
Synthesis, Physical Properties and Electrocatalytic Performance of Nickel Phosphides for Hydrogen Evolution Reaction of Water Electrolysis




Abstract
:1. Introduction
2. Experimental
2.1. Materials
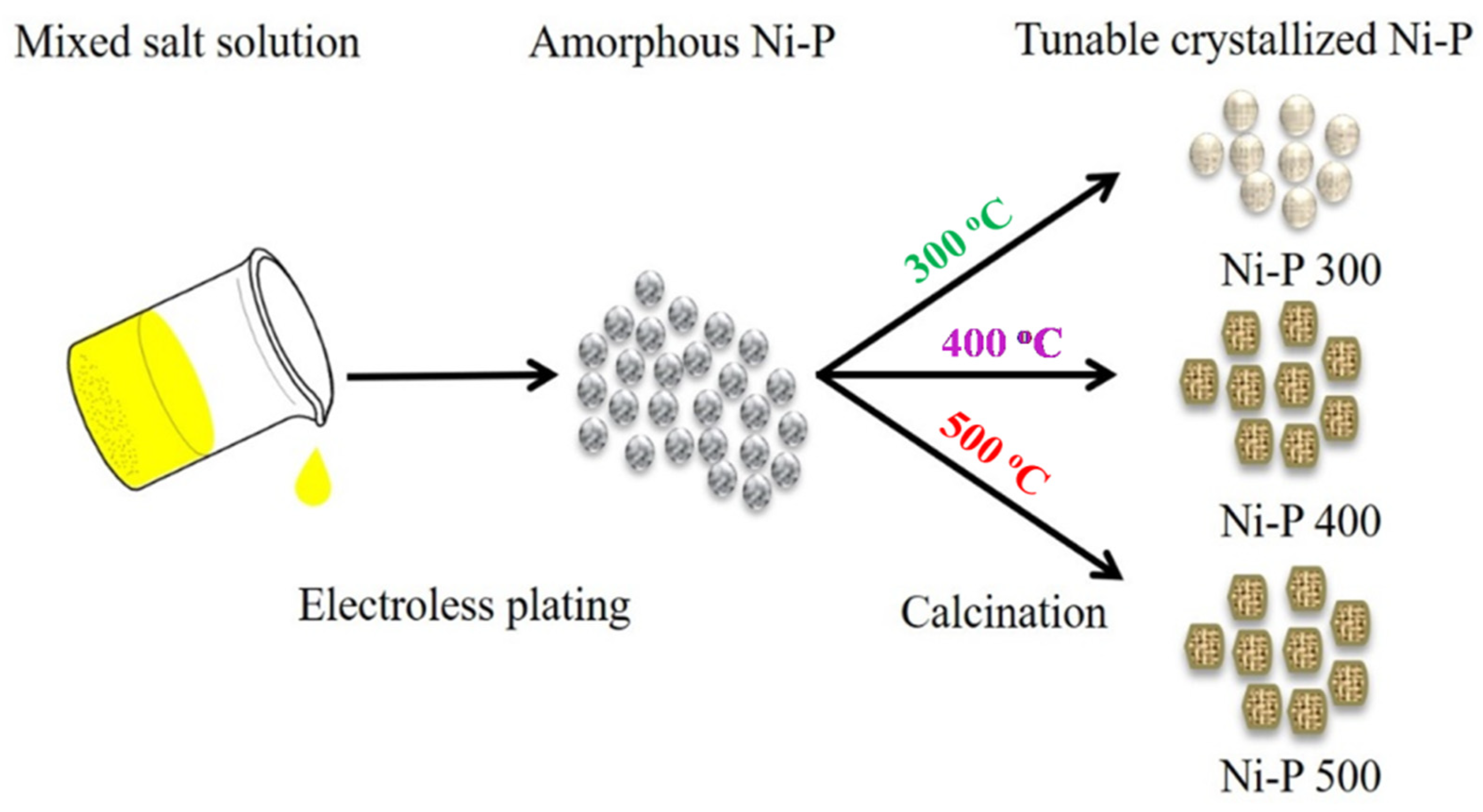
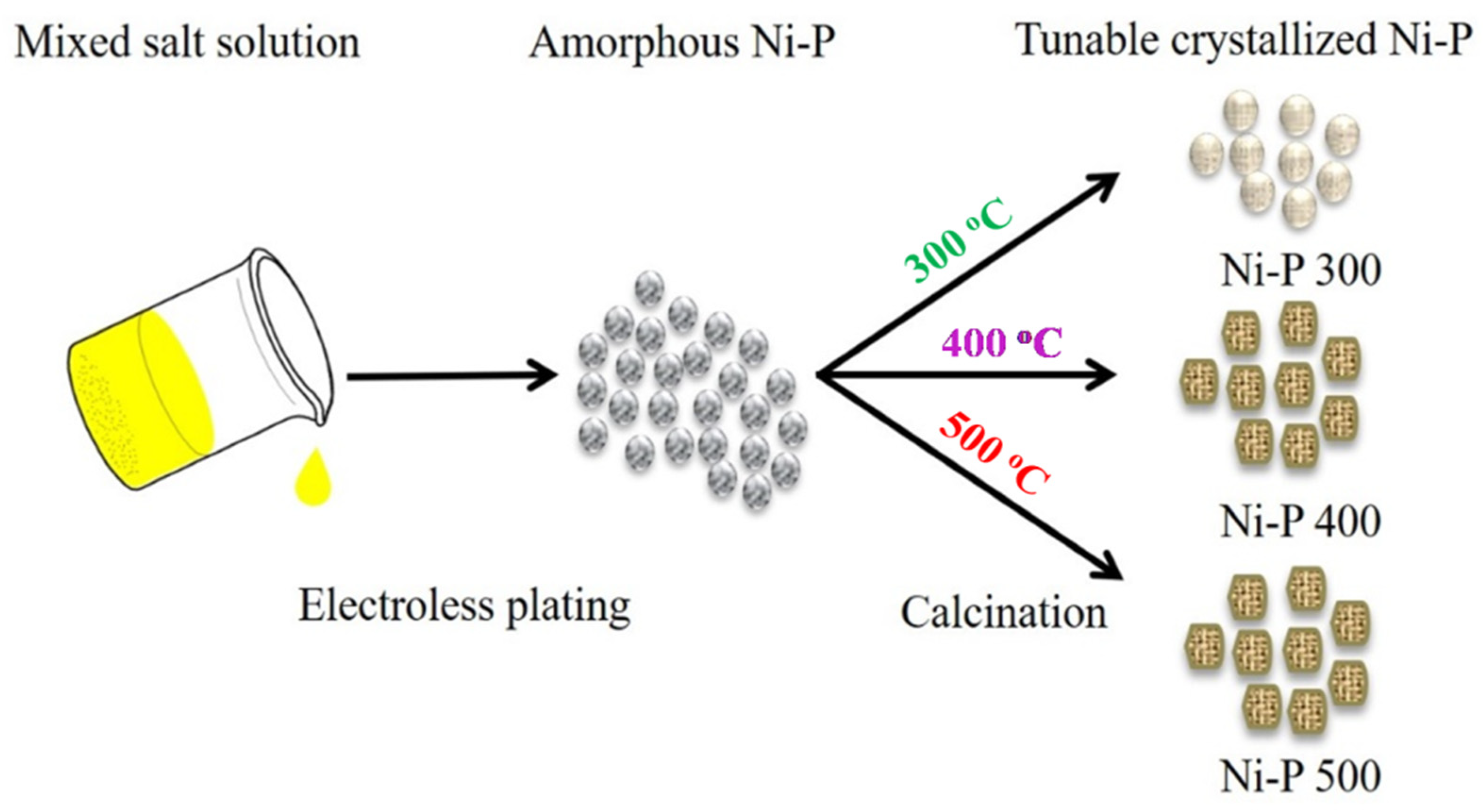
2.2. Synthesis of Amorphous Ni-P
2.3. Synthesis of Tunable Crystalline Ni-P
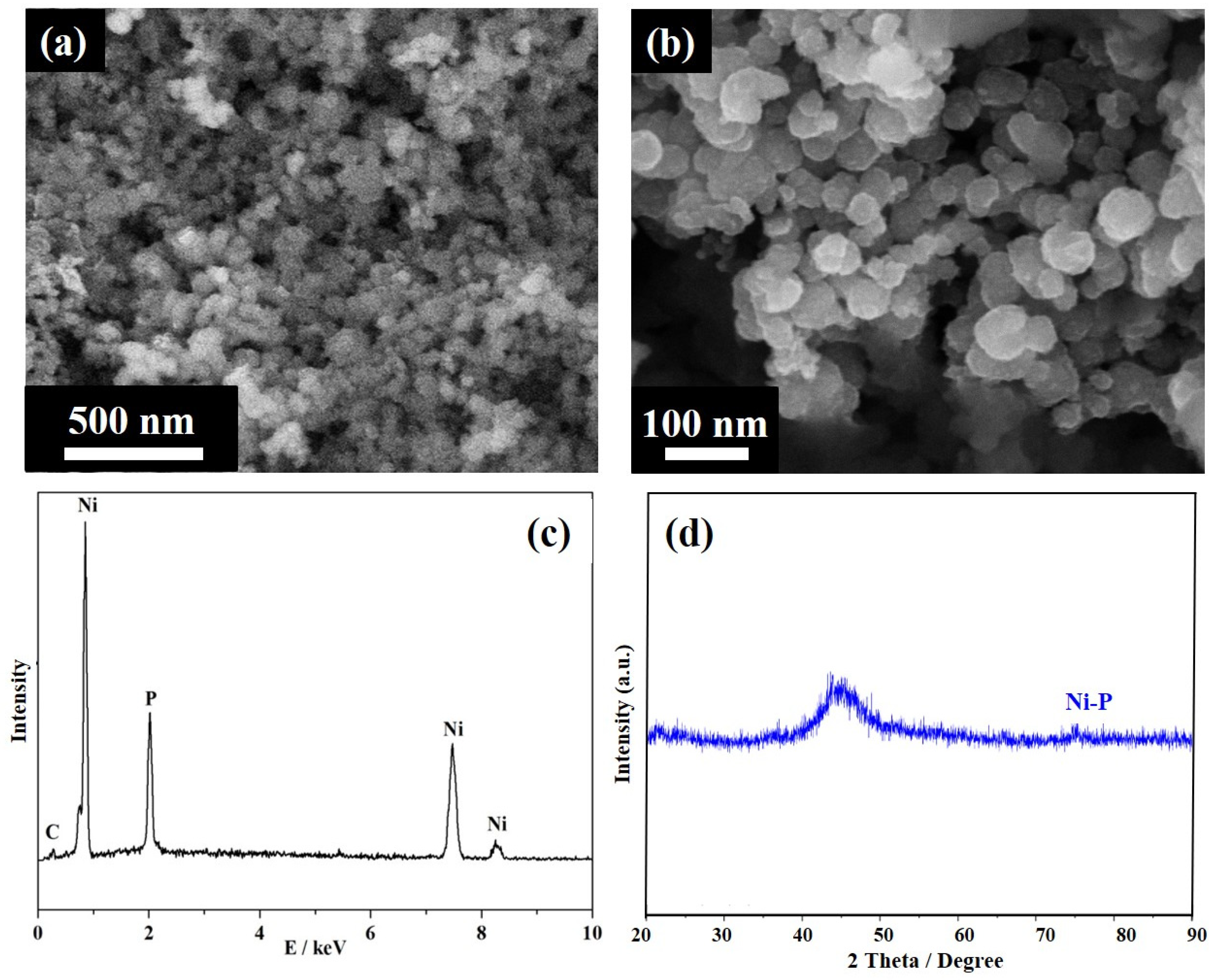
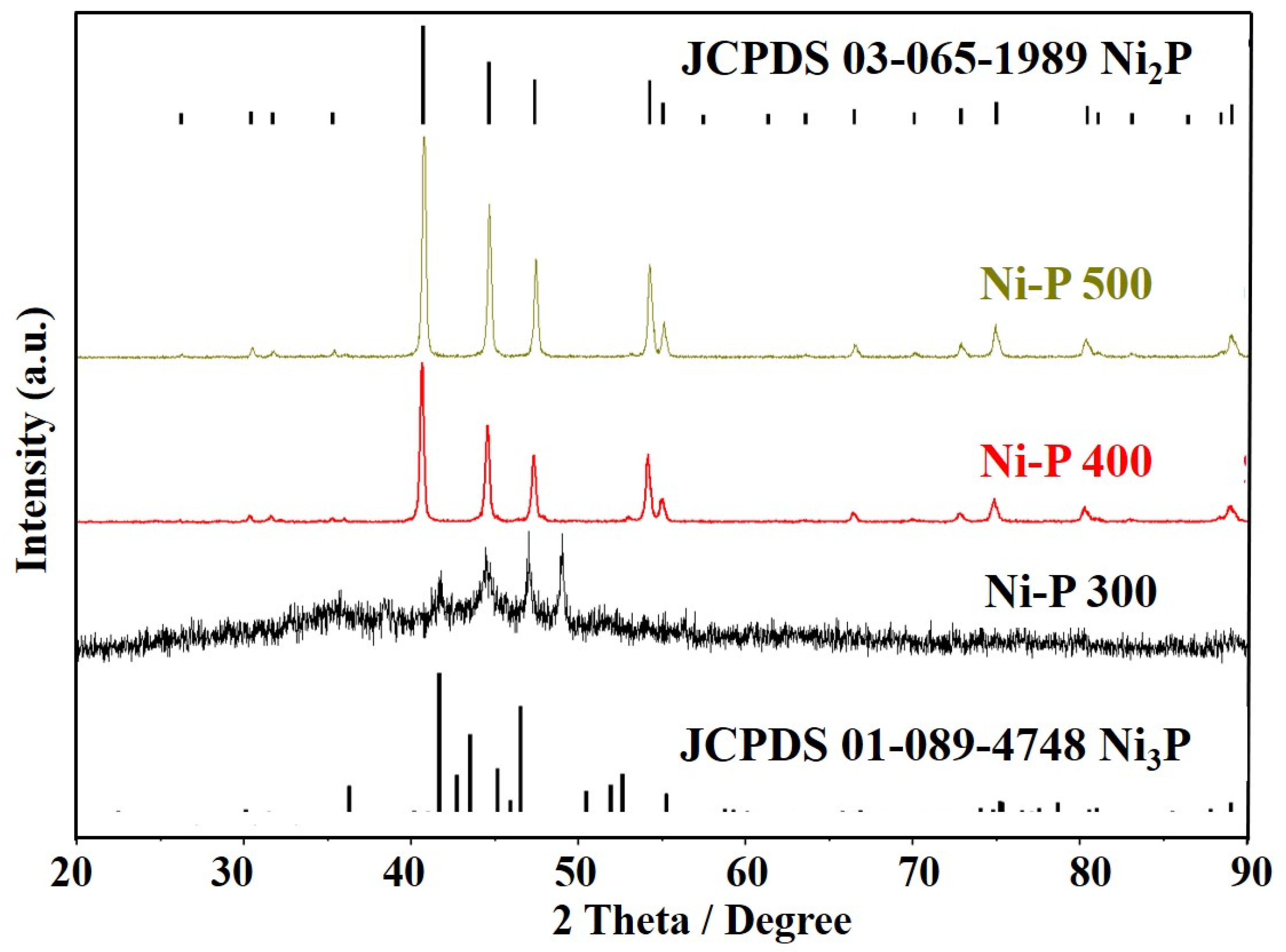
2.4. Physical and Chemical Characterization
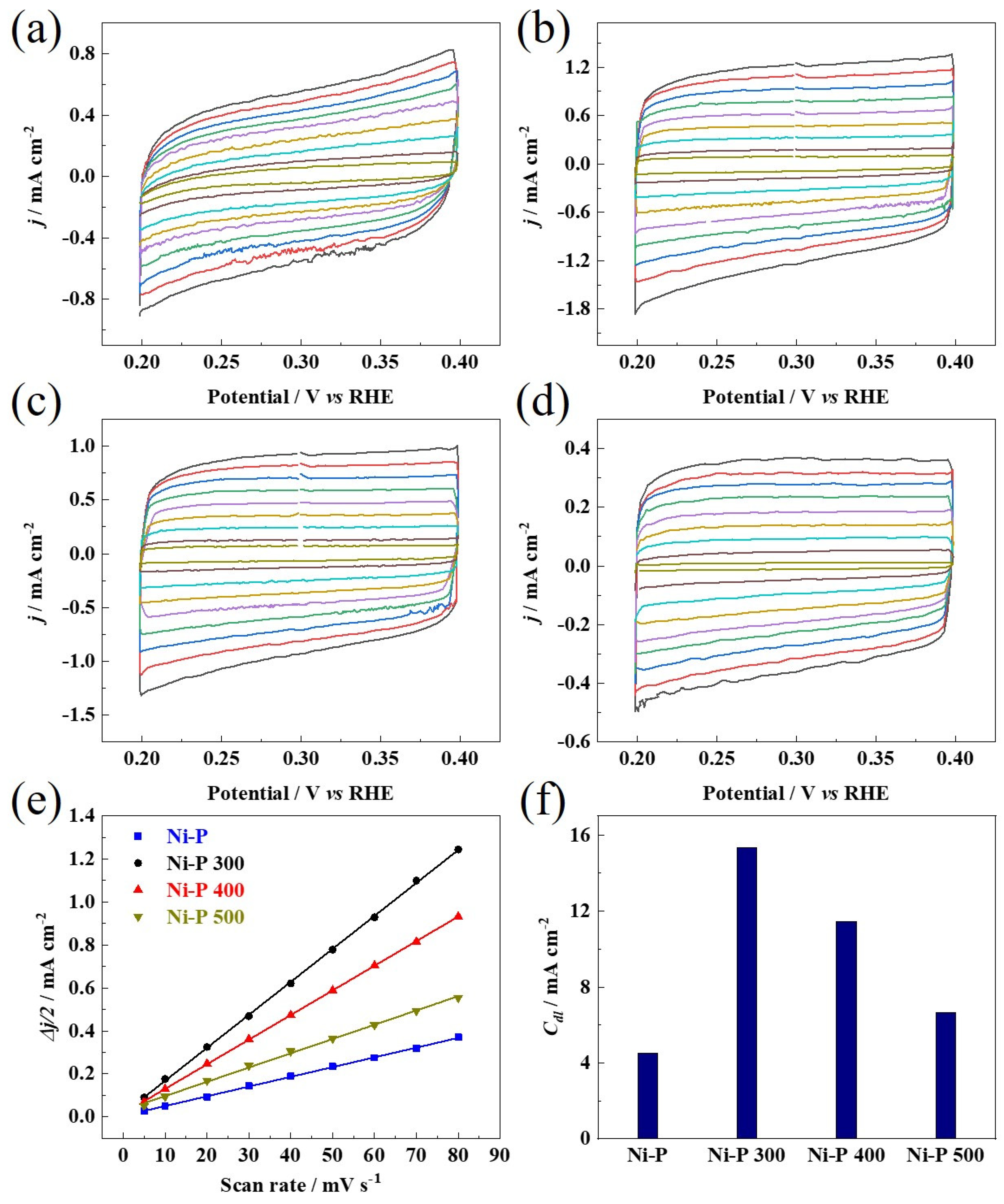
2.5. Electrochemical Characterization
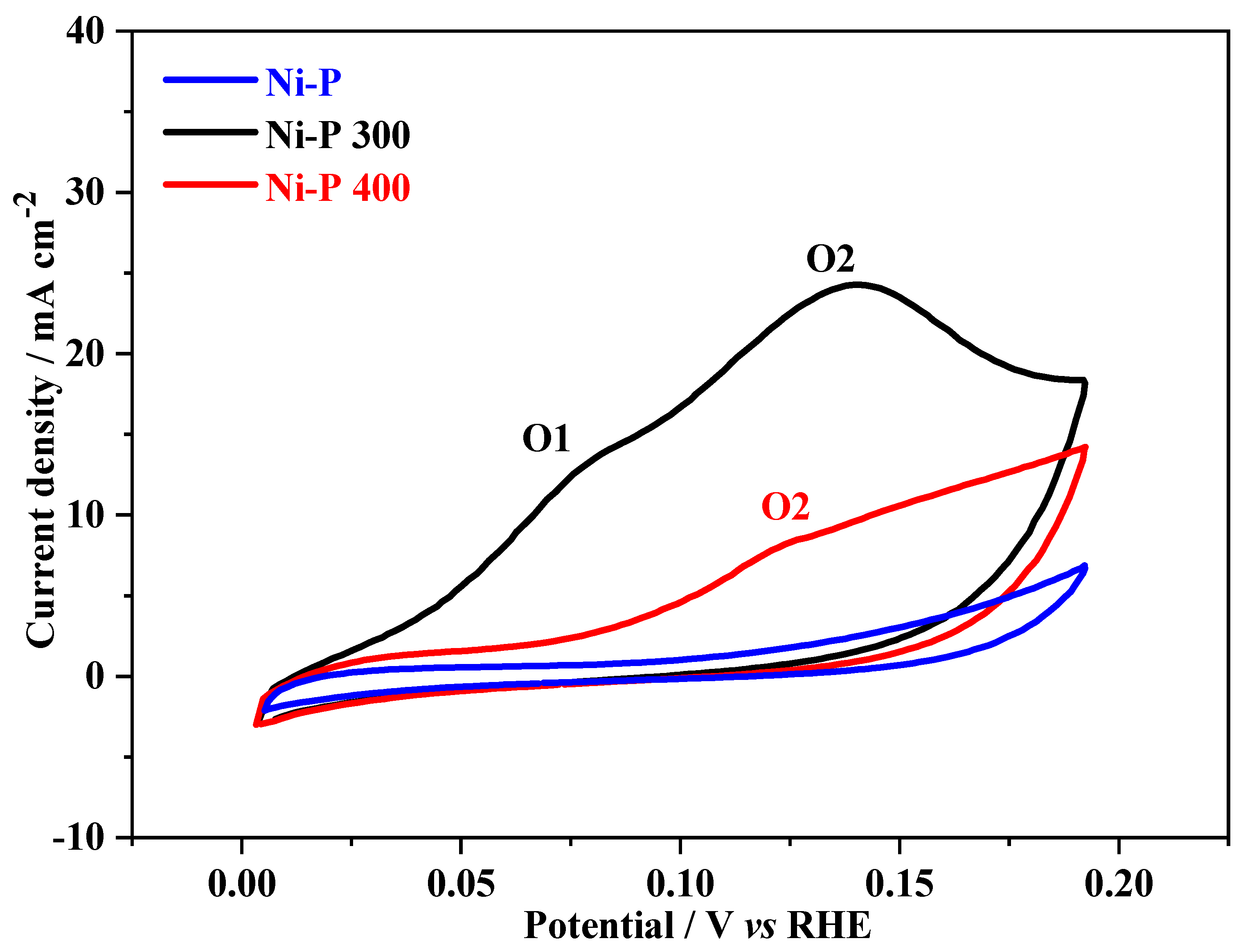
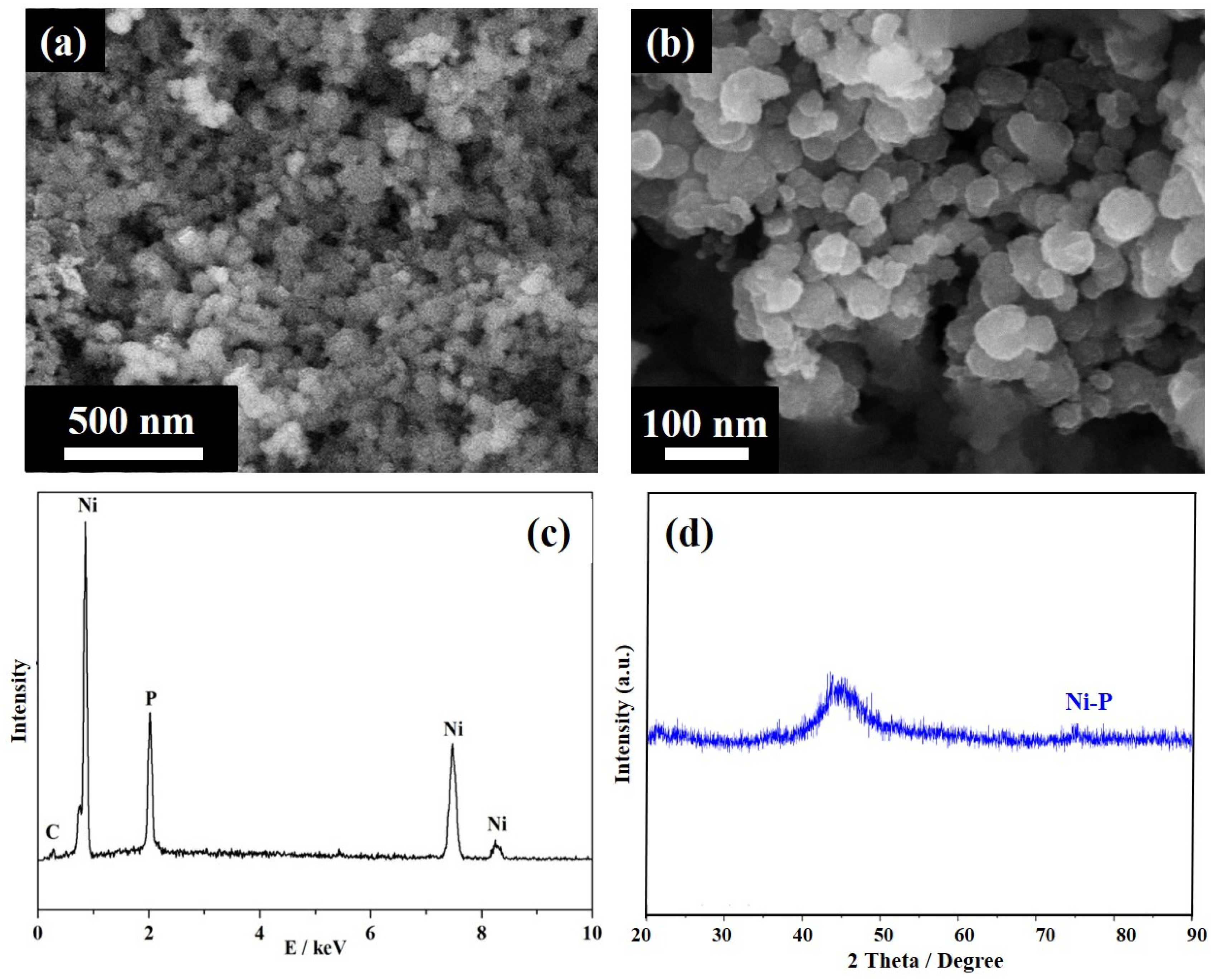
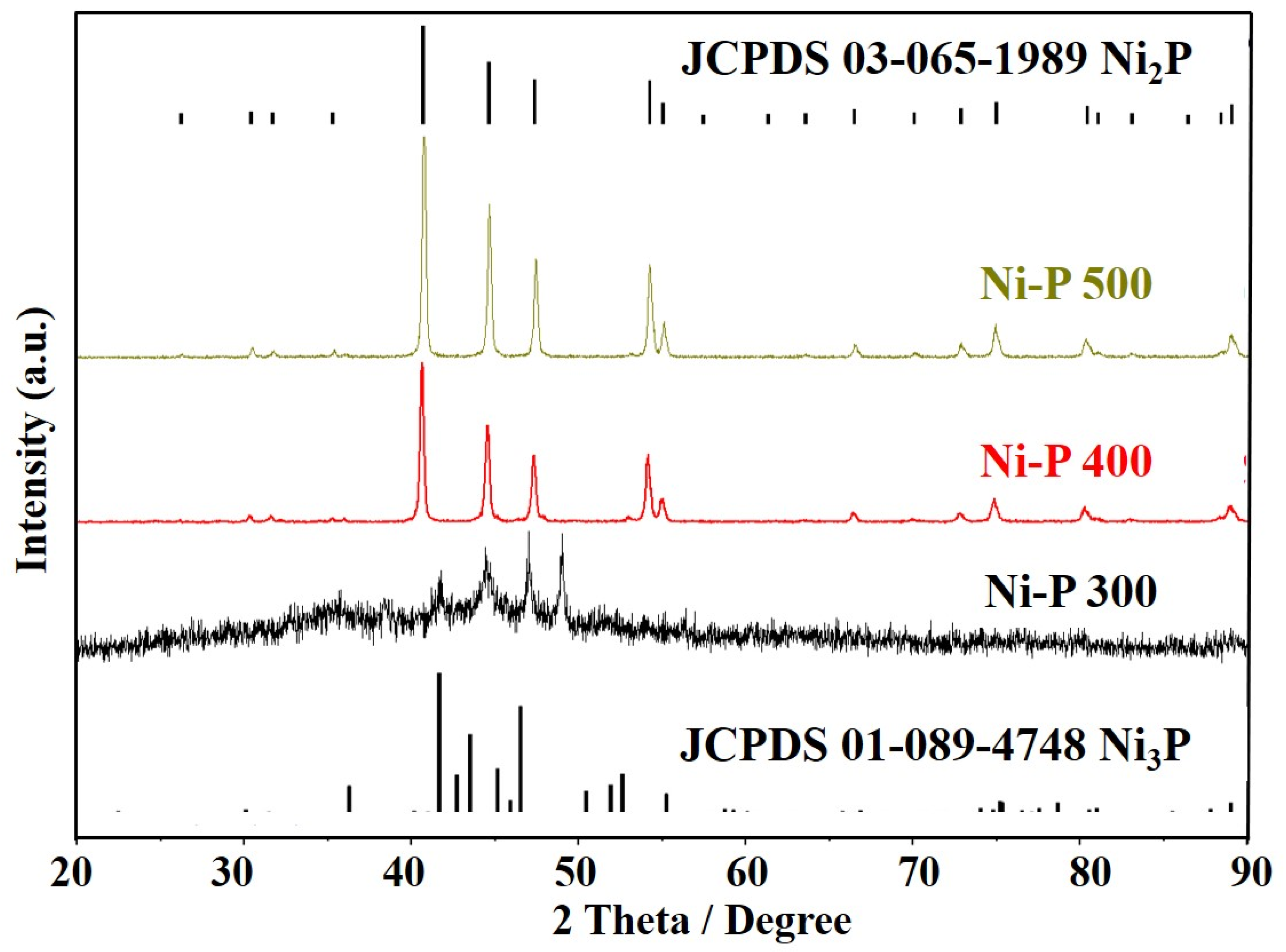
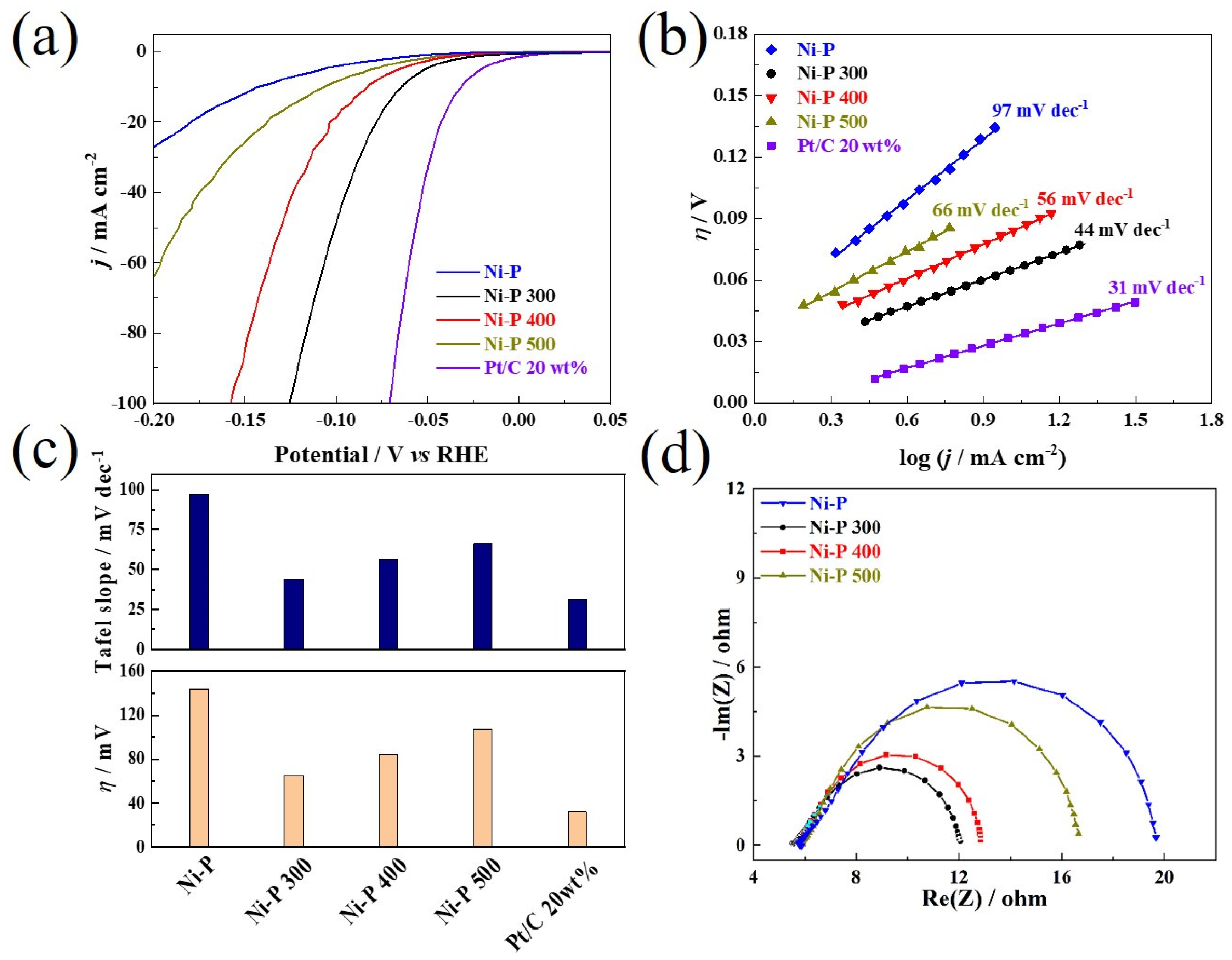
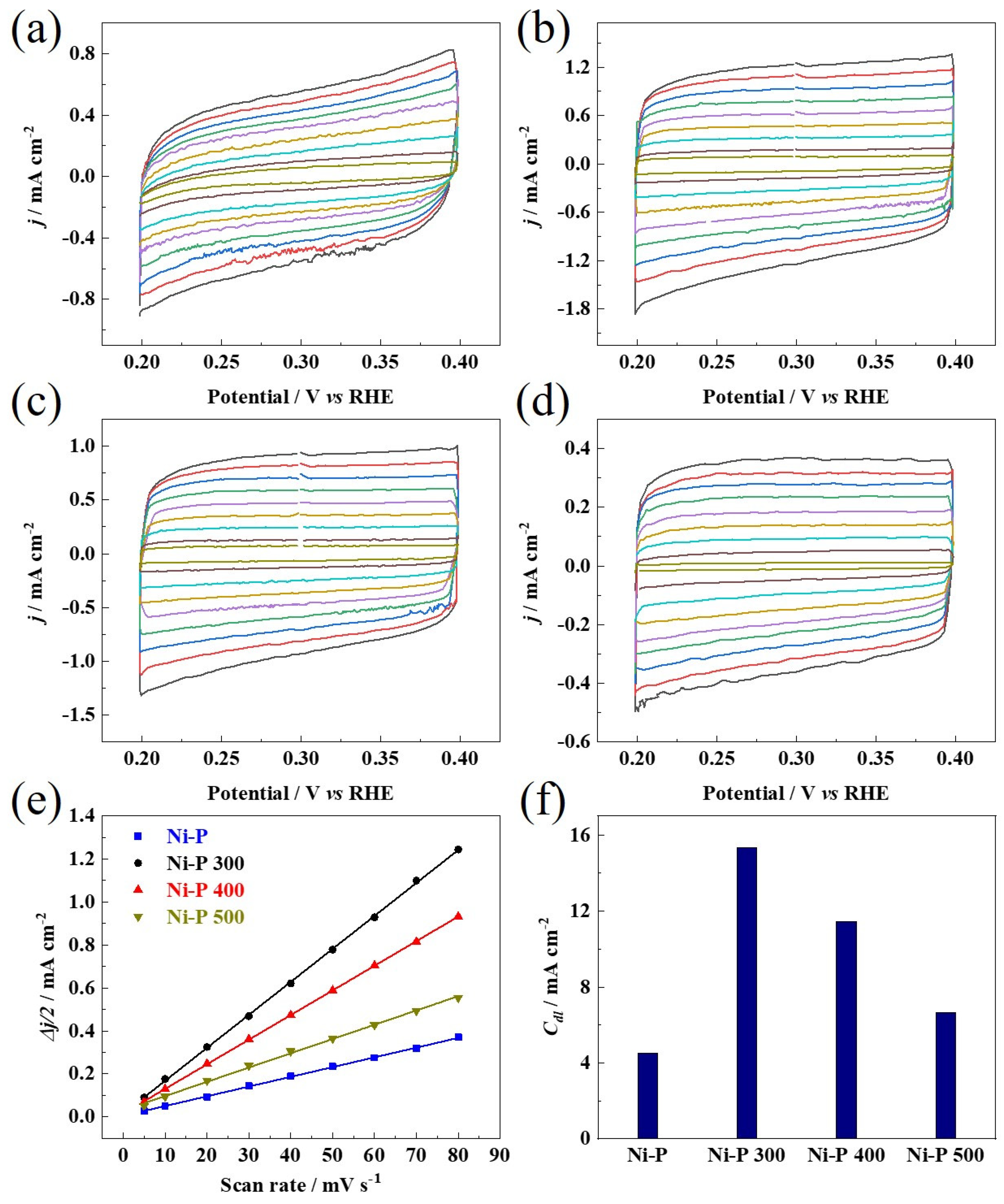
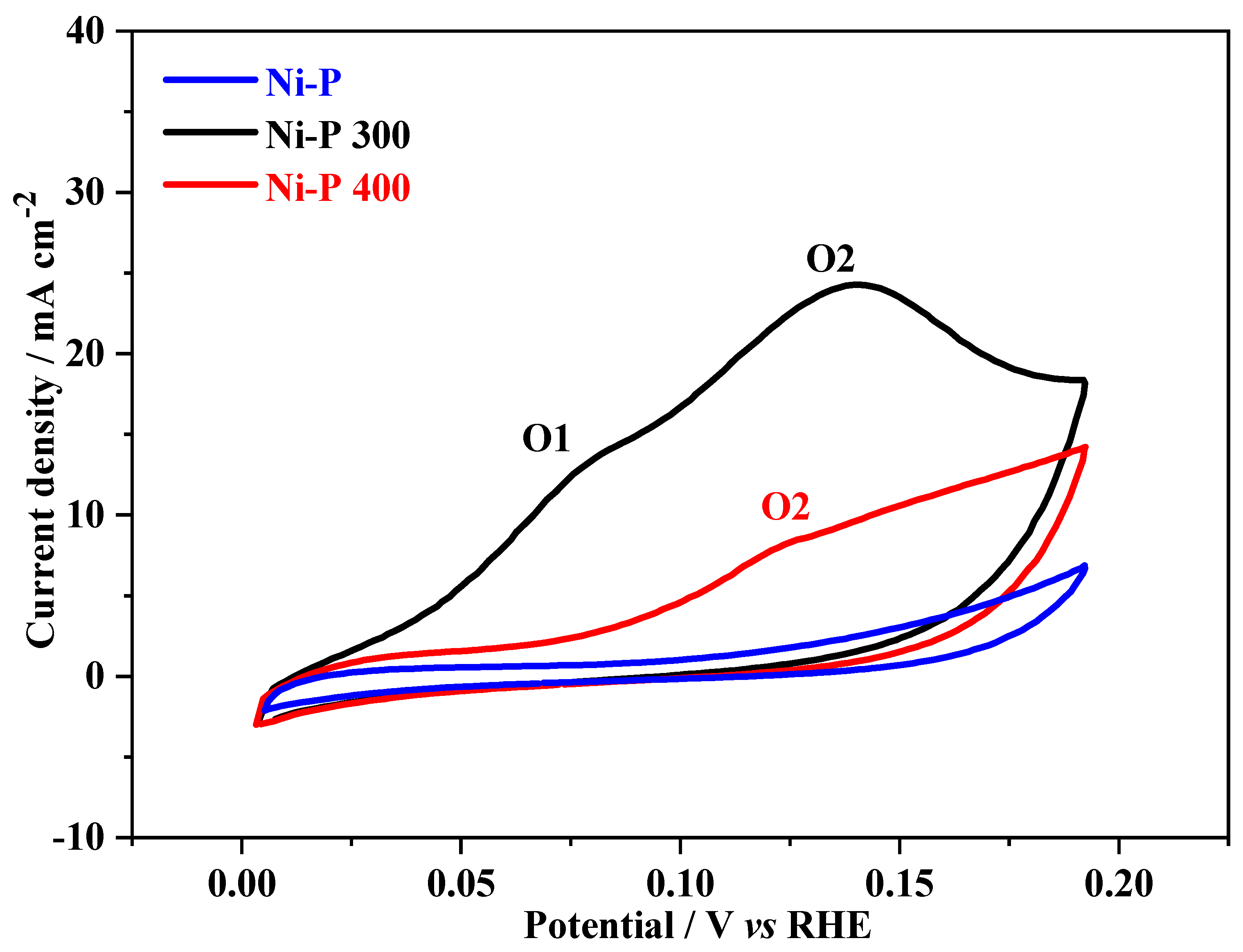
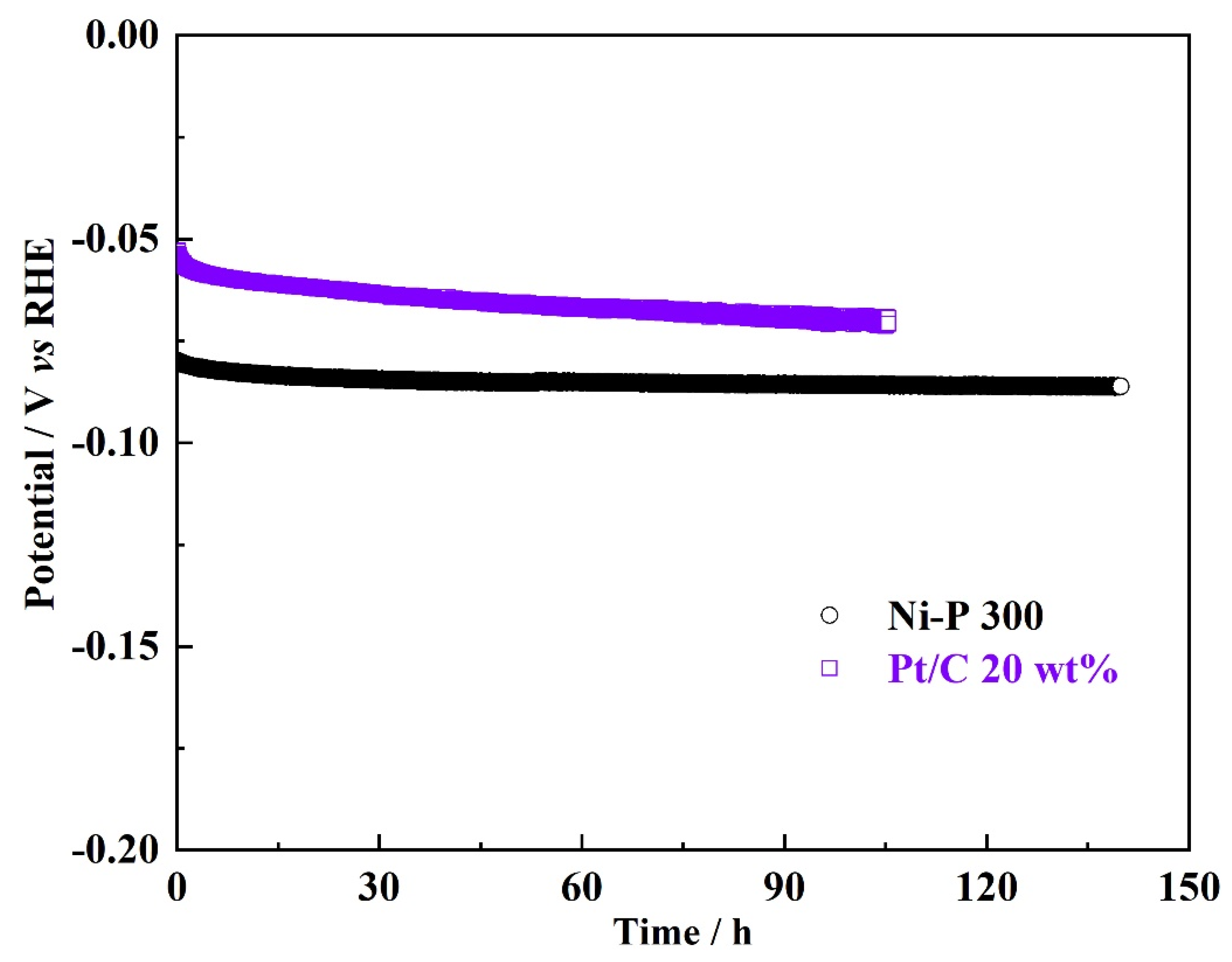
3. Results and Discussions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xing, Y.; Fang, B.; Bonakdarpour, A.; Zhang, S.; Wilkinson, D. Facile fabrication of mesoporous carbon nanofibers with unique hierarchical nanoarchitecture for electrochemical hydrogen storage. Int. J. Hydrogen. Energy 2014, 39, 7859–7867. [Google Scholar] [CrossRef]
- Liao, G.; Gong, Y.; Zhang, L.; Gao, H.; Yang, G.; Fang, B. Semiconductor polymeric graphitic carbon nitride photocatalysts: The “holy grail” for the photocatalytic hydrogen evolution reaction under visible light. Energy Environ. Sci. 2019, 12, 2080–2147. [Google Scholar] [CrossRef]
