
Prenatal Diagnosis, Course and Outcome of Patients with Truncus Arteriosus Communis
 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
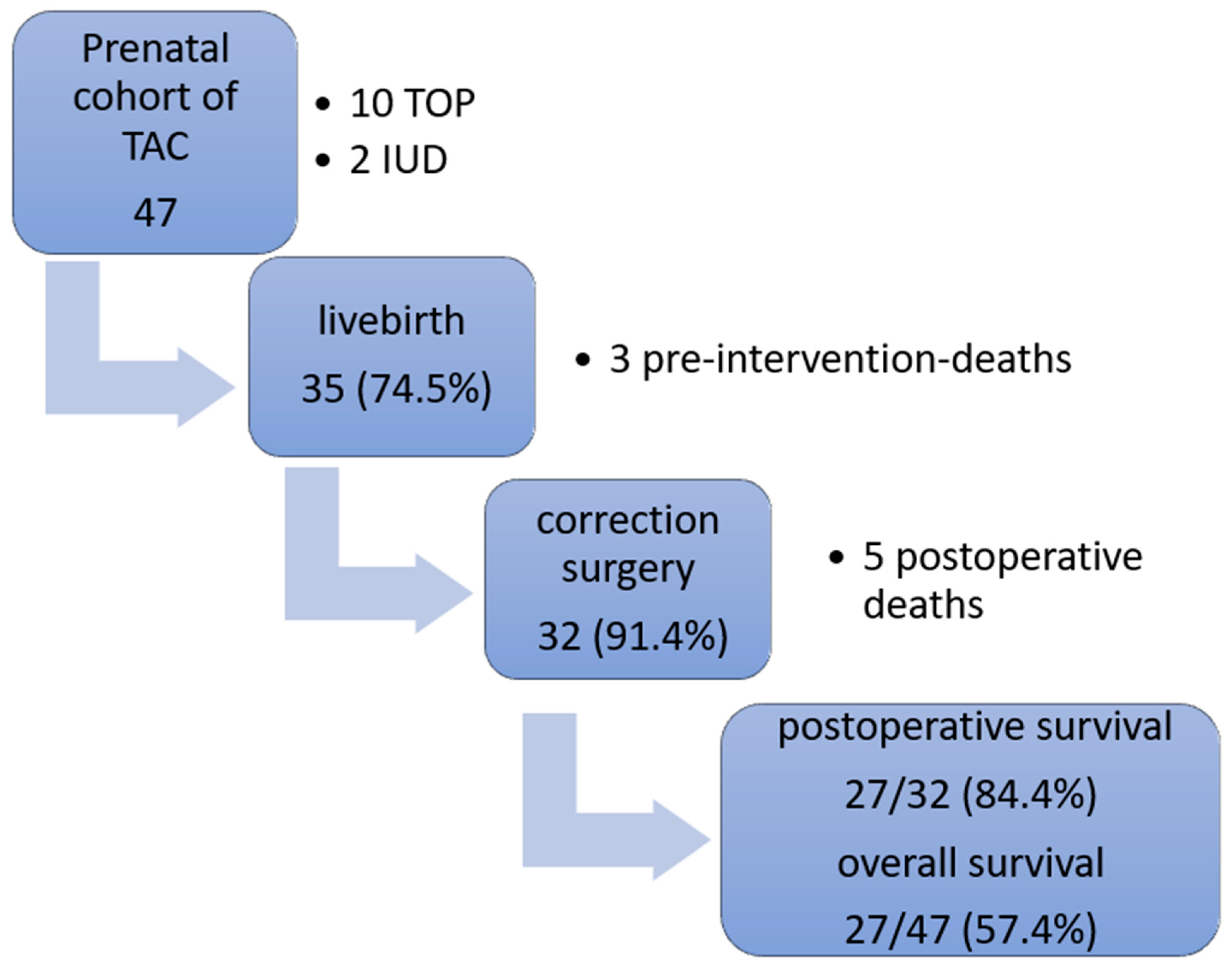
3. Results
3.1. Additional Anomalies
3.2. Outcome
3.3. Parameters of Outcome Prediction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Webb, S.; Qayyum, S.R.; Anderson, R.H.; Lamers, W.H.; Richardson, M.K. Septation and separation within the outflow tract of the developing heart. J. Anat. 2003, 202, 327–342. [Google Scholar] [CrossRef]
- Hoffman, J.I.E.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef]
- Jacobs, M.L. Congenital Heart Surgery Nomenclature and Database Project: Truncus arteriosus. Ann. Thorac. Surg. 2000, 69, 50–55. [Google Scholar] [CrossRef]
- Van Praagh, R.; Van Praagh, S. The anatomy of common aorticopulmonary trunk (truncus arteriosus communis) and its embryologic implications. A study of 57 necropsy cases. Am. J. Cardiol. 1965, 16, 406–425. [Google Scholar] [CrossRef]
- Van Praagh, R. Classification of Truncus Arteriosus Communis. Am. Heart J. 1976, 92, 129–132. [Google Scholar] [CrossRef]
- Patel, A.; Costello, J.; Backer, C.; Pasqualii, S.; Hill, K.; Wallace, A.; Jacob, J.; Jacobs, M. Prevalence of Noncardiac and Genetic Abnormalities in Neonates Undergoing Cardiac Operations: Analysis of The Society of Thoracic Surgeons Congenital Heart Surgery Database. Ann. Thorac. Surg. 2016, 102, 1607–1614. [Google Scholar] [CrossRef]
- Gómez, O.; Soveral, I.; Bennasar, M.; Crispi, F.; Masoller, N.; Marimon, E.; Bartrons, J.; Gratacós, E.; Martinez, J.M. Accuracy of Fetal Echocardiography in the Differential Diagnosis between Truncus Arteriosus and Pulmonary Atresia with Ventricular Septal Defect. Fetal Diagn. Ther. 2016, 39, 90–99. [Google Scholar] [CrossRef]
- Abel, J.S.; Berg, C.; Geipel, A.; Gembruch, U.; Herberg, U.; Breuer, J.; Brockmeier, K.; Gottschalk, I. Prenatal diagnosis, associated findings and postnatal outcome of fetuses with truncus arteriosus communis (TAC). Arch. Gynecol. Obstet. 2021, 304, 1455–1466. [Google Scholar] [CrossRef]
- Swanson, T.; Tierney, E.; Tworetzky, W.; Pigula, F.; McElhinney, D. Truncus Arteriosus: Diagnostic Accuracy, Outcomes, and Impact of Prenatal Diagnosis. Pediatr. Cardiol. 2009, 30, 256–261. [Google Scholar] [CrossRef]
