
DFT and TD-DFT Investigation of a Charge Transfer Surface Resonance Raman Model of N3 Dye Bound to a Small TiO2 Nanoparticle


Abstract
1. Introduction
1.1. Raman and Infrared Studies
1.2. Optical Studies, Theoretical Electronic Structure, and Charge Transfer Mechanisms
1.3. TiO2 Nanoparticle Models
2. Computational Methods and Models
3. Results and Discussion
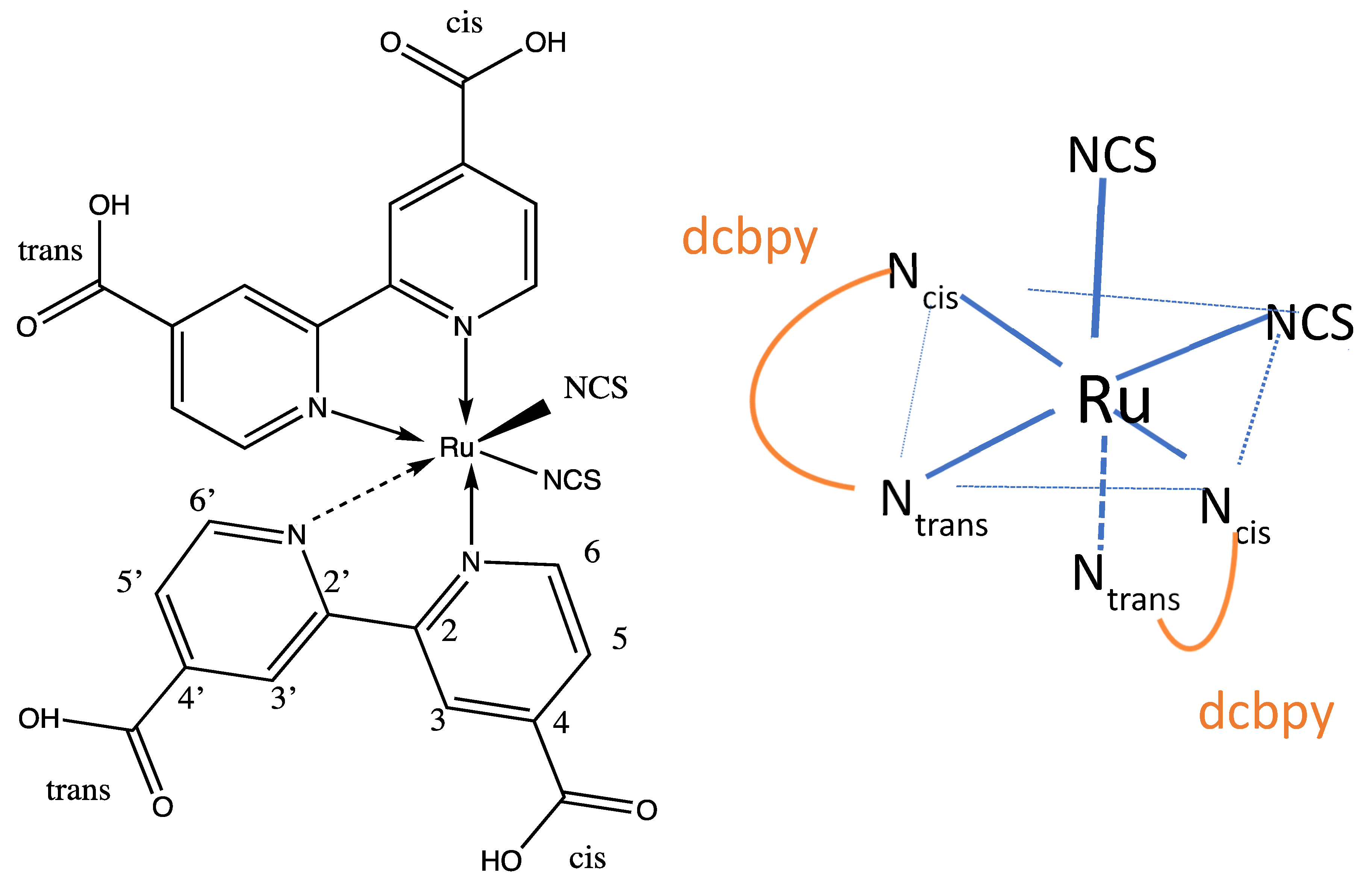
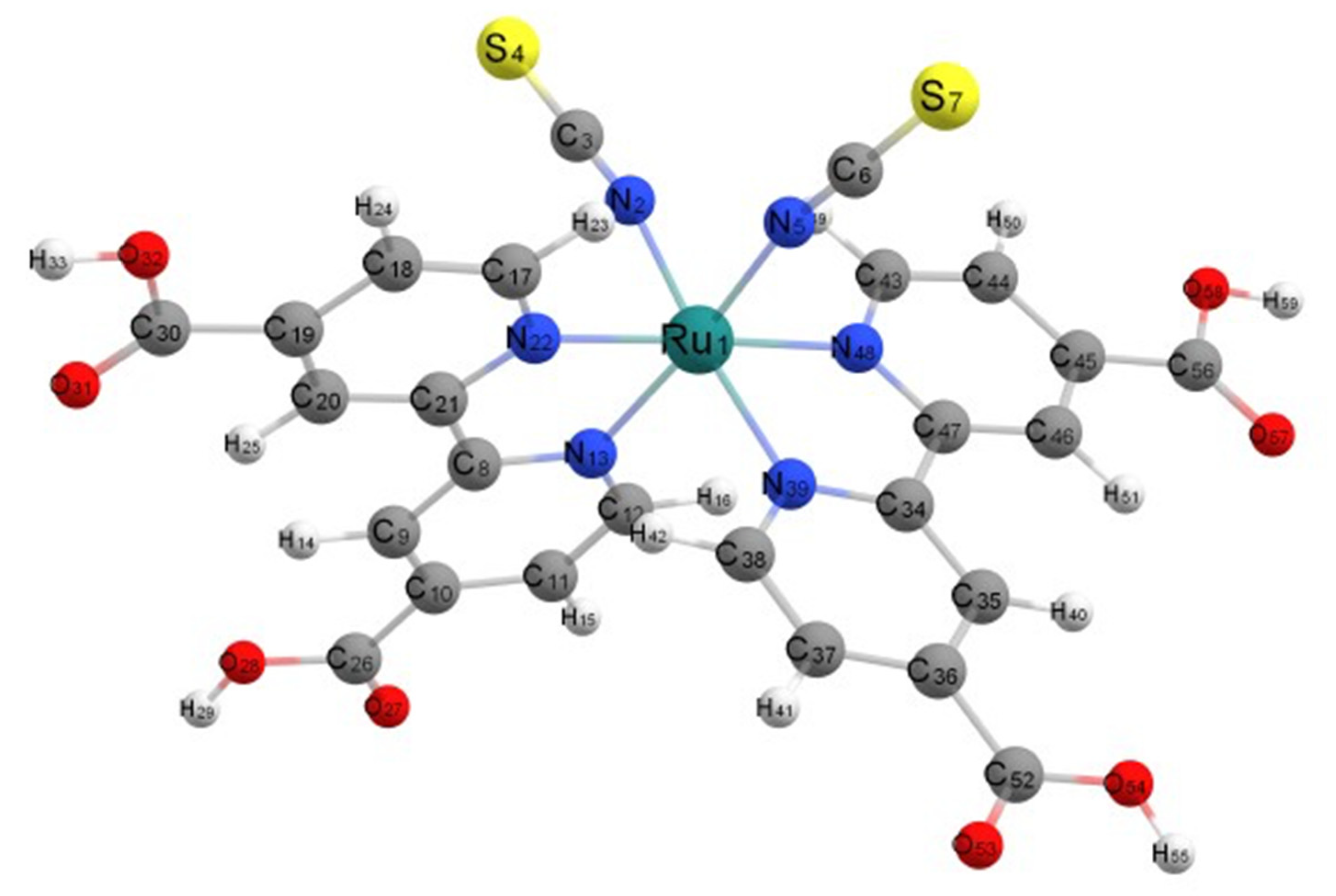
3.1. Optimized Geometry of Fully Protonated N3
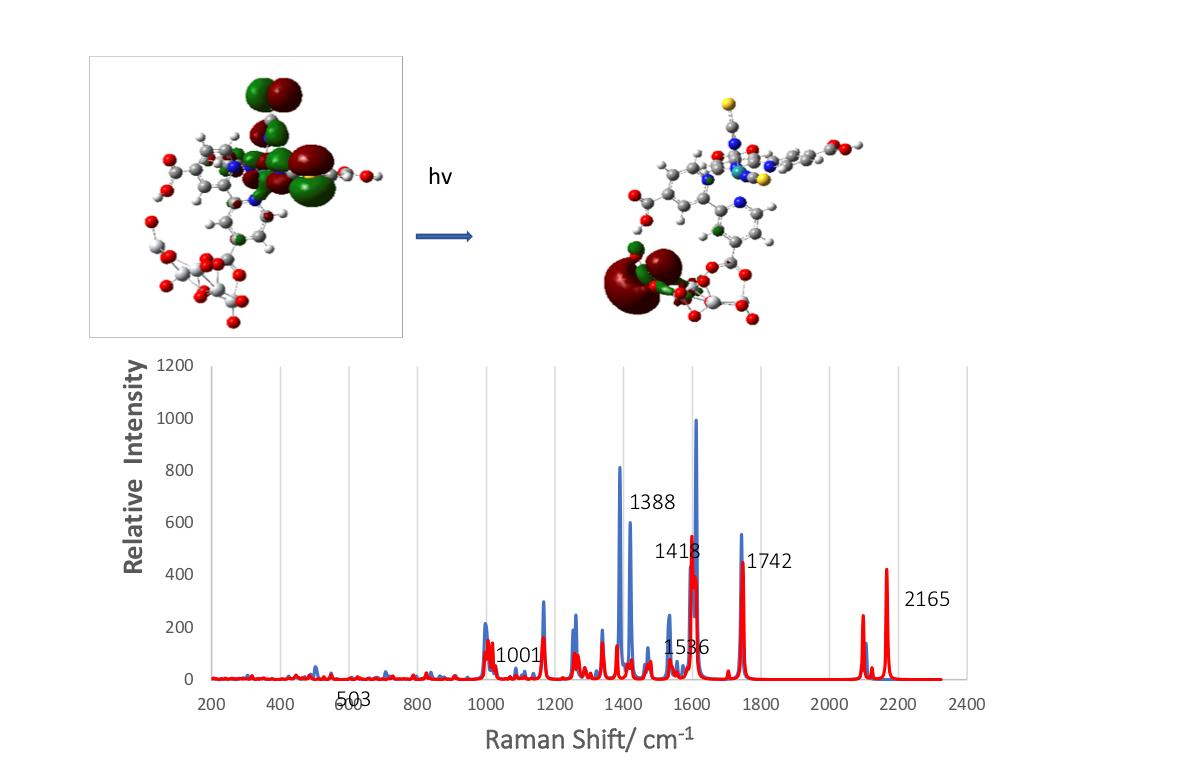
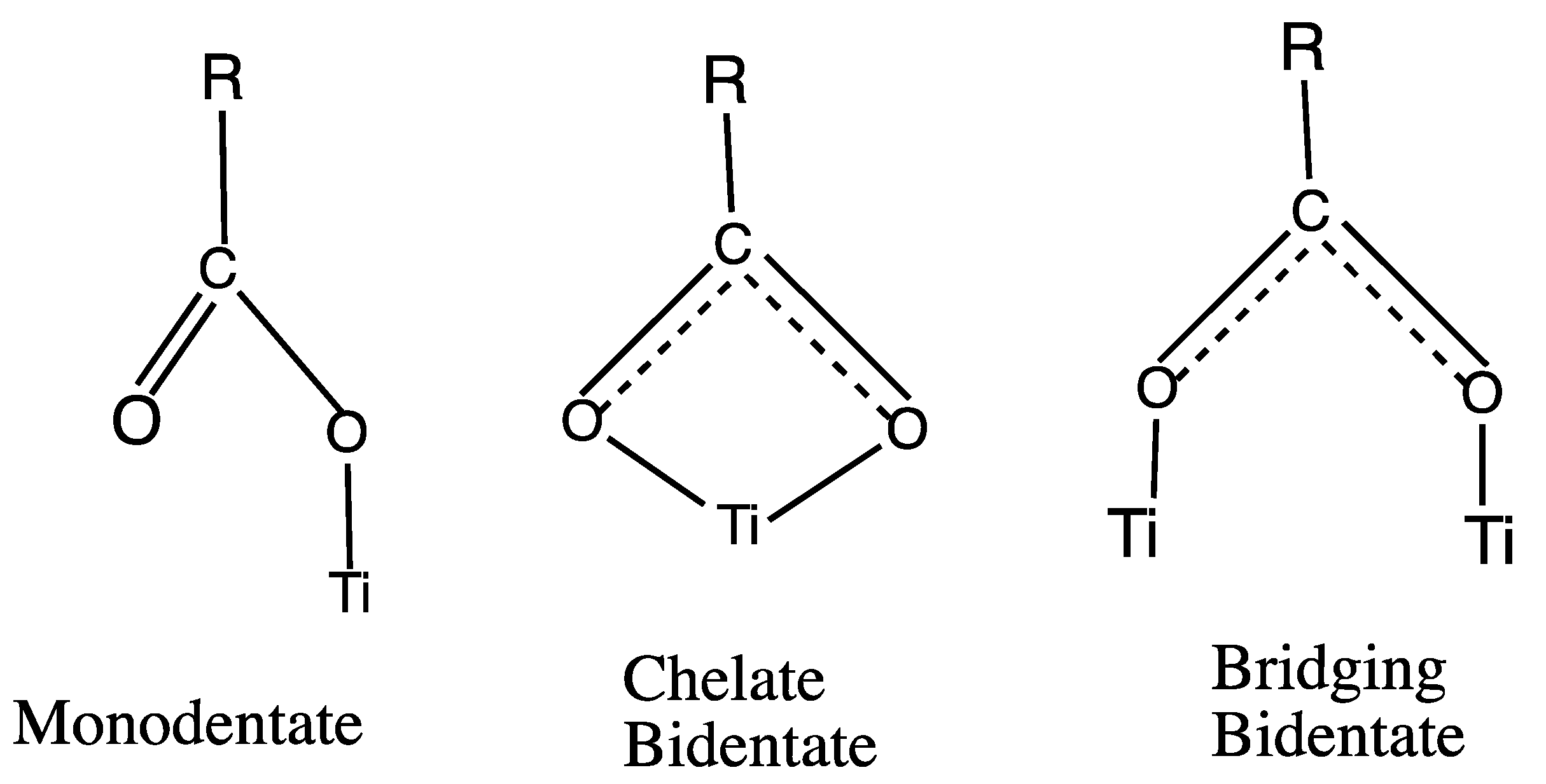
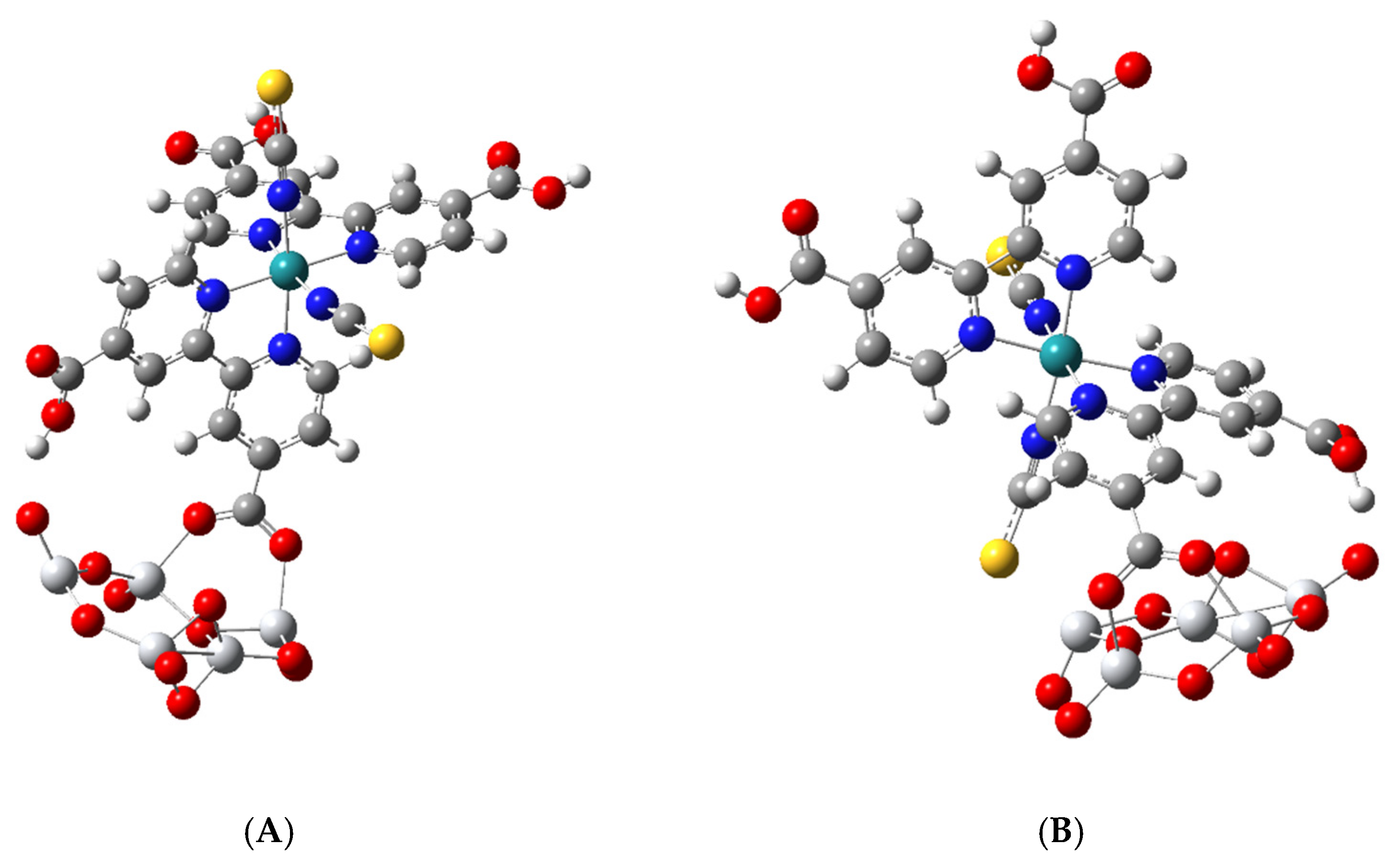
3.2. Geometric Optimization of the N3-Ti5O10 Complexes
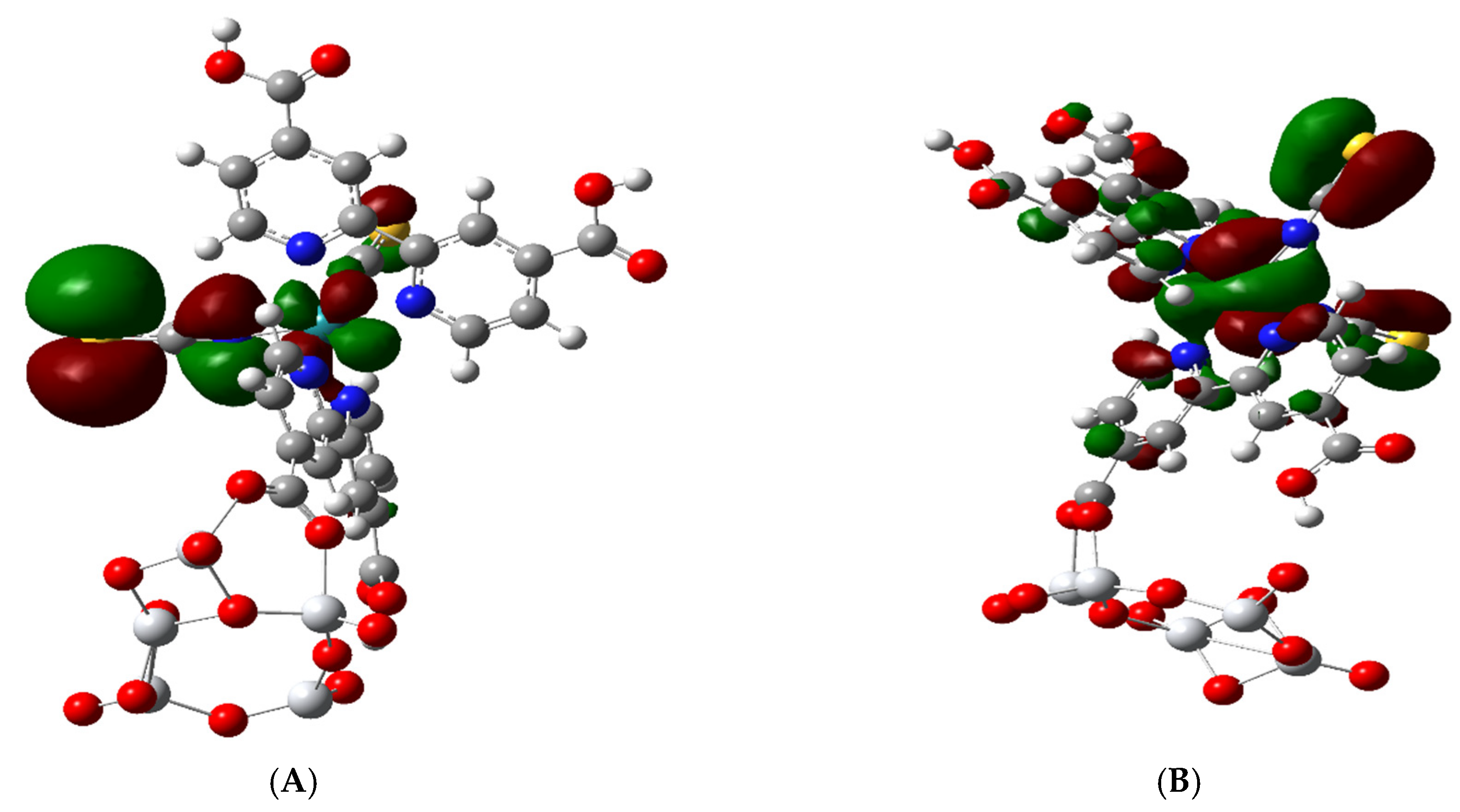
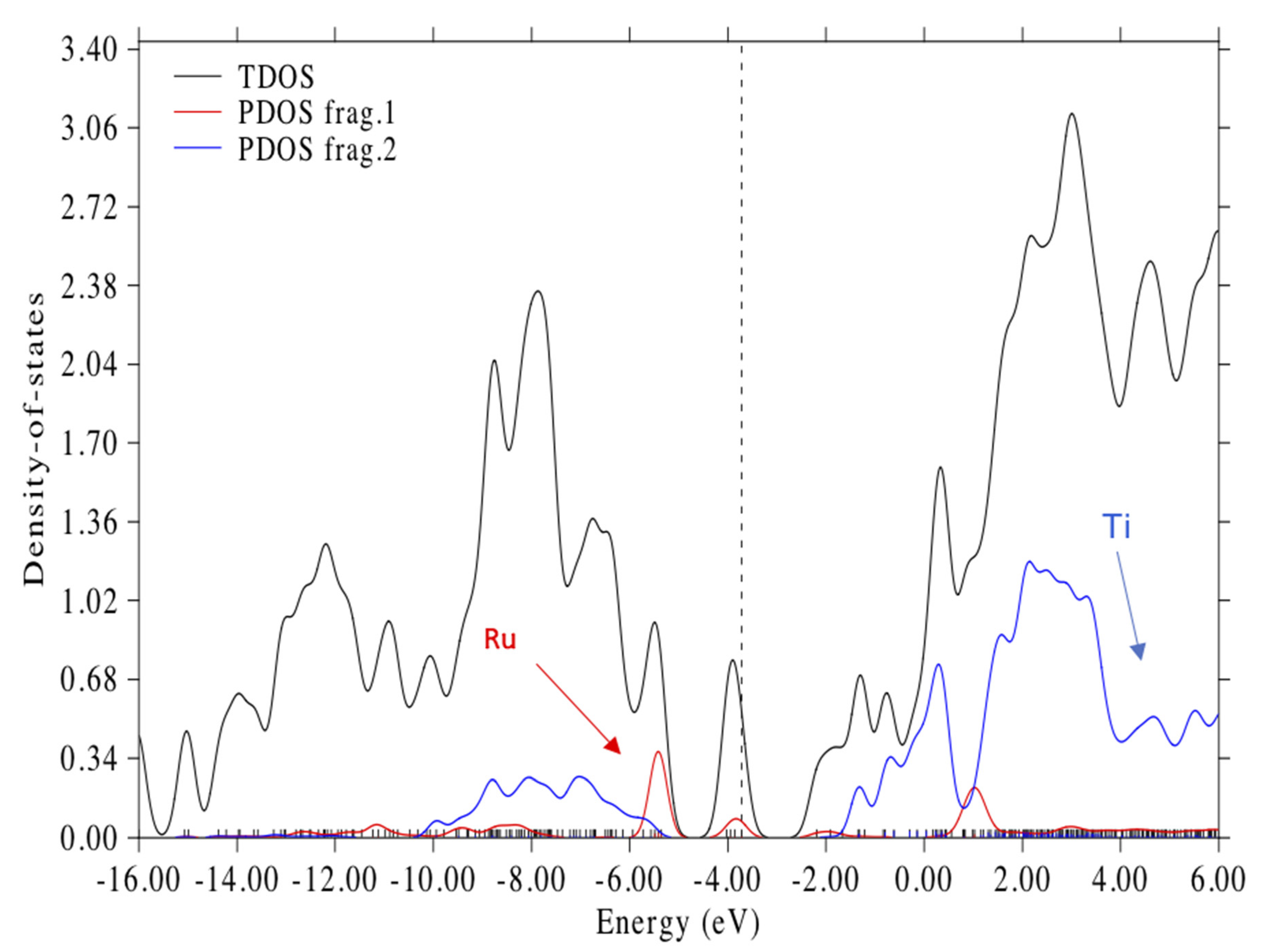
3.3. Electronic Structure of the Complex
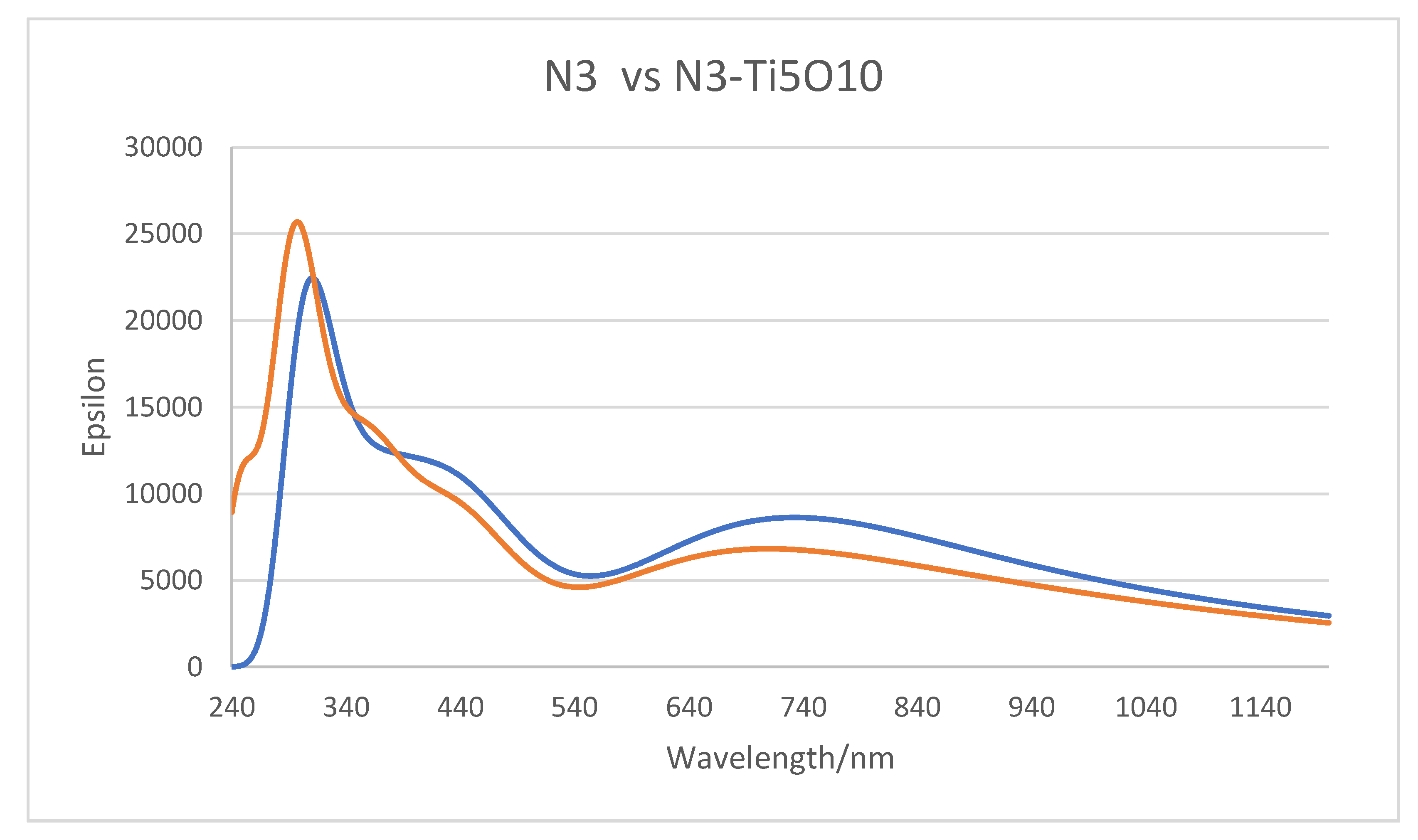
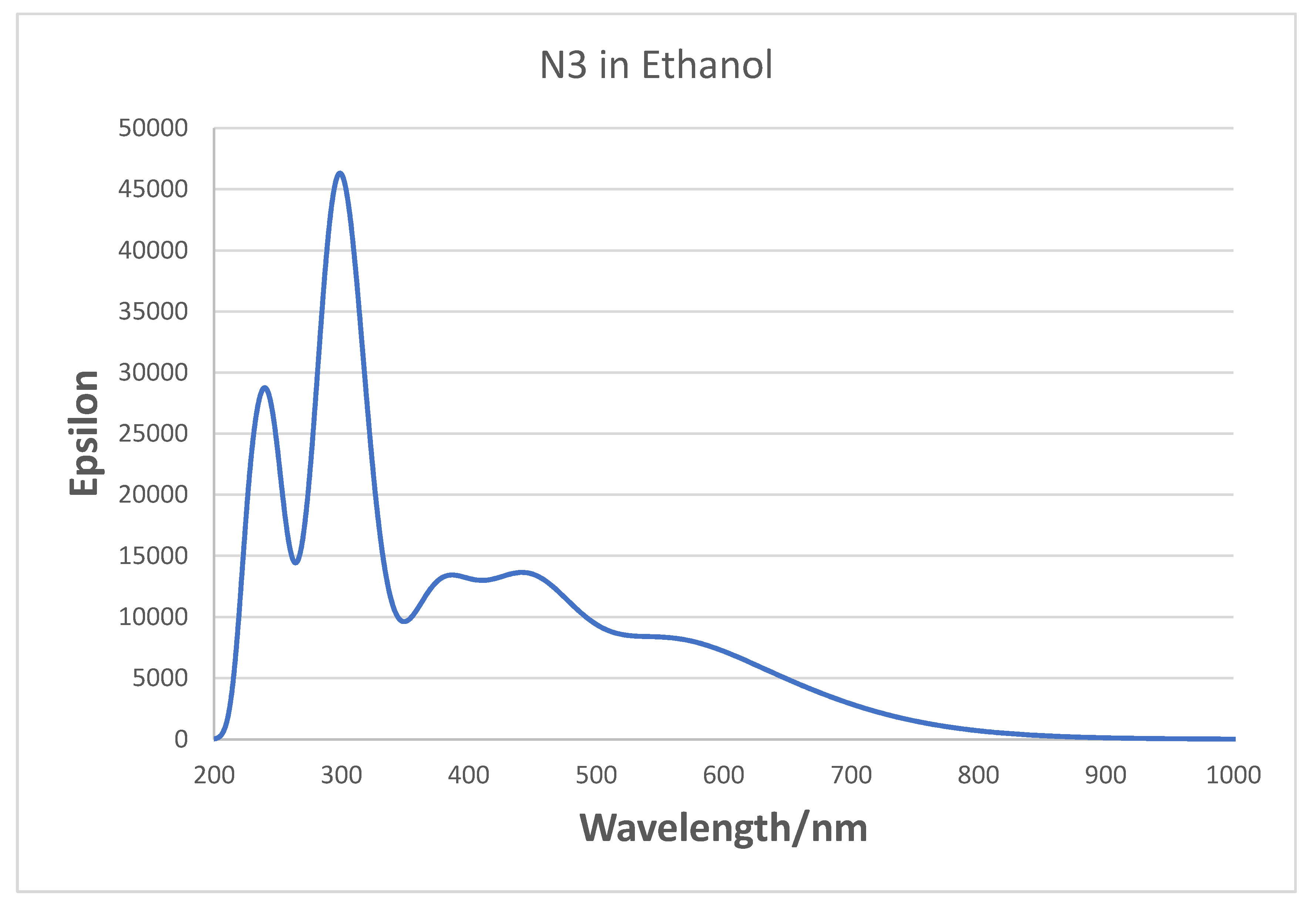
3.4. Simulated Optical Absorption Spectra
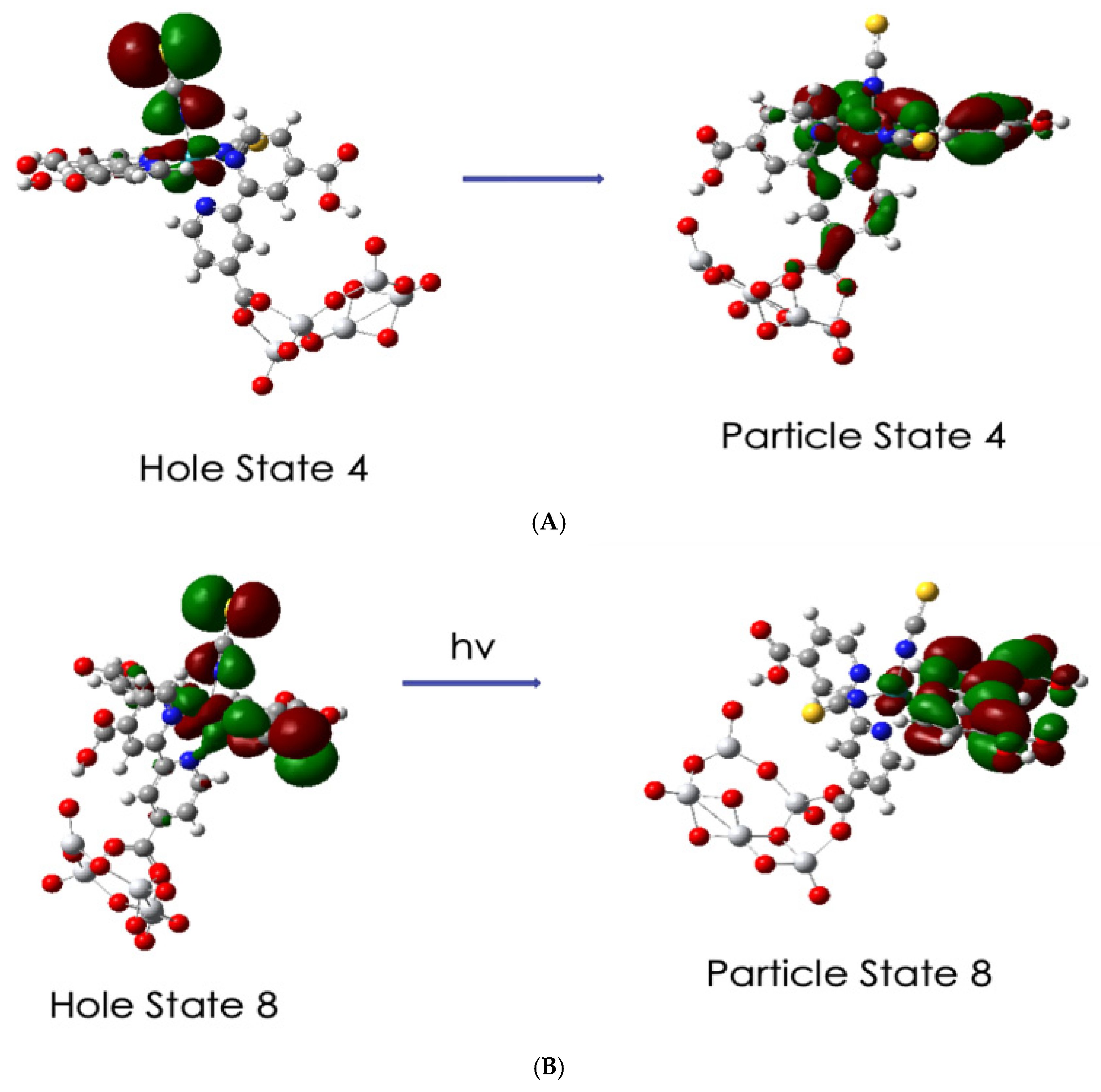
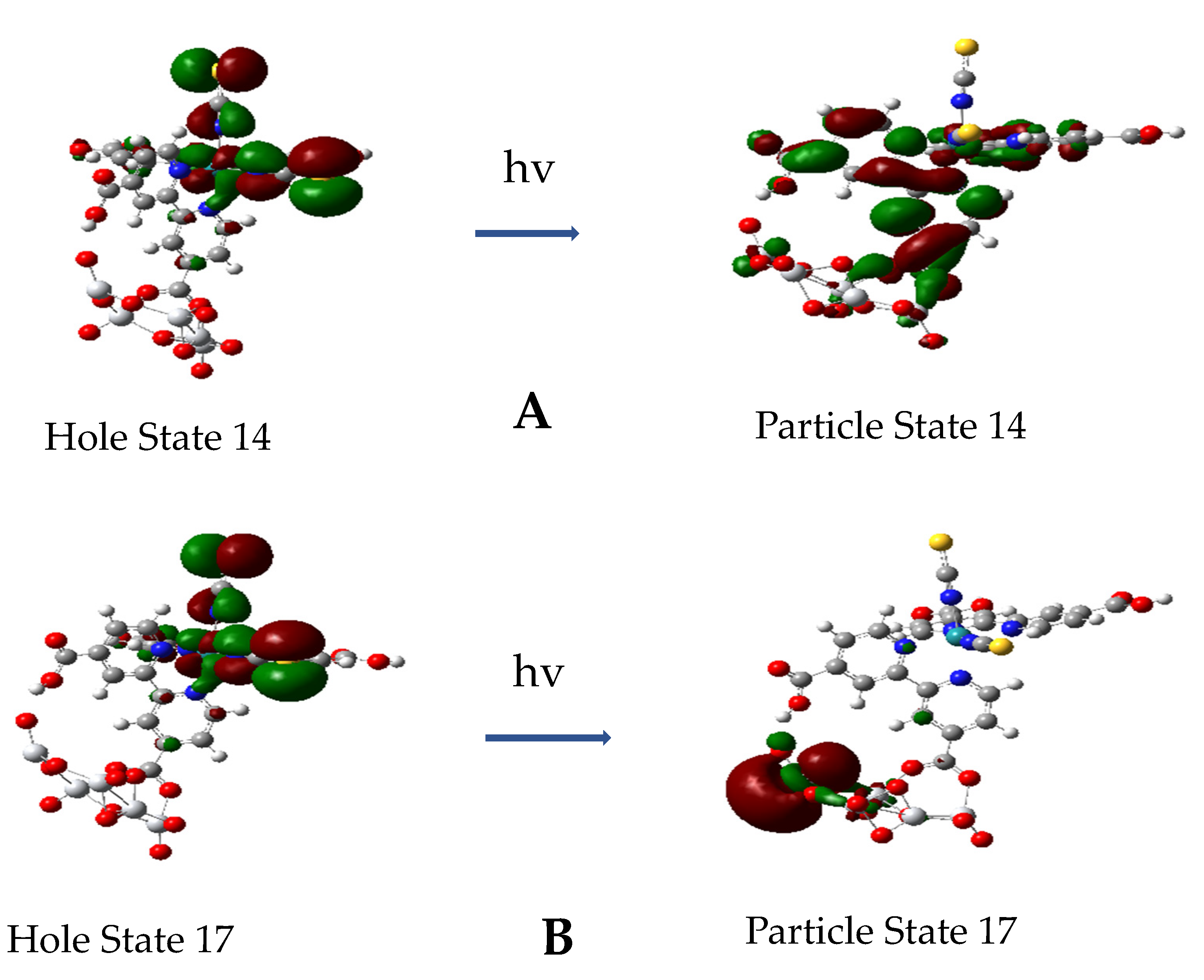
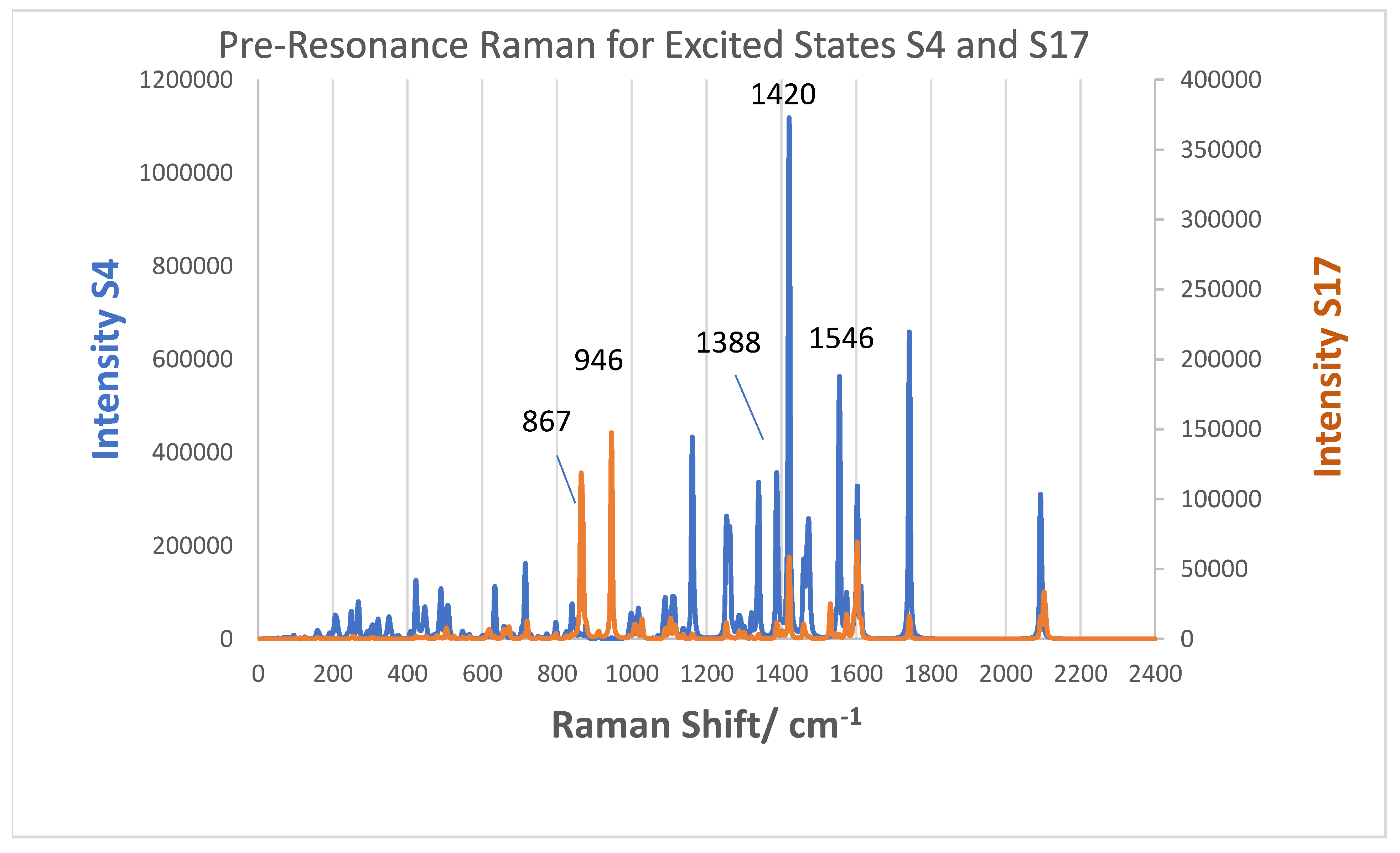
3.5. Excited State Transitions
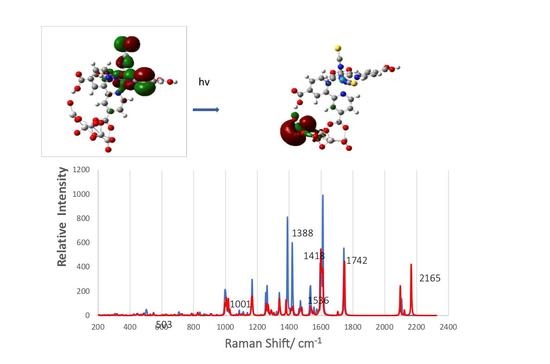
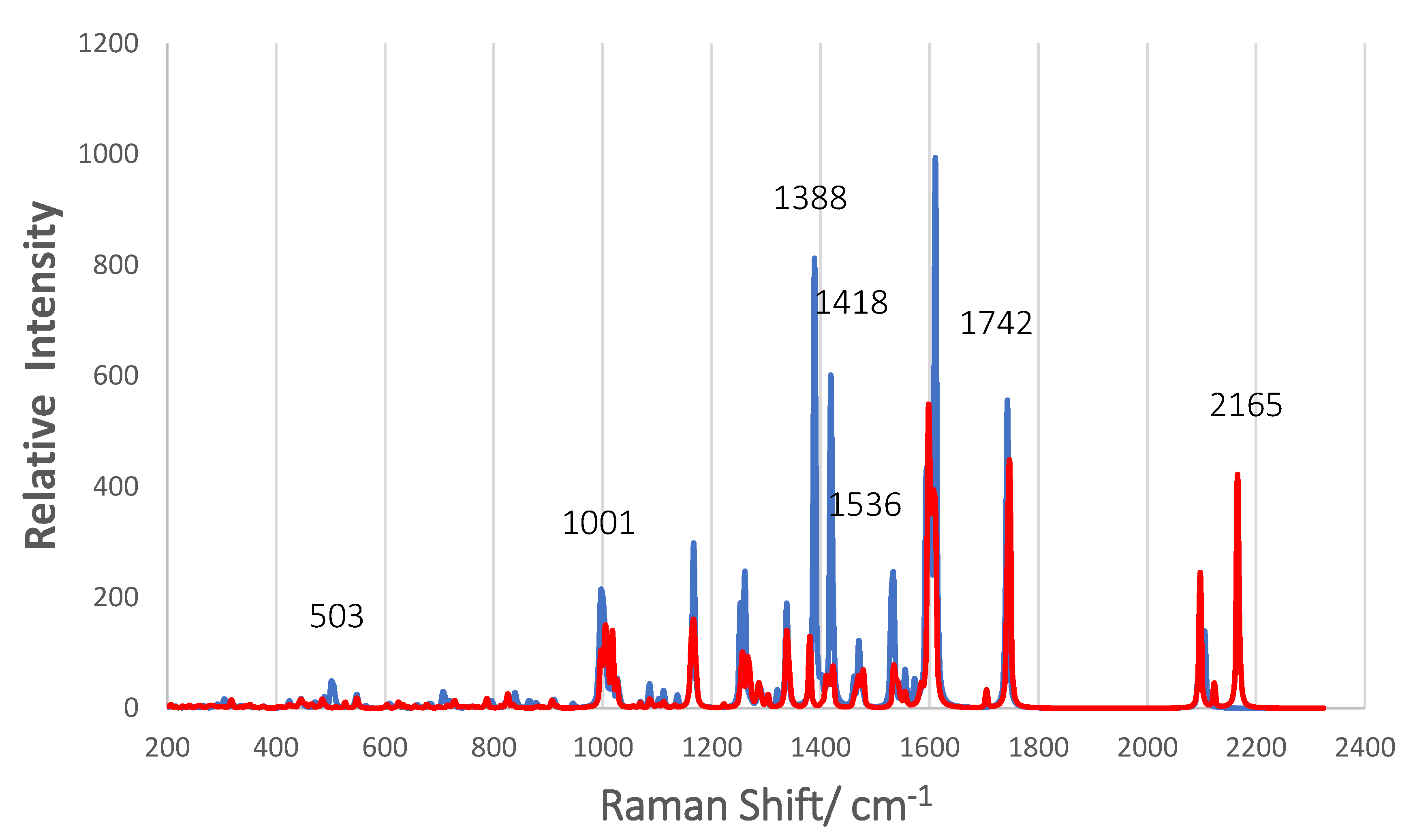
3.6. Simulated Raman Scattering Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- O’Regan, B.; Grätzel, A. Low-Cost, High-Efficiency Solar Cell Based on Dye-Sensitized Colloidal TiO2 Films. Nature 1991, 353, 737–740. [Google Scholar]
- Bai, Y.; Mora-Sero’, I.; De Angelis, F.; Biisquert, J.; Wang, P. Titanium Dioxide Nanomaterials for Photovoltaic Applications. Chem. Rev. 2014, 114, 10095–10130. [Google Scholar] [PubMed]
- De Angelis, F.; Fantacci, S.; Selloni, A.; Grätzel, M.; Nazeeruddin, M.K. Influence of the Sensitizer Adsorption Mode on the Open-Circuit Potential of Dye-Sensitized Solar Cells. Nano Lett. 2007, 7, 3189–3195. [Google Scholar] [CrossRef]
