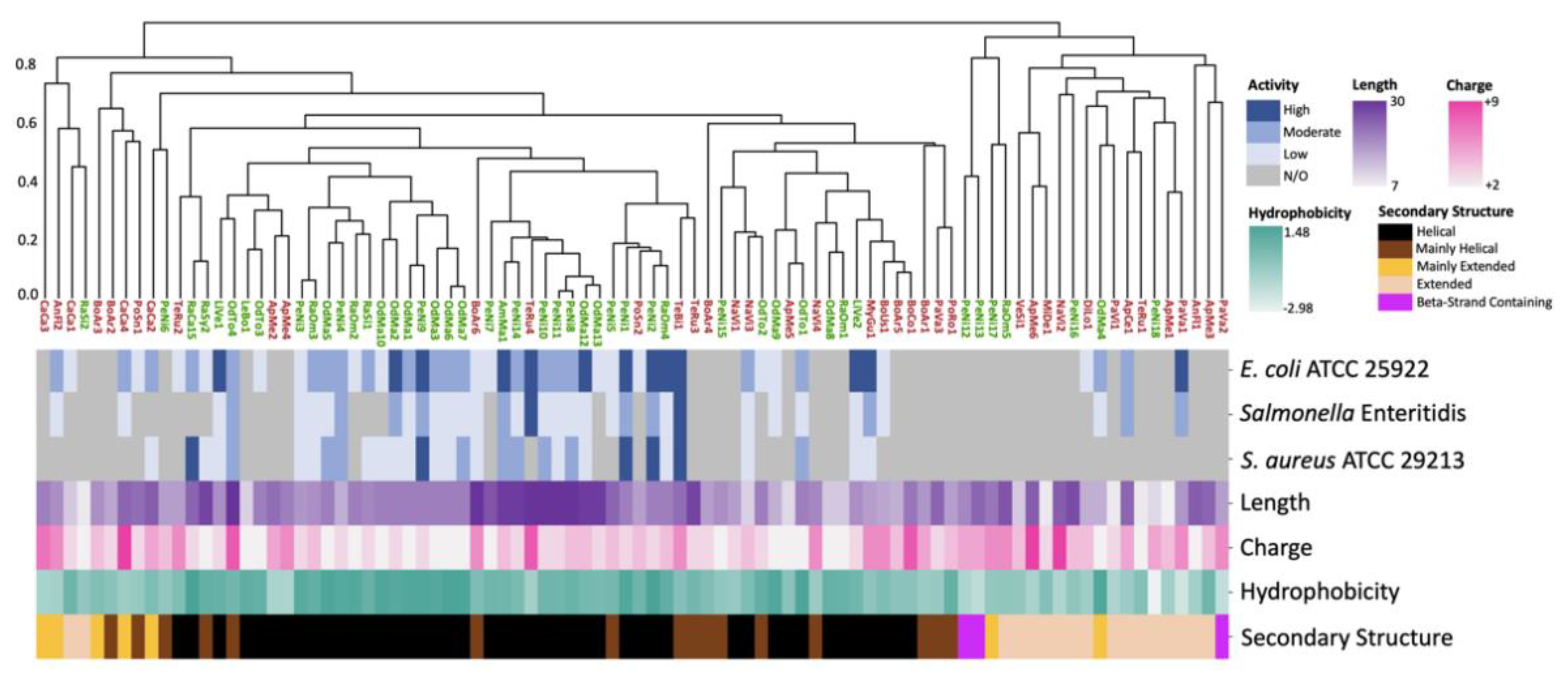
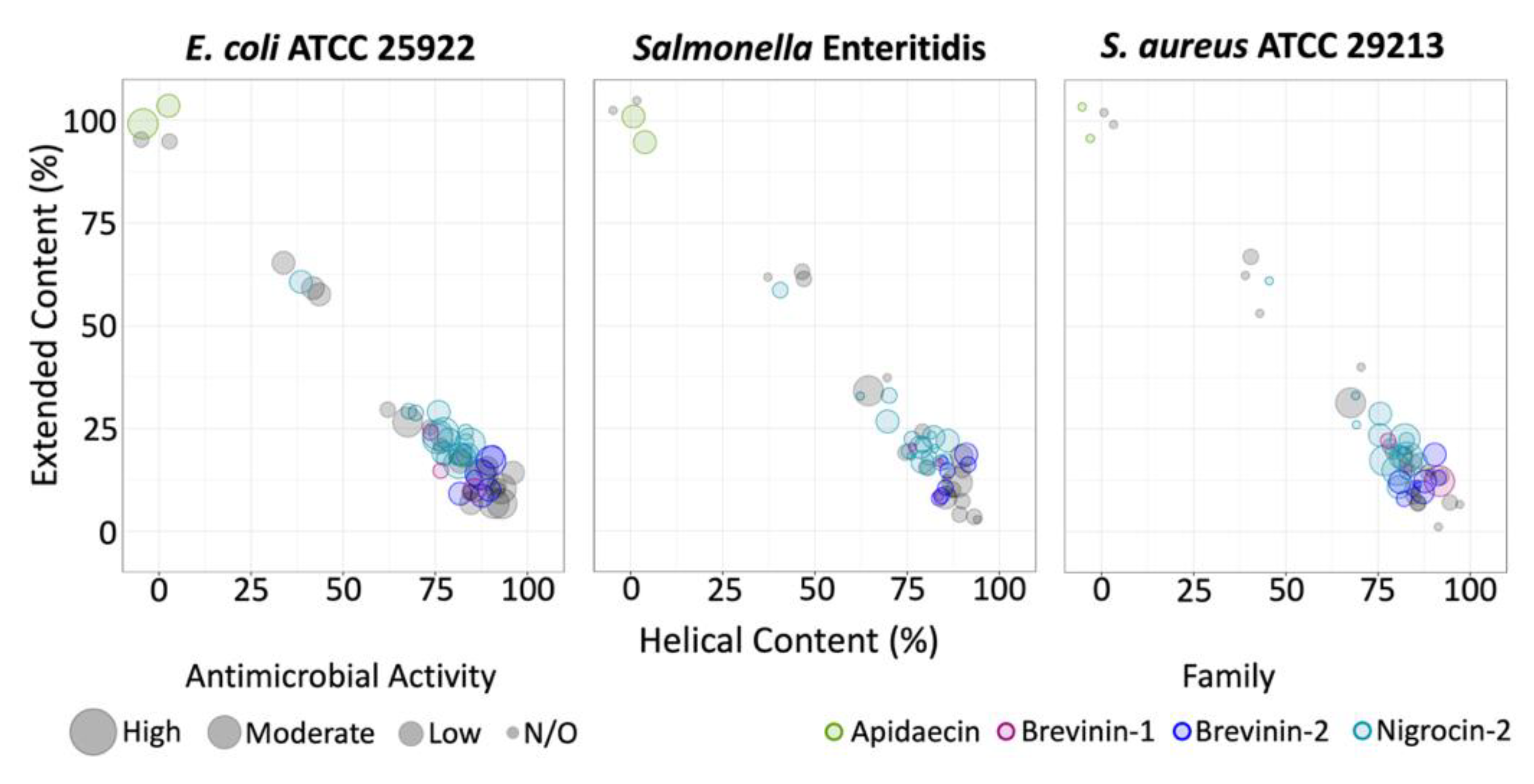
2.1. 3D Structure Prediction and Clustering
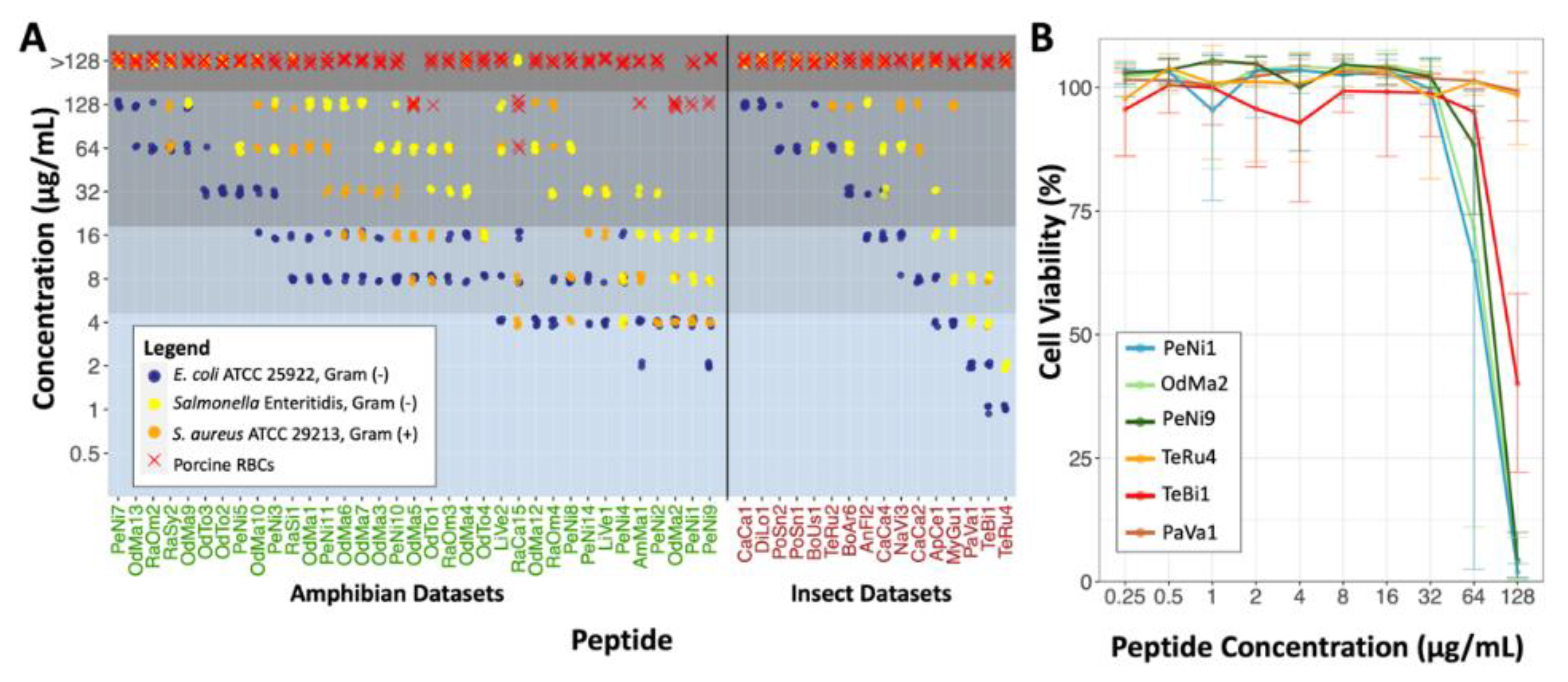
2.2. Antimicrobial Susceptibility Testing and Cytotoxicity