- Volpe, P.; Paladini, D.; Marasini, M.; Buonadonna, A.L.; Russo, M.G.; Caruso, G.; Marzullo, A.; Vassallo, M.; Martinelli, P.; Gentile, M. Common arterial trunk in the fetus: Characteristics, associations, and outcome in a multicentre series of 23 cases. Heart 2003, 89, 1437–1441. [Google Scholar] [CrossRef]
- Gul, A.; Corbacioglu, A.; Bakirci, I.T.; Ceylan, Y. Associated anomalies and outcome of fetal aberrant right subclavian artery. Arch. Gynecol. Obstet. 2012, 285, 27–30. [Google Scholar] [CrossRef]
- Duke, C.; Sharland, G.; Jones, A.; Simpson, J. Echocardiographic features and outcome of truncus arteriosus diagnosed during fetal life. Am. J. Cardiol. 2001, 88, 1379–1384. [Google Scholar] [CrossRef]
- van Nisselrooij, A.E.L.; Herling, L.; Clur, S.A.; Linskens, I.H.; Pajkrt, E.; Rammeloo, L.A.; ten Harkel, A.D.J.; Hazekamp, M.G.; Blom, N.A.; Haak, M.C. The prognosis of common arterial trunk from a fetal perspective: A prenatal cohort study and systematic literature review. Prenat. Diagn. 2021, 41, 754–765. [Google Scholar] [CrossRef]
- Lee, M.-Y.; Won, H.-S.; Lee, B.S.; Kim, E.A.-R.; Kim, Y.-H.; Park, J.-J.; Yun, T.-J. Prenatal diagnosis of common arterial trunk: A single-center’s experience. Fetal Diagn. Ther. 2013, 34, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Mărginean, C.; Gozar, L.; Mărginean, C.O.; Suciu, H.; Togănel, R.; Muntean, I.; Mureşan, M.C. Prenatal diagnosis of the fetal common arterial trunk. A case series. Med. Ultrason. 2018, 20, 100–104. [Google Scholar] [CrossRef]
- Thompson, L.; McElhinney, D.; Reddy, M.; Petrossian, E.; Silverman, N.; Hanley, F. Neonatal repair of truncus arteriosus: Continuing improvement in outcomes. Ann. Thorac. Surg. 2001, 72, 391–395. [Google Scholar] [CrossRef]
- Carvalho, J.S.; Allan, L.D.; Chaoui, R.; Copel, J.A.; DeVore, G.; Hecher, K.; Lee, W.; Munoz, H.; Paladini, D.; Tutschek, B.; et al. ISUOG Practice Guidelines (updated): Sonographic screening examination of the fetal heart. Ultrasound Obs. Gynecol. 2013, 41, 348–359. [Google Scholar] [CrossRef]
- McElhinney, D.B.; Clark, B.J.; Weinberg, P.M.; Kenton, M.L.; McDonald-McGinn, D.; Driscoll, D.A.; Zackai, E.H.; Goldmuntz, E. Association of chromosome 22q11 deletion with isolated anomalies of aortic arch laterality and branching. J. Am. Coll. Cardiol. 2001, 37, 2114–2119. [Google Scholar] [CrossRef]