- Kim, J.; Fang, B.; Song, M.; Yu, J. Topological transformation of thioether-bridged organosilicas into nanostructured functional materials. Chem. Mater. 2012, 24, 2256–2264. [Google Scholar] [CrossRef]
- Fang, B.; Kim, J.; Yu, J. Colloid-imprinted carbon with superb nanostructure as highly efficient cathode electrocatalsyt support in proton exchange membrane fuel cell. Electrochem. Commun. 2008, 10, 659–662. [Google Scholar]
- Fang, B.; Chaudhari, N.; Kim, M.; Kim, J.; Yu, J. Homogeneous deposition of platinum nanoparticles on carbon black for proton exchange membrane fuel cell. J. Am. Chem. Soc. 2009, 131, 15330–15338. [Google Scholar] [CrossRef]
- Fang, B.; Daniel, L.; Bonakdarpour, A.; Govindarajan, R.; Sharman, J.; Wilkinson, D. Dense Pt nanowire electrocatalysts for improved fuel cell performance using a graphitic carbon nitride-decorated hierarchical nanocarbon support. Small 2021, 17, 2102288. [Google Scholar] [CrossRef]
- Liao, G.; Tao, X.; Fang, B. An innovative synthesis strategy for highly efficient and defects-switchable hydrogenated TiO2 photocatalysts. Matter 2022, 5, 377–379. [Google Scholar] [CrossRef]
- Lu, L.; Zou, S.; Fang, B. The critical impacts of ligands on heterogeneous nanocatalysis: A review. ACS Catal. 2021, 11, 6020–6058. [Google Scholar] [CrossRef]
- Liao, G.; Li, C.; Li, X.; Fang, B. Emerging polymeric carbon nitride Z-scheme systems for photocatalysis. Cell Rep. Phys. Sci. 2021, 2, 100355. [Google Scholar] [CrossRef]
- Liao, G.; Li, C.; Fang, B. Donor-acceptor organic semiconductor heterojunction nanoparticles for efficient photocatalytic H2 evolution. Matter 2022, 5, 1635–1637. [Google Scholar] [CrossRef]