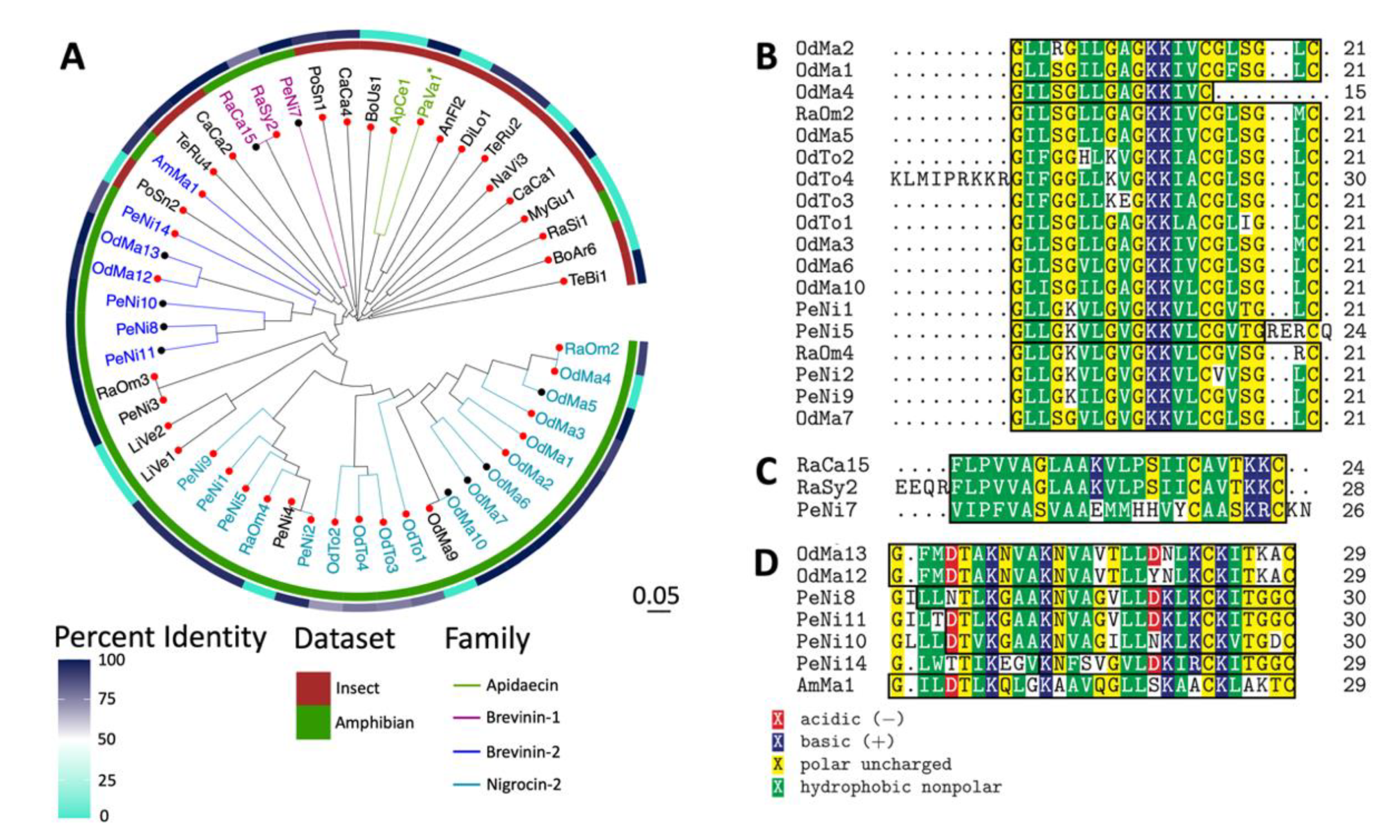
2.3. Sequence and Structural Characterization of AMPs Discovered Using rAMPage
3. Discussion
4. Materials and Methods
4.1. Peptide Discovery Using rAMPage
- “Species Count”—peptides identified in two or more species;
- “Insect Peptide,”—insect-derived peptides chosen using a reduced AMPlify prediction score threshold; and
- “AMPlify Score”—the top-scoring peptides with the highest net positive charge.