- Lee, C.-P.; Li, C.-T.; Ho, K.-C. Use of organic materials in dye-sensitized solar cells. Mater. Today 2017, 20, 267–283. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Zakeeruddin, S.M.; Humphry-Baker, R.; Jirousek, M.; Liska, P.; Vlachopoulos, N.; Shklover, V.; Fischer, C.-H. GrätzeAcid-Base Equilibria of (2,2′-Bipyridyl-4,4′-dicarboxylic acid) ruthenium(II) Complexes and the Effect of Protonation on Charge-Transfer Sensitization of Nanocrystalline Titania. Inorg. Chem. 1999, 38, 6298–6305. [Google Scholar] [CrossRef]
- Yang, L.; Jiang, X.; Ruan, W.; Zhao, B.; Xu, W.; Lombardi, J.R. Observation of Enhance Raman Scattering for Molecules Adsorbed on TiO2 Nanoparticles: Charge-Transfer Contribution. J. Phys. Chem. C 2008, 112, 20095–20098. [Google Scholar] [CrossRef]
- Musumeci, A.; Gosztola, D.; Schiller, T.; Dimitrijevic, N.M.; Mujica, V.; Martin, D.; Rajh, T. SERS of Semiconducting Nanoparticles (TiO2 Hybrid Composites). J. Am. Chem. Soc. 2009, 131, 6040–6041. [Google Scholar] [CrossRef]
- Lombardi, J.R.; Birke, R.L. Theory of Surface-Enhanced Raman Scattering in Semiconductors. J. Phys. Chem. C 2014, 118, 11120–11130. [Google Scholar] [CrossRef]
- Lombardi, J.; Birke, R.L. A Unified View of Surface-Enhanced Raman Scattering. Acc. Chem. Res. 2009, 42, 734–742. [Google Scholar] [CrossRef]
- Wang, X.; Shi, W.; Wang, S.; Zhao, H.; Lin, J.; Yang, Z.; Chen, M.; Guo, L. Two-Dimensional Amorphous TiO2 Nanosheets Enabling High-Efficiency Photoinduced Charge Transfer for Excellent SERS Activity. J. Am. Chem. Soc. 2019, 141, 5856–5862. [Google Scholar] [CrossRef]
- Maznichenko, D.; Venkatakrishnan, K.; Tan, B. Stimulating Multiple SERS Mechanisms by a Nanofibrous Three-Dimensional Network Structure of Titanium Dioxide (TiO2). J. Phys. Chem. C 2013, 117, 578–583. [Google Scholar] [CrossRef]
- Theil, F.; Zedler, L.; März, A.; Xie, W.; Csáki, A.; Fritzsche, W.; Cialla, D.; Schmitt, M.; Popp, J.; Dietzek, B. Evidence for SERRS Enhancement in the Spectra of Ruthenium Dye−Metal Nanoparticle Conjugates. J. Phys. Chem. C 2013, 117, 1121–1129. [Google Scholar] [CrossRef]