- Liao, G.; Li, C.; Liu, S.; Fang, B.; Yang, H. Emerging frontiers of Z-scheme photocatalytic systems. Trends Chem. 2022, 4, 111–127. [Google Scholar] [CrossRef]
- Rodríguez-Varela, J.; Alonso-Lemus, I.; Savadogo, O.; Palaniswamy, K. Overview: Current trends in green electrochemical energy conversion and storage. J. Mater. Res. 2021, 36, 4071–4083. [Google Scholar] [CrossRef]
- Carmo, M.; Fritz, D.; Mergel, J.; Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrogen Energy 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- McCrory, C.; Jung, S.; Ferrer, I.; Chatman, S.; Peters, J.; Jaramillo, T. Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting Devices. J. Am. Chem. Soc. 2015, 137, 4347–4357. [Google Scholar] [CrossRef]
- Wang, Z.; Xiao, B.; Lin, Z.; Xu, Y.; Lin, Y.; Meng, F.; Zhang, Q.; Gu, L.; Fang, B.; Guo, S. PtSe2/Pt heterointerface with reduced coordination for boosted hydrogen evolution reaction. Angew. Chem. Int. Ed. 2021, 60, 23388–23393. [Google Scholar] [CrossRef]
- Wei, J.; Zhou, M.; Long, A.; Xue, M.; Liao, H.; Wei, C.; Xu, Z.J. Heterostructured electrocatalysts for hydrogen evolution reaction under alkaline conditions. Nano-Micro Lett. 2018, 10, 75. [Google Scholar] [CrossRef]
- Zou, X.; Zhang, Y. Noble metal-free hydrogen evolution catalysts for water splitting. Chem. Soc. Rev. 2015, 44, 5148–5180. [Google Scholar] [CrossRef]
- Cao, S.; Wang, C.; Fu, W.; Chen, Y. Metal phosphides as co-catalysts for photocatalytic and photoelectrocatalytic water splitting. ChemSusChem 2017, 10, 4306–4323. [Google Scholar] [CrossRef]