4.2. Bacterial Isolates
4.3. Antimicrobial Susceptibility Testing (AST)
4.4. Structure Prediction and Clustering
4.5. Hemolysis Assay
4.6. Cytotoxicity
4.7. BLAST and Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. The Review on Antimicrobial Resistance. 2014. Available online: https://amr-review.org/sites/default/files/AMR%20Review%20Paper%20-%20Tackling%20a%20crisis%20for%20the%20health%20and%20wealth%20of%20nations_1.pdf (accessed on 31 October 2022).
- Gillings, M.R.; Paulsen, I.T.; Tetu, S.G. Genomics and the Evolution of Antibiotic Resistance: Genomics and Antibiotic Resistance. Ann. N.Y. Acad. Sci. 2017, 1388, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Llor, C.; Bjerrum, L. Antimicrobial Resistance: Risk Associated with Antibiotic Overuse and Initiatives to Reduce the Problem. Ther. Adv. Drug Saf. 2014, 5, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Reardon, S. WHO Warns against “post-Antibiotic” Era. Nature 2014. [Google Scholar] [CrossRef]
- Durand, G.A.; Raoult, D.; Dubourg, G. Antibiotic Discovery: History, Methods and Perspectives. Int. J. Antimicrob. Agents 2019, 53, 371–382. [Google Scholar] [CrossRef]
- Cruz, J.; Ortiz, C.; Guzmán, F.; Fernández-Lafuente, R.; Torres, R. Antimicrobial Peptides: Promising Compounds against Pathogenic Microorganisms. CMC 2014, 21, 2299–2321. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial Peptides of Multicellular Organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Zhang, L.; Gallo, R.L. Antimicrobial Peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef]
- Petchiappan, A.; Chatterji, D. Antibiotic Resistance: Current Perspectives. ACS Omega 2017, 2, 7400–7409. [Google Scholar] [CrossRef]
- Rima, M.; Rima, M.; Fajloun, Z.; Sabatier, J.-M.; Bechinger, B.; Naas, T. Antimicrobial Peptides: A Potent Alternative to Antibiotics. Antibiotics 2021, 10, 1095. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and Host-Defense Peptides as New Anti-Infective Therapeutic Strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Predicting Drug Resistance Evolution: Insights from Antimicrobial Peptides and Antibiotics. Proc. R. Soc. B 2018, 285, 20172687. [Google Scholar] [CrossRef]
- Andersson, D.I.; Hughes, D.; Kubicek-Sutherland, J.Z. Mechanisms and Consequences of Bacterial Resistance to Antimicrobial Peptides. Drug Resist. Updates 2016, 26, 43–57. [Google Scholar] [CrossRef]
- Meylan, S.; Andrews, I.W.; Collins, J.J. Targeting Antibiotic Tolerance, Pathogen by Pathogen. Cell 2018, 172, 1228–1238. [Google Scholar] [CrossRef]
- Koehbach, J.; Craik, D.J. The Vast Structural Diversity of Antimicrobial Peptides. Trends Pharmacol. Sci. 2019, 40, 517–528. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic, α-Helical Antimicrobial Peptides. Biopolymers 2000, 55, 4–30. [Google Scholar] [CrossRef]
- Chen, C.H.; Bepler, T.; Pepper, K.; Fu, D.; Lu, T.K. Synthetic Molecular Evolution of Antimicrobial Peptides. Curr. Opin. Biotechnol. 2022, 75, 102718. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Lin, D.; Sutherland, D.; Aninta, S.I.; Louie, N.; Nip, K.M.; Li, C.; Yanai, A.; Coombe, L.; Warren, R.L.; Helbing, C.C.; et al. Mining Amphibian and Insect Transcriptomes for Antimicrobial Peptide Sequences with RAMPage. Antibiotics 2022, 11, 952. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Priority Pathogens List for R&D of New Antibiotics; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Helbing, C.C.; Hammond, S.A.; Jackman, S.H.; Houston, S.; Warren, R.L.; Cameron, C.E.; Birol, I. Antimicrobial Peptides from Rana [Lithobates] Catesbeiana: Gene Structure and Bioinformatic Identification of Novel Forms from Tadpoles. Sci. Rep. 2019, 9, 1529. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Patočka, J.; Kuča, K. Insect Antimicrobial Peptides, a Mini Review. Toxins 2018, 10, 461. [Google Scholar] [CrossRef] [PubMed]