- Perez León, C.; Kador, L.; Peng, B.; Thelakkat, M. Influence of the Solvent on Ruthenium(II) Bipyridyl Complexes. J. Phys. Chem. B 2005, 109, 5783–5789. [Google Scholar] [CrossRef] [PubMed]
- Schade, L.; Franzka, S.; Biener, M.; Biener, J.; Hartmann, N. Surface-enhanced Raman spectroscopy on laser-engineered ruthenium dye-functionalized nanoporous gold. App. Surf. Sci. 2013, 347, 19–22. [Google Scholar] [CrossRef]
- Qiu, Z.; Zhang, M.; Wu, D.-Y.; Ding, S.-Y.; Zuo, Q.-Q.; Huang, Y.-F.; Shen, W.; Lin, X.-D.; Tian, Z.-Q.; Mao, B.-W. Raman Spectroscopic Investigation on TiO2–N719 Dye Interfaces Using Ag@TiO2 Nanoparticles and Potential Correlation Strategies. Chem. Phys. Chem. 2013, 14, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Q.-Q.; Feng, Y.-L.; Chen, S.; Qiu, Z.; Xie, L.-Q.; Xiao, Z.-Y.; Yang, Z.-L.; Mao, B.-W.; Tian, Z.-Q. Dimeric Core−Shell Ag2@TiO2 Nanoparticles for Off-Resonance Raman Study of the TiO2−N719 Interface. J. Phys. Chem. C 2015, 119, 18396–18403. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Sui, H.; Zhang, X.; Su, H.; Cheng, W.; Han, X.-X.; Zhao, B. Investigation of Charge Transfer in Ag/N719/TiO2 Interface by Surface-Enhanced Raman Spectroscopy. J. Phys. Chem. C 2016, 120, 13078–13086. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.; Li, P.; Han, X.X.; Ozaki, Y. Charge Transfer at the TiO2/N3/Ag Interface Monitored by Surface-Enhanced Raman Spectroscopy. J. Phys. Chem. C 2017, 121, 5145–5153. [Google Scholar] [CrossRef]
- Finnie, K.S.; Bartlett, J.R.; Woolfrey, J.L. Vibrational Spectroscopic Study of the Coordination of (2,2¢-Bipyridyl-4,4′-dicarboxylic acid) ruthenium(II) Complexes to the Surface of Nanocrystalline Titania. Langmuir 1998, 14, 2744–2749. [Google Scholar] [CrossRef]