- Weng, C.; Ren, J.; Yuan, Z. Transition metal phosphide-based materials for efficient electrochemical hydrogen evolution: A critical review. ChemSusChem 2020, 13, 3357–3375. [Google Scholar] [CrossRef]
- Li, Y.; Dong, Z.; Jiao, L. Multifunctional transition metal-based phosphides in energy-related electrocatalysis. Adv. Energy Mater. 2020, 10, 1902104. [Google Scholar] [CrossRef]
- Zhang, C.; Xie, Y.; Deng, H.; Zhang, C.; Su, J.-W.; Dong, Y.; Lin, J. Ternary nickel iron phosphide supported on nickel foam as a high-efficiency electrocatalyst for overall water splitting. Int. J. Hydrogen Energy 2018, 43, 7299–7306. [Google Scholar] [CrossRef]
- Popczun, E.; Read, C.; Roske, C.; Lewis, N.; Schaak, R. Highly active electrocatalysis of the HER by CoP. Angew. Chem. Int. Ed. 2014, 53, 5427–5430. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, C.; He, X.; White, T.; Demaree, J.; Griep, M.; Lin, J. Monolithic electrochemical cells for overall water splitting. J. Power Sources 2018, 397, 37–43. [Google Scholar] [CrossRef]
- Yan, H.; Xie, Y.; Jiao, Y.; Wu, A.; Tian, C.; Zhang, X.; Wang, L.; Fu, H. Holey reduced graphene oxide coupled with an Mo2N-Mo2C heterojunction for efficient hydrogen evolution. Adv. Mater. 2018, 30, 1704156. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Fang, B.; Wang, Z.; Wang, C.; Liu, Z.; Liu, F.; Wang, W.; Alfantazi, A.; Wang, D.; Wilkinson, D.P. MoS2 nanosheets: A designed structure with high active site density for the hydrogen evolution reaction. ACS Catal. 2013, 3, 2101–2107. [Google Scholar] [CrossRef]