- Frishman, D.; Argos, P. Knowledge-Based Protein Secondary Structure Assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically: Approved Standards; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015. [Google Scholar]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and Broth Dilution Methods to Determine the Minimal Inhibitory Concentration (MIC) of Antimicrobial Substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef]
- Li, W.-F.; Ma, G.-X.; Zhou, X.-X. Apidaecin-Type Peptides: Biodiversity, Structure–Function Relationships and Mode of Action. Peptides 2006, 27, 2350–2359. [Google Scholar] [CrossRef]
- Greco, I.; Molchanova, N.; Holmedal, E.; Jenssen, H.; Hummel, B.D.; Watts, J.L.; Håkansson, J.; Hansen, P.R.; Svenson, J. Correlation between Hemolytic Activity, Cytotoxicity and Systemic in Vivo Toxicity of Synthetic Antimicrobial Peptides. Sci. Rep. 2020, 10, 13206. [Google Scholar] [CrossRef]
- Maher, S.; McClean, S. Investigation of the Cytotoxicity of Eukaryotic and Prokaryotic Antimicrobial Peptides in Intestinal Epithelial Cells in Vitro. Biochem. Pharmacol. 2006, 71, 1289–1298. [Google Scholar] [CrossRef]
- Maturana, P.; Martinez, M.; Noguera, M.E.; Santos, N.C.; Disalvo, E.A.; Semorile, L.; Maffia, P.C.; Hollmann, A. Lipid Selectivity in Novel Antimicrobial Peptides: Implication on Antimicrobial and Hemolytic Activity. Colloids Surf. B Biointerfaces 2017, 153, 152–159. [Google Scholar] [CrossRef]
- Ilić, N.; Novković, M.; Guida, F.; Xhindoli, D.; Benincasa, M.; Tossi, A.; Juretić, D. Selective Antimicrobial Activity and Mode of Action of Adepantins, Glycine-Rich Peptide Antibiotics Based on Anuran Antimicrobial Peptide Sequences. Biochim. Biophys. Acta (BBA)-Biomembr. 2013, 1828, 1004–1012. [Google Scholar] [CrossRef]
- Conlon, J.M. Reflections on a Systematic Nomenclature for Antimicrobial Peptides from the Skins of Frogs of the Family Ranidae. Peptides 2008, 29, 1815–1819. [Google Scholar] [CrossRef]
- Savelyeva, A.; Ghavami, S.; Davoodpour, P.; Asoodeh, A.; Łos, M.J. An Overview of Brevinin Superfamily: Structure, Function and Clinical Perspectives. In Anticancer Genes; Grimm, S., Ed.; Advances in Experimental Medicine and Biology; Springer: London, UK, 2014; Volume 818, pp. 197–212. ISBN 978-1-4471-6457-9. [Google Scholar]
- Park, S.; Park, S.-H.; Ahn, H.-C.; Kim, S.; Kim, S.S.; Lee, B.J.; Lee, B.-J. Structural Study of Novel Antimicrobial Peptides, Nigrocins, Isolated from Rana Nigromaculata. FEBS Lett. 2001, 507, 95–100. [Google Scholar] [CrossRef]
- Conlon, J.M.; Ahmed, E.; Condamine, E. Antimicrobial Properties of Brevinin-2-Related Peptide and Its Analogs: Efficacy Against Multidrug-Resistant Acinetobacter Baumannii. Chem. Biol. Drug Des. 2009, 74, 488–493. [Google Scholar] [CrossRef]
- Wang, G. Post-Translational Modifications of Natural Antimicrobial Peptides and Strategies for Peptide Engineering. CBIOT 2012, 1, 72–79. [Google Scholar] [CrossRef]