- Cox, K.; Husain, N.; Jhaveri, S.; Geiger, M.; Berhane, H.; Patel, S. Fetal Echocardiographic Variables Associated with Pre-Surgical Mortality in Truncus Arteriosus: A Pilot Study. Pediatr. Cardiol. 2023, 44, 1397–1405. [Google Scholar] [CrossRef]
- Naimo, P.S.; Bell, D.; Fricke, T.A.; d’Udekem, Y.; Brizard, C.P.; Alphonso, N.; Konstantinov, I.E. Truncus arteriosus repair: A 40-year multicenter perspective. J. Thorac. Cardiovasc. Surg. 2021, 161, 230–240. [Google Scholar] [CrossRef]
- Rodefeld, M.D.; Hanley, F.L. Neonatal truncus arteriosus repair: Surgical techniques and clinical management. Pediatr. Card. Surg. Annu. 2002, 5, 212–217. [Google Scholar] [CrossRef]
- Hanley, F.; Heinemann, M.; Jonas, R.; Mayer, J.J.; Cook, N.; Wessel, D.; Castaneda, A. Repair of truncus arteriosus in the neonate. J. Thorac. Cardiovasc. Surg. 1993, 105, 1047–1056. [Google Scholar] [CrossRef]
- Calder, L.; Van Praagh, R.; Van Praagh, S.; Sears, W.P.; Corwin, R.; Levy, A.; Keith, J.D.; Paul, M.H. Truncus arteriosus communis. Clinical, angiocardiographic, and pathologic findings in 100 patients. Am. Heart J. 1976, 92, 23–38. [Google Scholar] [CrossRef]
- Miyamoto, T.; Sinzobahamvya, N.; Kumpikaite, D.; Asfour, B.; Photiadis, J.; Brecher, A.M.; Urban, A.E. Repair of truncus arteriosus and aortic arch interruption: Outcome analysis. Ann. Thorac. Surg. 2005, 79, 2077–2082. [Google Scholar] [CrossRef]
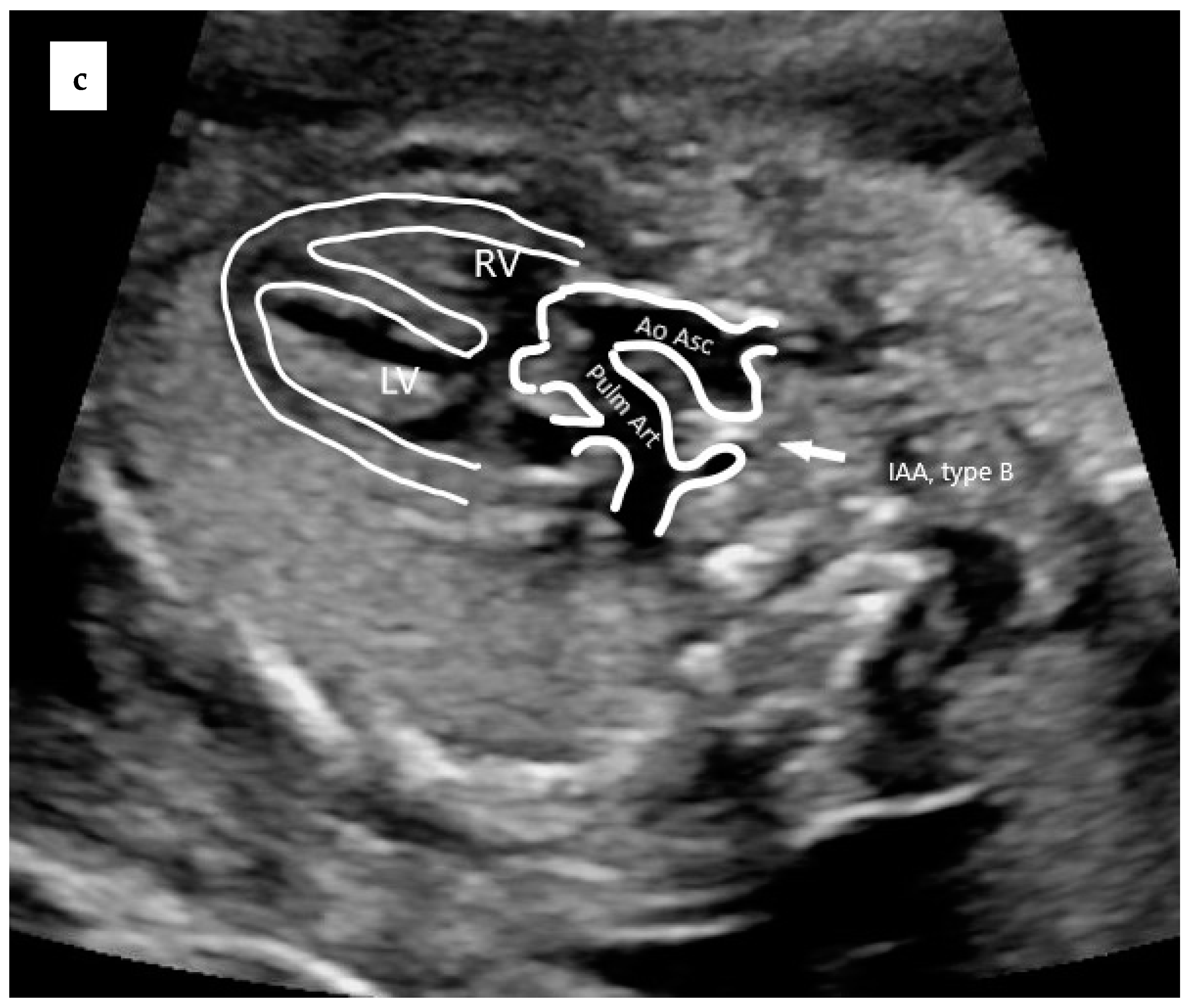
- Konstantinov, I.E.; Karamlou, T.; Blackstone, E.H.; Mosca, R.S.; Lofland, G.K.; Caldarone, C.A.; Williams, W.G.; Mackie, A.S.; McCrindle, B.W. Truncus arteriosus associated with interrupted aortic arch in 50 neonates: A congenital heart surgeons society study. Ann. Thorac. Surg. 2006, 81, 214–222. [Google Scholar] [CrossRef]
- Alfieris, G.M.; Swartz, M.F. The Initial Glimpse at Long-term Outcomes Following the Repair of Truncus Arteriosus. Semin. Thorac. Cardiovasc. Surg. 2016, 28, 512–513. [Google Scholar] [CrossRef]
- Parikh, R.; Eisses, M.; Latham, G.; Joffe, D.; Ross, F. Perioperative and Anesthetic Considerations in Truncus Arteriosus. Semin. Cardiothorac. Vasc. Anesth. 2018, 22, 285–293. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
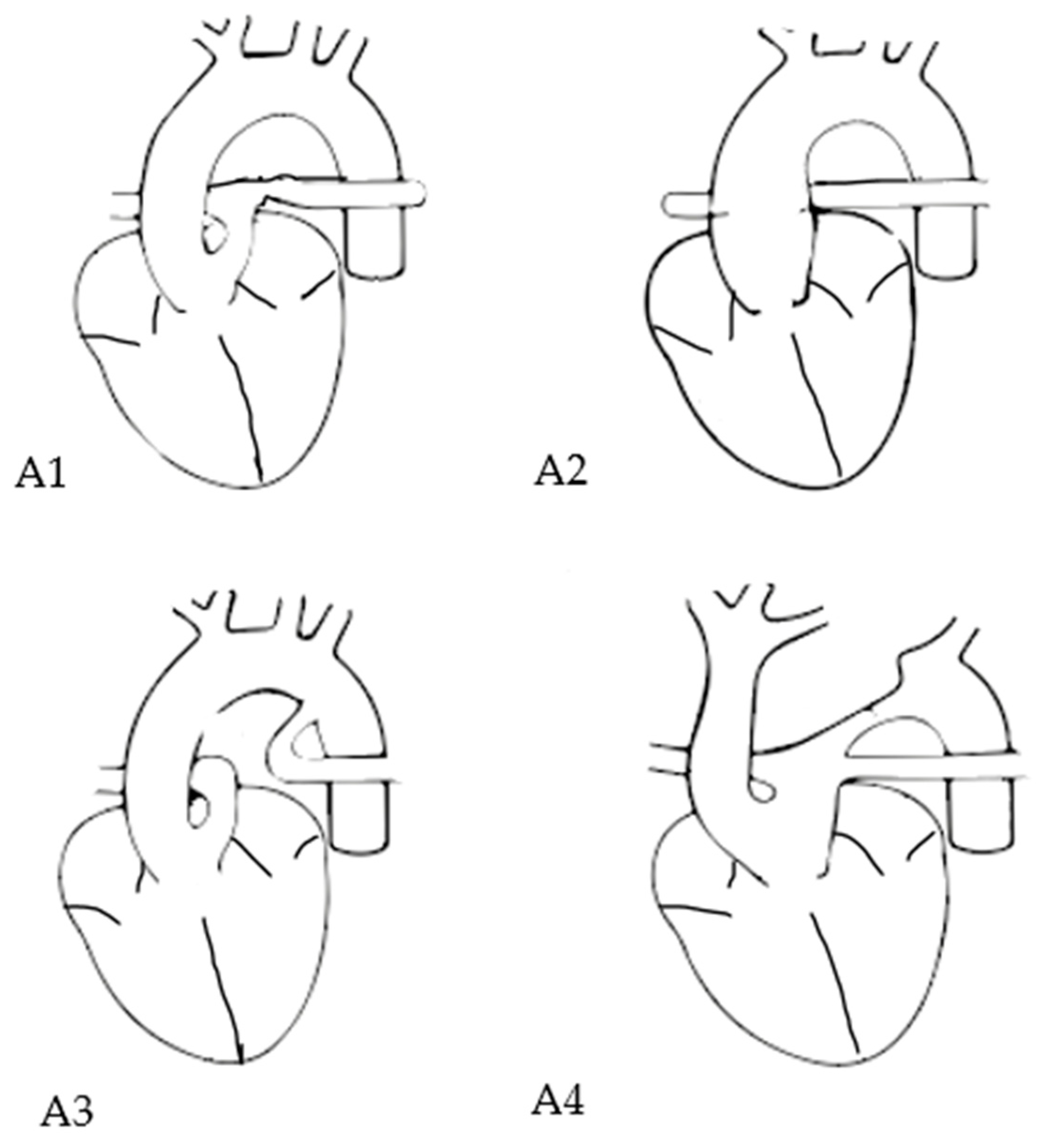
| Each type may include a modifier: “A” (with VSD) or “B” (intact ventricular septum). | |