- Wang, H.; Fu, W.; Yang, X.; Huang, Z.; Li, J.; Zhang, H.; Wang, Y. Hydrogen evolution reaction electrocatalysis. J. Mater. Chem. A 2020, 8, 6926–6956. [Google Scholar] [CrossRef]
- Giolando, D.M.; Wasalathanthri, R.N.; Jeffrey, S.; Su, N.; Sun, K. Stoichiometric control of electrocatalytic amorphous nickel phosphide to incre-ase hydrogen evolution reaction activity and stability in acidic medium Stoichiometric. ChemistrySelect 2017, 2, 8020–8027. [Google Scholar]
- Xu, J.; Liu, T.; Li, J.; Li, B.; Liu, Y.; Zhang, B.; Xiong, D.; Amorim, I.; Li, W.; Liu, L. Boosting the hydrogen evolution performance of ruthenium clusters through synergistic coupling with cobalt phosphide. Energy Environ. Sci. 2018, 11, 1819–1827. [Google Scholar] [CrossRef]
- Xu, H.; Shang, H.; Wang, C.; Du, Y. Surface and interface engineering of noble metal-free electrocatalysts for efficient overall water splitting. Coord. Chem. Rev. 2020, 418, 213374. [Google Scholar] [CrossRef]
- Xu, J.; Wei, X.-K.; Costa, J.D.; Lado, J.L.; Owens-Baird, B.; Gonçalves, L.; Fernandes, S.; Heggen, M.; Petrovykh, D.; Dunin-Borkowski, R.; et al. Interface engineering in nanostructured nickel phosphide catalyst for efficient and stable water oxidation. ACS Catal. 2017, 7, 5450–5455. [Google Scholar] [CrossRef]
- Gao, J.; Wu, Y.; Liu, L.; Shen, B.; Hu, W. Crystallization temperature of amorphous electroless nickel–phosphorus alloys. Mater. Lett. 2005, 59, 1665–1669. [Google Scholar]