- Strandberg, E.; Tiltak, D.; Ieronimo, M.; Kanithasen, N.; Wadhwani, P.; Ulrich, A.S. Influence of C-Terminal Amidation on the Antimicrobial and Hemolytic Activities of Cationic α-Helical Peptides. Pure Appl. Chem. 2007, 79, 717–728. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Papo, N.; Mignogna, G.; Andreu, D.; Shai, Y.; Barra, D.; Simmaco, M. Ranacyclins, a New Family of Short Cyclic Antimicrobial Peptides: Biological Function, Mode of Action, and Parameters Involved in Target Specificity. Biochemistry 2003, 42, 14023–14035. [Google Scholar] [CrossRef]
- Rifflet, A.; Gavalda, S.; Téné, N.; Orivel, J.; Leprince, J.; Guilhaudis, L.; Génin, E.; Vétillard, A.; Treilhou, M. Identification and Characterization of a Novel Antimicrobial Peptide from the Venom of the Ant Tetramorium Bicarinatum. Peptides 2012, 38, 363–370. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Nip, K.M.; Chiu, R.; Yang, C.; Chu, J.; Mohamadi, H.; Warren, R.L.; Birol, I. RNA-Bloom Enables Reference-Free and Reference-Guided Sequence Assembly for Single-Cell Transcriptomes. Genome Res. 2020, 30, 1191–1200. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov Model Speed Heuristic and Iterative HMM Search Procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef] [PubMed]
- Duckert, P.; Brunak, S.; Blom, N. Prediction of Proprotein Convertase Cleavage Sites. Protein Eng. Des. Sel. 2004, 17, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sutherland, D.; Hammond, S.A.; Yang, C.; Taho, F.; Bergman, L.; Houston, S.; Warren, R.L.; Wong, T.; Hoang, L.M.N.; et al. AMPlify: Attentive Deep Learning Model for Discovery of Novel Antimicrobial Peptides Effective against WHO Priority Pathogens. BMC Genom. 2022, 23, 77. [Google Scholar] [CrossRef] [PubMed]
- Hart, A.J.; Ginzburg, S.; Xu, M.; Fisher, C.R.; Rahmatpour, N.; Mitton, J.B.; Paul, R.; Wegrzyn, J.L. EnTAP : Bringing Faster and Smarter Functional Annotation to Non-model Eukaryotic Transcriptomes. Mol. Ecol. Resour. 2020, 20, 591–604. [Google Scholar] [CrossRef]
- Slater, G.; Birney, E. Automated Generation of Heuristics for Biological Sequence Comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef]
- Adamczak, R.; Porollo, A.; Meller, J. Combining Prediction of Secondary Structure and Solvent Accessibility in Proteins. Proteins 2005, 59, 467–475. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Dong, R.; Peng, Z.; Zhang, Y.; Yang, J. MTM-Align: An Algorithm for Fast and Accurate Multiple Protein Structure Alignment. Bioinformatics 2018, 34, 1719–1725. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Palme, J.; Hochreiter, S.; Bodenhofer, U. KeBABS: An R Package for Kernel-Based Analysis of Biological Sequences. Bioinformatics 2015, 31, 2574–2576. [Google Scholar] [CrossRef]
- Charif, D.; Lobry, J.R. SeqinR 1.0-2: A Contributed Package to the R Project for Statistical Computing Devoted to Biological Sequences Retrieval and Analysis. In Structural Approaches to Sequence Evolution; Bastolla, U., Porto, M., Roman, H.E., Vendruscolo, M., Eds.; Biological and Medical Physics, Biomedical Engineering; Springer: Berlin/Heidelberg, Germany, 2007; pp. 207–232. ISBN 978-3-540-35305-8. [Google Scholar]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T. ggtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Yu, G.; Lam, T.T.-Y.; Zhu, H.; Guan, Y. Two Methods for Mapping and Visualizing Associated Data on Phylogeny Using Ggtree. Mol. Biol. Evol. 2018, 35, 3041–3043. [Google Scholar] [CrossRef]