- Deacon, G.B.; Phillips, R.J. Relationships between the carbon-oxygen stretching frequencies of carboxylato complexes and the type of carboxylate coordination. Coord. Chem. Rev. 1980, 33, 227–250. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Humphry-Baker, R.; Liska, P.; Grätzel, M. Investigation of Sensitizer Adsorption and the Influence of Protons on Current and Voltage of a Dye-Sensitized Nanocrystalline TiO2 Solar Cell. J. Phys. Chem. B 2003, 107, 8981–8987. [Google Scholar] [CrossRef]
- Greijer, H.; Lindgren, J.; Hagfeldt, A. Resonance Raman Scattering of a Dye-Sensitized Solar Cell: Mechanism of Thiocyanato Ligand Exchange. J. Phys. Chem. B 2001, 105, 6314–6320. [Google Scholar] [CrossRef]
- Shoute, L.C.T.; Loppnow, G.R. Excited-State Metal-to-Ligand Charge Transfer Dynamics of a Ruthenium(II) Dye in Solution and Adsorbed on TiO2 Nanoparticles from Resonance Raman Spectroscopy. J. Am. Chem. Soc. 2003, 125, 15636–15646. [Google Scholar] [CrossRef]
- Perez León, C.; Kador, L.; Peng, B.; Thelakkat, M. Characterization of the Adsorption of Ru-bpy Dyes on Mesoporous TiO2 Films with UV-Vis, Raman, and FTIR Spectroscopies. J. Phys. Chem. B 2006, 110, 8723–8730. [Google Scholar] [CrossRef]
- Lee, K.E.; Gomez, M.A.; Elouatik, S. Further Understanding of the Adsorption Mechanism of N719 Sensitizer on Anatase TiO2 Films for DSSC Applications Using Vibrational Spectroscopy and Confocal Raman Imaging. Langmuir 2010, 26, 9575–9583. [Google Scholar] [CrossRef]
- Schiffmann, F.; VandeVondele, J.; Hutter, J.; Wirz, R.; Urakawa, A.; Baiker, A. Protonation-Dependent Binding of Ruthenium Bipyridyl Complexes to the Anatase (101) Surface. J. Phys. Chem. C 2010, 114, 8398–8404. [Google Scholar] [CrossRef]
- Shklover, V.; Ovchinnikov, Y.E.; Braginsky, L.S.; Zakeeruddin, S.M.; Grätzel, M. Structure of Organic/Inorganic Interface in Assembled Materials Comprising Molecular Components. Crystal Structure of the Sensitizer Bis [(4,4′-carboxy-2,2¢-bipyridine) (thiocyanato)] ruthenium(II). Chem. Mater. 1998, 10, 2533–2541. [Google Scholar] [CrossRef]