- Kucernak, A.R.J.; Naranammalpuram, S. Nickel phosphide: The effect of phosphorus content on hydrogen evolution activity and corrosion resistance in acidic medium. J. Mater. Chem. A 2014, 2, 17435–17445. [Google Scholar] [CrossRef]
- Popczun, E.J.; McKone, J.R.; Read, C.G.; Biacchi, A.J.; Wiltrout, A.M.; Lewis, N.S.; Schaak, R.E. Nanostructured nickel phosphide as an electrocatalyst for the hydrogen evolution reaction. J. Am. Chem. Soc. 2013, 135, 9267–9270. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Jiang, Y.; An, T.; Cao, M. Increasing the active sites and intrinsic activity of transition metal chalcogenide electrocatalysts for enhanced water splitting. J. Mater. Chem. A 2020, 8, 25465–25498. [Google Scholar] [CrossRef]
- Fujii, S.; Hamasaki, H.; Takeoka, H.; Tsuruoka, T.; Akamatsu, K.; Nakamura, Y. Electroless nickel plating on polymer particles. J. Colloid. Interface Sci. 2014, 430, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhao, R.; Xiang, H.; Wu, J.; Zhong, W.; Li, W.; Zhang, Q.; Yang, N.; Li, X. Ni-activated transition metal carbides for efficient hydrogen evolution in acidic and alkaline Solutions. Adv. Energy Mater. 2020, 10, 2002260. [Google Scholar] [CrossRef]
- Moreau, L.; Ha, D.; Zhang, H.; Hovden, R.; Muller, D.; Robinson, R. Defining crystalline/amorphous phases of nanoparticles through X-ray absorption spectroscopy and X-ray diffraction: The case of nickel phosphide. Chem. Mater. 2013, 25, 2394–2403. [Google Scholar] [CrossRef]