- Yu, G. Using Ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Yu, G. Data Integration, Manipulation and Visualization of Phylogenetic Trees, 1st ed.; Chapman and Hall/CRC: Boca Raton, FL, USA, 2022. [Google Scholar]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name | Source Organism | Sequence | Length | Charge * | AMPlify Score | MW (Da) |
|---|---|---|---|---|---|---|
| AmMa1 | Amolops mantzorum | GILDTLKQLGKAAVQGLLSKAACKLAKTC | 29 | 4 | 80.0 | 2943.59 |
| AnFl2 | Anterhynchium flavomarginatum | GILRSLGWIQMPRSRRRHR | 19 | 6 | 31.8 | 2375.82 |
| ApCe1 | Apis cerana | GIYTGRLLPVYIPQPRPPHPRLRR | 24 | 5 | 38.6 | 2853.39 |
| BoAr6 | Bombus ardens | GILRLVTRRFRFSPTNLNRYTVARLVSGVP | 30 | 6 | 22.1 | 3460.06 |
| BoUs1 | Bombus ussurensis | RKIIAVSVHKLCRVKR | 16 | 6 | 29.9 | 1906.40 |
| CaCa1 | Camponotus castaneus, Odontomachus monticola, Polistes rothneyi, Polistes snelleni, Sphecidae sp. KJ-8906, Vespa dybowskii | FACPIGFFRLKR | 12 | 3 | 7.27 | 1454.79 |
| CaCa2 | Camponotus castaneus, Odontomachus monticola, Temnothorax rugatulus | FIKTQVLKHLVAGVRVARGLDWKWR | 25 | 5 | 28.7 | 2977.57 |
| CaCa4 | Camponotus castaneus | RRFFFATAPCGYSRKFCKITRRKR | 24 | 9 | 23.6 | 2996.58 |
| DiLo | Diachasmimorpha longicaudata | GAFVLWGPTPRPRRR | 15 | 4 | 26.0 | 1766.07 |
| LiVe1 | Litoria verreauxii | GWLDIAKKVASVVAGIVKR | 19 | 3 | 80.0 | 2010.44 |
| LiVe2 | Litoria verreauxii | GWLDIAKKVASVVAGLGKR | 19 | 3 | 70.0 | 1968.36 |
| MyGu1 | Myrmecia gulosa | RRAIFASIRGYLGLRKR | 17 | 6 | 25.3 | 2033.44 |
| NaVi3 | Nasonia vitripennis x Nasonia giraulti F1 | KLFLTLWKLKR | 11 | 4 | 30.5 | 1445.84 |
| OdMa1 | Odorrana margaretae | GLLSGILGAGKKIVCGFSGLC | 21 | 2 | 80.0 | 1993.45 |
| OdMa2 | Odorrana margaretae | GLLRGILGAGKKIVCGLSGLC | 21 | 3 | 67.0 | 2028.54 |
| OdMa3 | Odorrana margaretae | GLLSGLLGAGKKIVCGLSGMC | 21 | 2 | 80.0 | 1977.47 |
| OdMa4 | Odorrana margaretae | GILSGLLGAGKKIVC | 15 | 2 | 70.0 | 1428.79 |
| OdMa5 | Odorrana margaretae | GILSGLLGAGKKIVCGLSGLC | 21 | 2 | 80.0 | 1959.43 |
| OdMa6 | Odorrana margaretae | GLLSGVLGVGKKIVCGLSGLC | 21 | 2 | 80.0 | 1973.46 |
| OdMa7 | Odorrana margaretae | GLLSGVLGVGKKVLCGLSGLC | 21 | 2 | 80.0 | 1973.46 |
| OdMa9 | Odorrana margaretae | GLISGILGAGKKVLC | 15 | 2 | 67.0 | 1428.79 |
| OdMa10 | Odorrana margaretae | GLISGILGAGKKVLCGLSGLC | 21 | 2 | 70.0 | 1959.43 |
| OdMa12 | Odorrana margaretae | GFMDTAKNVAKNVAVTLLYNLKCKITKAC | 29 | 4 | 70.0 | 3158.82 |
| OdMa13 | Odorrana margaretae | GFMDTAKNVAKNVAVTLLDNLKCKITKAC | 29 | 3 | 67.0 | 3110.73 |
| OdTo1 | Odorrana tormota | GILSGLLGAGKKLACGLIGLC | 21 | 2 | 80.0 | 1957.46 |
| OdTo2 | Odorrana tormota | GIFGGHLKVGKKIACGLSGLC | 21 | 3 | 67.0 | 2058.52 |
| OdTo3 | Odorrana tormota | GIFGGLLKEGKKIACGLSGLC | 21 | 2 | 48.5 | 2064.53 |
| OdTo4 | Odorrana tormota | KLMIPRKKRGIFGGLLKVGKKIACGLSGLC | 30 | 8 | 47.4 | 3186.06 |
| PaVa1 | Partula varia | RPRPQQVPPRPPHPRLRR | 18 | 6 | 27.5 | 2240.63 |
| PeNi1 | Pelophylax nigromaculatus | GLLGKVLGVGKKVLCGVTGLC | 21 | 3 | 70.0 | 2014.55 |
| PeNi2 | Pelophylax nigromaculatus | GLLGKVLGVGKKVLCVVSGLC | 21 | 3 | 70.0 | 2042.61 |
| PeNi3 | Pelophylax nigromaculatus | GIFSLIKGAAKVVAKGLG | 18 | 3 | 65.2 | 1729.12 |
| PeNi4 | Pelophylax nigromaculatus | GLLGKVLGVGKKVLC | 15 | 3 | 67.0 | 1483.91 |
| PeNi5 | Pelophylax nigromaculatus | GLLGKVLGVGKKVLCGVTGRERCQ | 24 | 4 | 57.7 | 2471.01 |