- Fantacci, S.; De Angelis, F.; Selloni, A. Absorption Spectrum and Solvatochromism of the [Ru(4,4-COOH-2,2-bpy)2(NCS)2] Molecular Dye by Time Dependent Density Functional Theory. J. Am. Chem. Soc. 2003, 125, 4381–4387. [Google Scholar] [CrossRef] [PubMed]
- Nazeeruddin, M.K.; De Angelis, F.; Fantacci, S.; Selloni, A.; Viscardi, G.; Liska, P.; Ito, S.; Takeru, B.; Grätzel, M. Combined Experimental and DFT-TDDFT Computational Study of Photoelectrochemical Cell Ruthenium Sensitizers. J. Am. Chem. Soc. 2005, 127, 16835–16847. [Google Scholar] [CrossRef]
- Persson, P.; Lundqvist, M.J. Calculated Structural and Electronic Interactions of the Ruthenium Dye N3 with a Titanium Dioxide Nanocrystal. J. Phys. Chem. B 2005, 109, 11918–11924. [Google Scholar] [CrossRef]
- De Angelis, F.; Fantacci, S.; Selloni, A.; Nazeeruddin, M.K.; Grätzel, M. Time-Dependent Density Functional Theory Investigations on the Excited States of Ru(II)-Dye-Sensitized TiO2 Nanoparticles: The Role of Sensitizer Protonation. J. Am. Chem. Soc. 2007, 129, 14156–114157. [Google Scholar] [CrossRef]
- De Angelis, F.; Fantacci, S.; Selloni, A.; Nazeeruddin, M.K.; Grätzel, M. First-Principles Modeling of the Adsorption Geometry and Electronic Structure of Ru(II) Dyes on Extended TiO2 Substrates for Dye-Sensitized Solar Cell Applications. J. Phys. Chem. C 2010, 114, 6054–6061. [Google Scholar] [CrossRef]
- De Angelis, F.; Fantacci, S.; Moscani, E.; Nazeeruddin, M.K.; Grätzel, M. Absorption Spectra and Excited State Energy Levels of the N719 Dye on TiO2 in Dye-Sensitized Solar Cell Models. J. Phys. Chem. C 2011, 115, 8825–88316. [Google Scholar] [CrossRef]
- Prezhdo, O.V.; Duncan, W.R.; Prezhdo, V.V. Dynamics of the Photoexcited Electron at the Chromophore–Semiconductor Interface. Acc. Chem. Res. 2008, 41, 339–348. [Google Scholar] [CrossRef]