- Henkes, A.; Vasquez, Y.; Schaak, R. Converting metals into phosphides: A general strategy for the synthesis of metal phosphide nanocrystals. J. Am. Chem. Soc. 2007, 129, 1896–1897. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, Y.; Zhang, H.; Xiao, L.; Wu, W. Novel C@Ni3P nanoparticles for highly selective hydrogenation of furfural to furfuryl alcohol. Catal. Lett. 2022, 152, 883–894. [Google Scholar] [CrossRef]
- Li, R.; Xu, J.; Lu, C.; Huang, Z.; Wu, Q.; Ba, J.; Tang, T.; Meng, D.; Luo, W. Amorphous NiFe phosphides supported on nanoarray-structured nitrogen-doped carbon paper for high-performance overall water splitting. Electrochim. Acta 2020, 357, 136873. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Zheng, B.; Yang, D.; Zhang, W.; Chen, Y. Self-assembled Ni2P/FeP heterostructural nanoparticles embedded in N-doped graphene nanosheets as highly efficient and stable multifunctional electrocatalyst for water splitting. Electrochim. Acta 2019, 318, 449–459. [Google Scholar] [CrossRef]
- Song, F.; Li, W.; Yang, J.; Han, G.; Liao, P.; Sun, Y. Interfacing nickel nitride and nickel boosts both electrocatalytic hydrogen evolution and oxidation reactions. Nat. Commun. 2018, 9, 4531. [Google Scholar] [CrossRef] [PubMed]
- Anantharaj, S.; Ede, S.; Karthick, K.; Sankar, S.S.; Sangeetha, K.; Karthik, P.; Kundu, S. Precision and correctness in the evaluation of electrocatalytic water splitting: Revisiting activity parameters with a critical assessment. Energy Environ. Sci. 2018, 11, 744–771. [Google Scholar] [CrossRef]