| PeNi7 | Bufo gargarizans, Leptobrachium boringii, Megophrys sangzhiensis, Polypedates megacephalus, Pelophylax nigromaculatus, Rhacophorus dennysi, Rana omeimontis | VIPFVASVAAEMMHHVYCAASKRCKN | 26 | 2 | 43.2 | 2863.42 |
| PeNi8 | Bufo gargarizans, Megophrys sangzhiensis, Polypedates megacephalus, Pelophylax nigromaculatus, Rhacophorus dennysi, Rana omeimontis | GILLNTLKGAAKNVAGVLLDKLKCKITGGC | 30 | 4 | 63.0 | 3012.69 |
| PeNi9 | Leptobrachium boringii, Megophrys sangzhiensis, Polypedates megacephalus, Pelophylax nigromaculatus, Rhacophorus dennysi, Rana omeimontis | GLLGKILGVGKKVLCGVSGLC | 21 | 3 | 62.2 | 2014.55 |
| PeNi10 | Leptobrachium boringii, Polypedates megacephalus, Pelophylax nigromaculatus, Rhacophorus dennysi, Rana omeimontis | GLLLDTVKGAAKNVAGILLNKLKCKVTGDC | 30 | 3 | 61.8 | 3056.70 |
| PeNi11 | Leptobrachium boringii, Polypedates megacephalus, Pelophylax nigromaculatus, Rhacophorus dennysi, Rana omeimontis | GILTDTLKGAAKNVAGVLLDKLKCKITGGC | 30 | 3 | 61.8 | 3001.62 |
| PeNi14 | Bufo gargarizans, Polypedates megacephalus, Pelophylax nigromaculatus, Rana omeimontis | GLWTTIKEGVKNFSVGVLDKIRCKITGGC | 29 | 3 | 67.00 | 3123.71 |
| PoSn1 | Polistes snelleni | ISIKEALEHSFFHTVPRKWCKKH | 23 | 3 | 30.4 | 2822.31 |
| PoSn2 | Polistes snelleni | TALKSLSILKKLAKLNM | 17 | 4 | 23.7 | 1872.37 |
| RaCa15 | Rana catesbeiana | FLPVVAGLAAKVLPSIICAVTKKC | 24 | 3 | 67.0 | 2442.09 |
| RaOm2 | Rana omeimontis | GILSGLLGAGKKIVCGLSGMC | 21 | 2 | 80.0 | 1977.47 |
| RaOm3 | Rana omeimontis | GIFSLIKGAAKVVAKGLGK | 19 | 4 | 67.0 | 1857.30 |
| RaOm4 | Rana omeimontis | GLLGKVLGVGKKVLCGVSGRC | 21 | 4 | 67.0 | 2043.55 |
| RaSi1 | Allobates femoralis, Pristimantis toftae, Ranitomeya sirensis | GLVGKLVKGGLKLIGHVANG | 20 | 3 | 36.9 | 1930.35 |
| RaSy2 | Rana sylvatica | EEQRFLPVVAGLAAKVLPSIICAVTKKC | 28 | 2 | 21.9 | 2984.64 |
| TeBi1 | Tetramorium bicarinatum | KIKIPWGKVKDFLVGGMKAVGKK | 23 | 6 | 45.00 | 2528.17 |
| TeRu2 | Temnothorax rugatulus | AFVRILCYCCPRRIKRR | 17 | 6 | 31.9 | 2153.70 |
| TeRu4 | Temnothorax rugatulus | SWLSKSVKKLVNKKNYTRLEKLAKKKLFNE | 30 | 8 | 25.5 | 3622.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richter, A.; Sutherland, D.; Ebrahimikondori, H.; Babcock, A.; Louie, N.; Li, C.; Coombe, L.; Lin, D.; Warren, R.L.; Yanai, A.; et al. Associating Biological Activity and Predicted Structure of Antimicrobial Peptides from Amphibians and Insects. Antibiotics 2022, 11, 1710. https://doi.org/10.3390/antibiotics11121710
Richter A, Sutherland D, Ebrahimikondori H, Babcock A, Louie N, Li C, Coombe L, Lin D, Warren RL, Yanai A, et al. Associating Biological Activity and Predicted Structure of Antimicrobial Peptides from Amphibians and Insects. Antibiotics. 2022; 11(12):1710. https://doi.org/10.3390/antibiotics11121710
Chicago/Turabian StyleRichter, Amelia, Darcy Sutherland, Hossein Ebrahimikondori, Alana Babcock, Nathan Louie, Chenkai Li, Lauren Coombe, Diana Lin, René L. Warren, Anat Yanai, and et al. 2022. "Associating Biological Activity and Predicted Structure of Antimicrobial Peptides from Amphibians and Insects" Antibiotics 11, no. 12: 1710. https://doi.org/10.3390/antibiotics11121710
APA StyleRichter, A., Sutherland, D., Ebrahimikondori, H., Babcock, A., Louie, N., Li, C., Coombe, L., Lin, D., Warren, R. L., Yanai, A., Kotkoff, M., Helbing, C. C., Hof, F., Hoang, L. M. N., & Birol, I. (2022). Associating Biological Activity and Predicted Structure of Antimicrobial Peptides from Amphibians and Insects. Antibiotics, 11(12), 1710. https://doi.org/10.3390/antibiotics11121710