| subtype A1 | The main pulmonary artery is present and bifurcates into the left and right pulmonary arteries |
| subtype A2 | The right and left branch pulmonary arteries arise from the common trunk without a main pulmonary trunk |
| subtype A3 | One branch of the pulmonary artery (typically the right) arises from the common trunk, and the other arises from a ductus arteriosus or the aorta |
| subtype A4 | Presence of aortic arch hypoplasia, coarctation or interrupted aortic arch and a large ductus arteriosus. |
| prenatal classification into Van Praagh subtype possible | 37/47 (78.7%) |
| type A 1 | 18/37 (48.6%) |
| type A 2 | 13/37 (35.1%) |
| type A 3 | 2/37 (5.4%) |
| type A 4 | 4/37 (10.8%) |
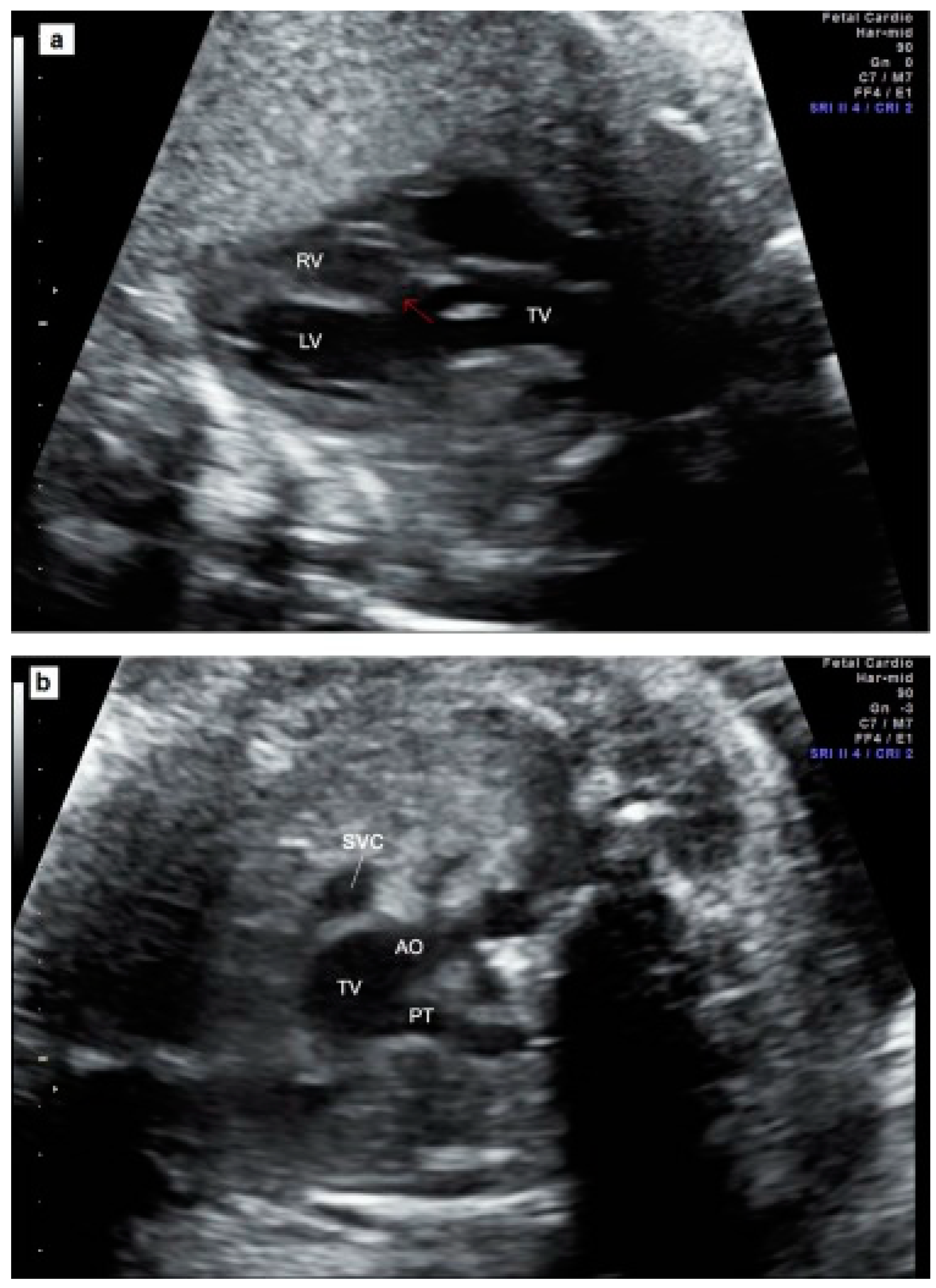
| existing data about common trunk valve details | 33/47 (70.2%) |
| unicuspid valve | 1/33 (3.0%) |
| bicuspid valve | 7/33 (21.2%) |
| tricuspid valve | 13/33 (39.4%) |
| quadricuspid valve | 12/33 (36.4%) |
| valve insufficiency | 14/47 (29.8%) |
| valve stenosis | 8/47 (17.0%) |
| Case | Chromosomal Anomaly | Additional Cardiac Malformation | Additional Extracardiac Malformation | Outcome |
|---|---|---|---|---|
| 1 | 22q11.2 microdeletion (del 22q11.2) | ASD + RAA | thymic aplasia (22q11.2 microdeletion) | alive |
| 2 | del 22q11.2 | ASD | thymic aplasia 34 | alive |
| 3 | - | SCA | anal atresia | alive |
| 4 | n.a. | RAA | esophageal atresia, biliary atresia and renal hypoplasia | died within 7 days postpartum |
| 5 | n.a. | ASD, RAA, high take-off of the RCA | spondylocostal dysostosis | alive |
| 6 | n.a. | - | thymic aplasia | alive |
| 7 | n.a. | - | thymic aplasia | alive |
| 8 | n.a. | - | thymic aplasia | alive |
| 9 | - | ASD+ RAA | microcephaly | alive |
| 10 | del 22q11.2 | ASD | thymic aplasia, clubfeet | alive |
| 11 | - | - | SUA + ductus venosus agenesis | IUD |
| 12 | trisomy 13 | ASD | central nervous system anomaly, spina bifida, hydronephrosis, hexadactyly | TOP |
| 13 | trisomy 13 | - | clubfeet, hexadactyly (trisomy 13) | TOP |
| 14 | trisomy 13 | AVSD | SUA, spina bifida, hexadactyly, microcephaly, cleft lip and palate (trisomy 13) | TOP |
| 15 | n.a. | ASD | thymic aplasia, hexadactyly, renal agenesis | TOP |
| 16 | n.a. | hypoplastic heart | anencephaly | TOP |