- Persson, P.; Bergström, R.; Lunell, J.S. Quantum Chemical Study of Photoinjection Processes in Dye-Sensitized TiO2 Nanoparticles. J. Phys. Chem. B 2000, 104, 10348–10351. [Google Scholar] [CrossRef]
- Tae, E.L.; Lee, S.H.; Lee, J.K.; Yoo, S.S.; Kang, J.E.; Yoon, K.B. A Strategy to Increase the Efficiency of the Dye-Sensitized TiO2 Solar Cells Operated by Photoexcitation of Dye-to-TiO2 Charge-Transfer Bands. J. Phys. Chem. B 2005, 109, 22513–22522. [Google Scholar] [CrossRef] [PubMed]
- Duncan, W.R.; Stier, W.M.; Prezhdo, O.V. Ab Initio Nonadiabatic Molecular Dynamics of the Ultrafast Electron Injection across the Alizarin-TiO2 Interface. J. Am. Chem. Soc. 2005, 127, 7941–7951. [Google Scholar] [CrossRef]
- Hannappel, T.; Burfeindt, B.; Storck, W.; Willig, F. Measurement of Ultrafast Photoinduced Electron Transfer from Chemically Anchored Ru-Dye Molecules into Empty Electronic States in a Colloidal Anatase TiO2 Film. J. Phys. Chem. B 1997, 101, 6799–6802. [Google Scholar] [CrossRef]
- Walters, A.; Gaal, D.A.; Hupp, J.T. Interfacial Charge Transfer and Colloidal Semiconductor Dye-Sensitization: Mechanism Assessment via Stark Emission Spectroscopy, J. Phys. Chem. B. 2002, 106, 5139–5142. [Google Scholar] [CrossRef]
- Zhao, Y.; Liang, W.-Z. Theoretical investigation of resonance Raman scattering of dye molecules absorbed on semiconductor surfaces, J. Chem. Phys. 2011, 135, 044108. [Google Scholar] [CrossRef]
- Ramakrishna, G.; Ghosh, H.N. Emission from the Charge Transfer State of Xanthene Dye-Sensitized TiO2 Nanoparticles: A New Approach to Determining Back Electron Transfer Rate and Verifying the Marcus Inverted Regime. J. Phys. Chem. B 2001, 105, 7000–7008. [Google Scholar] [CrossRef]