- Ni, W.; Krammer, A.; Hsu, C.; Chen, H.; Schüler, A.; Hu, X. Ni3N as an active hydrogen oxidation reaction catalyst in alkaline medium. Angew. Chemie. Int. Ed. 2019, 58, 7445–7449. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(mV vs. RHE) | Tafel Slope (mV dec−1) | Rct (Ω cm−2) | Cdl (mF cm−2) | |
|---|---|---|---|---|
| Ni-P | 144 | 97 | 14.1 | 4.5 |
| Ni-P 300 | 65 | 53 | 6.3 | 15.3 |
| Ni-P 400 | 84 | 56 | 6.8 | 11.5 |
| Ni-P 500 | 107 | 66 | 10.9 | 6.6 |
| Pt/C 20 wt% | 32 | 31 | / | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Hou, F.; Peng, S.; Wang, X.; Fang, B. Synthesis, Physical Properties and Electrocatalytic Performance of Nickel Phosphides for Hydrogen Evolution Reaction of Water Electrolysis. Nanomaterials 2022, 12, 2935. https://doi.org/10.3390/nano12172935
Liu G, Hou F, Peng S, Wang X, Fang B. Synthesis, Physical Properties and Electrocatalytic Performance of Nickel Phosphides for Hydrogen Evolution Reaction of Water Electrolysis. Nanomaterials. 2022; 12(17):2935. https://doi.org/10.3390/nano12172935
Chicago/Turabian StyleLiu, Gaoyang, Faguo Hou, Shanlong Peng, Xindong Wang, and Baizeng Fang. 2022. "Synthesis, Physical Properties and Electrocatalytic Performance of Nickel Phosphides for Hydrogen Evolution Reaction of Water Electrolysis" Nanomaterials 12, no. 17: 2935. https://doi.org/10.3390/nano12172935
APA StyleLiu, G., Hou, F., Peng, S., Wang, X., & Fang, B. (2022). Synthesis, Physical Properties and Electrocatalytic Performance of Nickel Phosphides for Hydrogen Evolution Reaction of Water Electrolysis. Nanomaterials, 12(17), 2935. https://doi.org/10.3390/nano12172935