| 17 | - | ASD | SUA, hydronephrosis, renal agenesis, hemivertebra, plantar cutaneous appendage | died |
| Case | Chromosomal Anomaly | Additional Cardiac Malformation | Additional Extracardiac Malformation | Outcome |
|---|---|---|---|---|
| 1 | 46, X,dup (y) (q12) {10} | LPSVC + ASD, high take-off of the right coronary artery | - | alive |
| 2 | n.a. | RAA + ASD, high take-off of the right coronary artery (RCA) | - | alive |
| 3 | del 22q11.2 | RAA | - | alive |
| 4 | n.a. | RAA | - | alive |
| 5 | del 22q11.2 | multiple VSD, RAA | - | alive |
| 6 | del 22q11.2 | RAA | - | alive |
| 7 | del 22q11.2 | ASD | - | alive |
| 8 | - | ASD, SCA | - | alive |
| 9 | n.a. | LPSVC, high take-off of the RCA and intramural LCA from RCA | - | died |
| 10 | - | RAA, high take-off of the RCA and intramural RCA | - | alive |
| 11 | - | anomalous origin of LCA | - | died |
| Case | Chromosomal Anomaly | Additional Cardiac Malformation | Additional Extracardiac Malformation | Outcome |
|---|---|---|---|---|
| 1 | del 22q11.2 | - | - | died |
| 2 | T13 | - | - | TOP |
| 3 | T18 | - | - | TOP |
| 4 | del 22q11.2 | - | - | TOP |
| Case | TAC- Type | Chromosomal Anomaly | Additional Cardiac Malformation | Additional Extracardiac Malformation | Outcome |
|---|---|---|---|---|---|
| 1 | 1 | - | - | - | alive |
| 2 | 1 | - | - | - | alive |
| 3 | 1 | - | - | - | alive |
| 4 | 2 | - | - | - | IUD |
| 5 | unknown | - | - | - | TOP |
| 6 | 1 | - | - | - | TOP |
| Case 1 | conduit re-operation |
| Case 2 | conduit re-operation + aortic valvuloplasty |
| Case 3 | conduit re-operation+ repeat atrioventricular valvuloplasty |
| Case 4 | conduit re-operation |
| Case 5 | conduit re-operation + aortic valvuloplasty |
| Case 6 | conduit re-operation + RPA angioplasty |
| Case 7 | conduit re-operation + aortic valvuloplasty |
| Case 8 | conduit re-operation + conduit balloon angioplasty |
| Case 9 | conduit re-operation |
| Case 10 | conduit and LPA balloon angioplasty |
| Case 11 | conduit re-operation + LPA & RPA angioplasty + conduit balloon angioplasty and pulmonary artery (PA) balloon angioplasty |
| Case 12 | conduit re-operation + aortic valvuloplasty + RPA & LPA balloon angioplasty |
| Case 13 | PA angioplasty |
| Case 14 | conduit re-operation |
| Case 15 | conduit re-operation + aortic valvuloplasty |