- Asbury, J.B.; Hao, E.; Wang, Y.; Ghosh, H.N.; Lian, T. Ultrafast Electron Transfer Dynamics from Molecular Adsorbates to Semiconductor Nanocrystalline Thin Films. J. Phys. Chem. B 2001, 105, 4545–4557. [Google Scholar] [CrossRef]
- Wang, H.-F.; Chen, L.-Y.; Su, W.-N.; Chung, J.-C.; Hwang, B.-J. Effect of the Compact TiO2 Layer on Charge Transfer between N3 Dyes and TiO2 Investigated by Raman Spectroscopy. J. Phys. Chem. C 2010, 114, 3185–3189. [Google Scholar] [CrossRef]
- Ma, S.; Livingstone, R.; Zhao, B.; Lombardi, J.R. Enhanced Raman Spectroscopy of Nanostructured Semiconductor Phonon Modes. J. Phys. Chem. Lett. 2011, 2, 671–674. [Google Scholar] [CrossRef]
- Nomoto, T.; Fujio, K.; Sasahara, A.; Okajima, H.; Koide, N.; Katayama, H.; Onishi, H. Phonon mode of TiO2 coupled with the electron transfer from N3 dye. J. Chem. Phys. 2013, 138, 224704. [Google Scholar] [PubMed]
- Lundqvist, M.J.; Nilsing, M.; Persson, P.; Lunell, S. DFT Study of Bare and Dye-SensitizedTiO2 Clusters and Nanocrystals. Int. J. Quantum Chem. 2006, 106, 3214–3234. [Google Scholar] [CrossRef]
- Oprea, C.I.; Gîrtu, M.A. Structure and Electronic Properties of TiO2 Nanoclusters and Dye-Nanocluster Systems Appropriate to Model Hybrid Photovoltaic or Photocatalytic Applications. Nanomaterials 2019, 9, 357. [Google Scholar] [CrossRef]
- Berardo, E.; Hu, H.-S.; Shevlin, S.A.; Woodley, S.M.; Kowalski, K.; Zwijnenburg, M.A. Modeling Excited States in TiO2 Nanoparticles: On the Accuracy of a TD-DFT Based Description. J. Chem. Theory Comput. 2014, 10, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Martin, R.L. Natural Transition Orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Le Bahers, T.; Adamo, C.; Ciofoni, I. A Qualitative Index of Spatial Extent in Charge-Transfer Excitations. J. Chem. Theory Comput. 2011, 7, 2498–2506. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.A.; Millam, J. GaussView, Version 6.0; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com (accessed on 5 May 2021).
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Kouki, N.; Trabelsi, S.; Seydou, M.; Maurel, F.; Tangour, B. Internal path investigation of the acting electrons during the photocatalysis of panchromatic ruthenium dyes in dye-sensitized solar cells. Comptes Rendus Chim. 2019, 22, 34–45. [Google Scholar] [CrossRef]
- Qu, Z.-W.; Kroes, G.-J. Theoretical Study of the Electronic Structure and Stability of Titanium Dioxide Clusters (TiO2)n with n =1–9. J. Phys. Chem. B 2006, 110, 8998–9007. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, F.; Fantacci, S.; Selloni, A. A Time-dependent density functional theory study of the absorption spectrum of [Ru(4,40-COOH-2,20-bpy)2(NCS)2]in water solution: Influence of the pH. Chem. Phys. Lett. 2004, 389, 204–208. [Google Scholar] [CrossRef]
- Albright, T.A.; Burdett, J.K.; Whangobo, M.-H. Orbital Interactions in Chemistry, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 416–422. [Google Scholar]
- Sundari, C.D.D.; Martoprawiro, M.A.; Ivansyah, L. A DFT and TDDFT Study of PCM Effect on N3 Dye Absorption in Ethanol Solution. J. Phys. Conf. Ser. 2017, 812, 12068. [Google Scholar] [CrossRef]
- Neugebauer, J.; Reiher, M.; Kind, C.; Hess, B.A. Quantum Chemical Calculation of Vibrational Spectra of Large Molecules—Raman and IR Spectra for Buckminsterfullerene. J. Comp. Chem. 2002, 23, 895. [Google Scholar] [CrossRef]
- Lombardi, J.R.; Birke, R.L. A Unified Approach to Surface-Enhanced Raman Spectroscopy. J. Phys. Chem. C 2008, 112, 5605. [Google Scholar] [CrossRef]
- Albrecht, A.C. On the Theory of Raman Intensities. J. Chem. Phys. 1961, 34, 1476. [Google Scholar] [CrossRef]
- Birke, R.L.; Lombardi, J.R. Relative Contributions of Franck-Condon to Herzberg-Teller terms in charge transfer surface-enhanced Raman scattering spectroscopy. J. Chem. Phys. 2020, 152, 224107. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-K.; Coropceanu, V.; Brédas, J.-L. Assessing the nature of the charge-transfer electronic states in organic solar cells. Nat. Commun. 2018, 9, 5295. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Calc. B3LYP Vac. | Calc. B3LYP EtOH | Literature [58] B3LYP Vac. | Literature [28] BPW91 Vac. C2 | Experiment X-ray [27] |
|---|---|---|---|---|---|
| Ru-NCS | |||||
| Ru-NCS | 2.058 | 2.086 | 2.045 | 2.036 | 2.048 |
| Ru-NCS | 2.058 | 2.086 | 2.045 | 2.036 | 2.046 |
| Ru-N(bpytrans)A | 2.085 | 2.086 | 2.077 | 2.079 | 2.036 |
| Ru-N(bpytrans)B | 2.085 | 2.086 | 2.076 | 2.079 | 2.058 |
| Ru-N(bpycis)A | 2.080 | 2.094 | 2.074 | 2.056 | 2.030 |
| Ru-N(bpycis)B | 2.081 | 2.094 | 2.074 | 2.056 | 2.013 |
| N = C(NCS) | 1.185 | 1.179 | 1.178 | 1.162–1.103 | |
| C-S (NCS) | 1.628 | 1.647 | 1.626 | 1.615–1.685 | |
| <SCN-Ru-NSC | 92.6 | 90.5 | 90.2 | 88.7(5) | |
| <N(bpytrans)-Ru- N(bpycis) | 78.5 | 78.4 | 78.9 | 79.5(5) | |
| <N(bpycis)-Ru- N(bpycis) | 177.9 | 175.2 | 169.5 | 174.5(5) | |
| <SCN-Ru-N(bpyrans) | 172.7 | 173.8 | |||
| <N(bpytrans)-Ru-N(bpytrans) | 93.9 | 92.6 | 95.1 | 90.6(5) 90.6(5) |
| MO | Energy (eV) | Character |
|---|---|---|
| LUMO + 3 | −1.34 | Ti 3dZ2 and π* dcbpy on both dcbpy |
| LUMO + 2 | −1.67 | π* dcbpy not bound to TiO2 and Ru 4dxz |
| LUMO + 1 | −1.92 | π* dcbpy bound to TiO2 and Ru 4dZ2 |
| LUMO | −2.19 | π* dcbpy not bound, Ru 4dZ2, and on bind. π* pyr-COO |
| HOMO | −3.72 | S 3px, N 2px on both NCS, Ru 4dyz |
| HOMO-1 | −3.86 | S 3px, N 2px on both NCS, Ru 4dyz |
| HOMO-2 | −3.94 | S 3py, N 2py on both NCS, Ru 4dyz |
| HOMO-3 | −4.02 | S 3py, N 2py on one NCS, Ru 4dyz |
| HOMO-4 | −5.36 | S 3pz, N 2pz |
| HOMO-5 | −5.41 | C = S π on both NCS, Ru 4dZ2 |
| HOMO-6 | −5.49 | C = S π on both NCS, Ru 4dZ2 |
| HOMO-7 | −5.57 | C = S π on both NCS, Ru 4dZ2 |
| HOMO-8 | −5.73 | O 2p, Ti 3dxy |
| A | B | C | D | This Paper | Assignments |
|---|---|---|---|---|---|
| 2104 | 2104 | 2095 | 2105 | 2104 | (N = C) stretch in NCS |
| 1735 | 1727 | 1726 | 1742 | (C = O) stretch in COOH | |
| 1611 | 1610 | 1605 | 1610 | 1613 | bpy ring stretch in anchor dcbyp |
| 1544 | 1541 | 1542 | 1539 | 1534 | bpy ring stretch and anchor. O = C=O stretch |
| 1469 | 1474 | 1471 | 1471 | ring stretch in both bpy ligands | |
| 1433 | 1430 | 1419 | i.p. C–H wag on both bpy, C–CO stretch | ||
| 1388 | 1376 | 1367 | 1388 | i.p. C–H wag on anchored bpy, sym. stretch of anchored COO | |
| 1303 | 1315 | 1331 | 1338 | i.p. C-H wag on unbound dcbpy, O-H bend on both COOH of dcbpy | |
| 1290 | 1294 | 1288 | i.p. C-H wag on unbound dcbpy, O-H bend on both COOH of this dcbpy | ||
| 1260 | 1256 | 1268 | 1260 | 1261 | unsym. ring stretch on unbound dcbpy, O-H bend on one COOH |
| 1222 | 1252 | i.p. C–H wag on unbound dcbpy | |||
| 1130 | 1155 | 1167 | i.p. C–H wag on unbound dcbpy, O-H bend on both COOH of dcbpy | ||
| 1106 | 1102 | 1106 | 1102 | 1111 | sym. i.p. C-H wag on unbound dcbpy |
| 1021 | 1020 | 1001 | Trigonal ring stretch on both bpy | ||
| 920 | 839 | o. p. C–H wag on anchored bpy, Ti–O–Ti stretch, | |||
| 809 | 827 | C = S stretch, dcbpy deformation, anchored COO bend | |||
| 702 | 750 | 796 | unsym. Ti-O-Ti stretch | ||
| 698 | 699 | 719 | unsym. ring stretch on anchored bpy, O–H bend in COOH H-bond to surface | ||
| 454 | 512 | 503 | Ti–O = C stretch of anchored COO grps, o.p. bpy ring deformation. | ||
| 364 | 397 | 376 | Ru-NCS wag, unbound dcbpy rock | ||
| 318 | 320 | Ru-N(bpy)trans wag |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birke, R.L.; Lombardi, J.R. DFT and TD-DFT Investigation of a Charge Transfer Surface Resonance Raman Model of N3 Dye Bound to a Small TiO2 Nanoparticle. Nanomaterials 2021, 11, 1491. https://doi.org/10.3390/nano11061491
Birke RL, Lombardi JR. DFT and TD-DFT Investigation of a Charge Transfer Surface Resonance Raman Model of N3 Dye Bound to a Small TiO2 Nanoparticle. Nanomaterials. 2021; 11(6):1491. https://doi.org/10.3390/nano11061491
Chicago/Turabian StyleBirke, Ronald L., and John R. Lombardi. 2021. "DFT and TD-DFT Investigation of a Charge Transfer Surface Resonance Raman Model of N3 Dye Bound to a Small TiO2 Nanoparticle" Nanomaterials 11, no. 6: 1491. https://doi.org/10.3390/nano11061491
APA StyleBirke, R. L., & Lombardi, J. R. (2021). DFT and TD-DFT Investigation of a Charge Transfer Surface Resonance Raman Model of N3 Dye Bound to a Small TiO2 Nanoparticle. Nanomaterials, 11(6), 1491. https://doi.org/10.3390/nano11061491