| Case 16 | conduit and LPA & RPA balloon angioplasty |
| Case 17 | LPA balloon angioplasty |
| Case 18 | conduit re-operation + PA angioplasty |
| Case 19 | PA balloon angioplasty |
| Case 20 | conduit re-operation + PA balloon angioplasty |
| Case 21 | conduit re-operation |
| Case 22 | conduit re-operation, ASD reduction, various thorax openings and closings because of hematoma and ECMO |
| Comparison Parameter | Group 1 (Survivor) | Group 2 (Non-Survivor) | p Value | Cramer’s V |
|---|---|---|---|---|
| TAC subtype | 14 (subtype 1) | 4 (subtype 1) | 1.0 | 0.0645 |
| 9 (subtype 2) | 4 (subtype 2) | 0.8415 | 0.1 | |
| 1 (subtype 3) | 0 (subtype 3) | 0.5598 | 0.0972 | |
| 3 (subtype 4) | 1 (subtype 4) | 0.5376 | 0 | |
| 1 × unknown (IUD patient) | ||||
| number of valve leaflets | 1 unicuspid | 0 unicuspid | 0.4028 | 0.0835 |
| 6 bicuspid | 1 bicuspid | 0.8065 | 0.0522 | |
| 9 tricuspid | 4 tricuspid | 0.2943 | 0.2629 | |
| 11 quadricuspid | 1 quadricuspid | 0.522 | 0.1931 | |
| 4 unknown | ||||
| valve stenosis | 5/27 | 3/10 | 0.7642 | 0.1241 |
| valve insufficiency | 12/27 | 2/10 | 0.3272 | 0.2236 |
| presence of genetic anomaly | 8/27 | 1/10 | 0.4201 | 0.2034 |
| presence of extracardiac anomaly | 9/27 | 3/10 | 0.8415 | 0.0329 |
| presence of FGR | 4/27 | 3/10 | 0.5657 | 0.1724 |
| presence of additional intracardiac anomaly | 21/27 | 7/10 | 1.0 | 0.0805 |
| ASD | 14 | 3 | 0.4166 | 0.1945 |
| RAA | 11 | 2 | 0.431 | 0.1931 |
| cardiomegaly | 1 | 0 | 0.5967 | 0.1013 |
| prematurity | 1/27 | 3/10 | 0.0908 | 0.376 |
| Autor | Our Results | Cox et al., 2023 [19] Pediatr Cardiol. | Van Nisselroij et al., 2021 [13] Prenatal Diagnosis | Abel et al., 2021 [8] Archives of Gynecology and Obstetrics | Mărginean et al., 2018 [15] Medical Ultrasono-graphy | Gomez et al., 2016 [7] Fetal Diagn Ther | Lee et al., 2013 [14] Fetal Diagn Ther | Swanson et al., 2009 [9] Pediatr Cardiol. | Volpe et al., 2003 [10] Heart | Duke et al., 2001 [12] Am J Cardiol. |
|---|---|---|---|---|---|---|---|---|---|---|
| recruitment | 2008–2021 | 2010–2020 | 2002–2016 | 2010–2018 | 2009–2017 | 2006–2015 | 2003–2012 | 1992–2007 | 1993–2002 | 1990–1999 |
| cases | 47 | 23 | 38 | 34 confirmed cases | 17 | 10 | 12 | 43 prenatal cases | 23 | 17 |
| centers | 4 | 2 | 1 | 2 | 1 | 1 | 1 | 1 | 3 | 1 |
| diagnosis (pre-/postnatal) | pre | pre | pre | pre | pre | pre | pre | pre + post | pre | pre |
| TOP | 10/47 = 21.3% | 4/23 = 17.4% | 18/38 = 47.3% | 14/34 = 41.2% | 8/17 = 47.0% | 9/10 = 90.0% of confirmed TAC | 4/12 = 33.3% | 17/43 = 39.5% | 8/23 = 34.8% | 4/17 = 23.5% |
| IUD | 2/47 = 4.3% | 2/19 = 10.5% | 2/38 = 5.3% | 1/34 = 2.9% | - | - | - | 2/43 = 4.7% | 2/23 = 8.7% | - |
| live births | 35/47 = 74.5% | 17/23 = 73.9% | 18/38 = 47.3% | 19/34 = 55.8% | 4/17 = 23.5% | 1/10 = 10.0% | 8/12 = 66% | 24/43 = 55.8% 19/43 = 44.1% with confirmed TAC | 13/23 = 56.5% | 13/17 = 76.5% |
| overall survival | 27/47 = 57.4% | 14/23 = 93.3% | 12/38 = 31.6% | 14/34 = 41.2% | 1/17 = 5.9% | 1/10 = 10.0% | 6/12 = 50.0% | 13/43 = 30.2% 13/38= 34.2% when considered postnatal confirmed TAC) | 8/23 = 34.8% | 5/17 = 29.4% |
| intention-to-treat survival | 27/32 = 84.4% 3 pre surgical deaths in non-isolated cases | 14/17 = 82.4% 2 pre-surgical deaths | 12/16 = 75.0% 2/18 prenatal death (without active treatment) and 2 other pre-surgical deaths with ITT) | 14/19 = 73.7% | 1/3 = 33.3% | 1/1 = 100% | 6/7 = 85.7% 1 presurgical death in non-isolated form) | 13/19 = 68.4% of confirmed TAC cases | 8/10 = 80.0% 3 pre-surgical deaths) | 5/8 = 62.5% 4 presurgical deaths |
| peri-/postoperative mortality | 5/32 = 15.6% | 1/15 = 0.7% | 2/14 = 14.3% | 5/19 = 26.3% | 2/3 = 66.7% | 0 | 1/7 = 14.3% | 4/17 = 23.5% | 2/8 = 25.0% | 3/8 = 37.5% but one palliative operation 2/7 = 28.6% with repair operation |
| associated anomalies (genetic) | 15/47 = 34.1% of prenatal cases 15/37 = 40.5% with invasive diagnostics | 1/23 = 47.8% of prenatal cases 11/23 = 47.8% with invasive diagnostics | 15/38 = 39.5% of prenatal cases 15/38 = 39.5% with invasive diagnostics | 13/34 = 38.2% of prenatal cases 13/25 = 52.0% with invasive diagnostics | 1/17 = 5.9% of prenatal cases 1/5 = 20.0% with invasive diagnostics | 4/10= 40.0% of prenatal cases 4/10= 40.0% with invasive diagnostics | 2/12 = 16.7% of prenatal cases 2/12 = 16.7% with invasive diagnostics | 5/43 = 11.6% of prenatal cases 5/17 = 29.4% with invasive diagnostics | 8/23 = 34.8% of prenatal cases 8/22 = 36.4% with invasive diagnostics | 3/17 = 17.6% of prenatal cases 3/10 = 30.0% with invasive diagnostics for microdel |
| microdel 22q11.2 | 9 /37 = 24.3% with invasive diagnostics | 4/21 = 19.0% with invasive diagnostics | 8/38 = 21.1% with invasive diagnostics | 6/25 = 24.0% with invasive diagnostics | 0/5 = 0% with invasive diagnostics | 1/10 = 10.0% of confirmed cases with invasive diagnostics | 0/9 = 0% with invasive diagnostics for microdel | 5/17 = 29.4% with invasive diagnostics | 6/19 = 31.6% with invasive diagnostics for microdel | 3/10 = 30.0% with invasive diagnostics for microdel |
| extra- cardiac anomalies | 17/47 = 36.2% | 10/23 = 43.5% | 20/38 = 52.6% | 20/34 = 58.8% | 5/17 = 29.4% | 4/10 = 40.0% | 2/12 = 16.7% | 6/19 = 31.6% | 10/23 = 43.5% | 4/17 = 23.5% |
| additional intra-cardiac anomalies | 30/47 = 63.8% excluding coronary anomalies, 32/47 = 68.1% including coronary anomalies 4/47 = 8.5% “major” cardiac anomalies | not reported | 14/38 = 36.8% | 15/34 = 44.1% including minor defects 3/34 = 8.8% “major” cardiac anomalies | not reported | 2/10 = 20.0% | 9/12 = 75.0% | not reported | 8/23 = 34.8% | not reported |
| surgical repair | 32 | 15 | 14 | 13 done, 1 planned | 3 (1 of them only first step) | 1 | 7 (+1 intraoperative death) | 17 (of prenatal cases) | 6 (+2 palliative and +2 awaiting surgery) | 7 (+1 palliative) |
| FU (months) | 52 (median) | not reported (FU until discharge from hospital) | 72 (mean) | 42 (mean) | 4 and 8 months (both died), one alive 2 months | 10 (only one patient) | 38.5 (mean) 41 (median) | not reported | 10 (median) | 41 (median) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolter, A.; Haessig, A.; Kurkevych, A.; Weichert, J.; Bosselmann, S.; Mielke, G.; Bedei, I.A.; Schenk, J.; Widriani, E.; Axt-Fliedner, R. Prenatal Diagnosis, Course and Outcome of Patients with Truncus Arteriosus Communis. J. Clin. Med. 2024, 13, 4465. https://doi.org/10.3390/jcm13154465
Wolter A, Haessig A, Kurkevych A, Weichert J, Bosselmann S, Mielke G, Bedei IA, Schenk J, Widriani E, Axt-Fliedner R. Prenatal Diagnosis, Course and Outcome of Patients with Truncus Arteriosus Communis. Journal of Clinical Medicine. 2024; 13(15):4465. https://doi.org/10.3390/jcm13154465
Chicago/Turabian StyleWolter, Aline, Annika Haessig, Andrii Kurkevych, Jan Weichert, Stephan Bosselmann, Gunther Mielke, Ivonne Alexandra Bedei, Johanna Schenk, Ellydda Widriani, and Roland Axt-Fliedner. 2024. "Prenatal Diagnosis, Course and Outcome of Patients with Truncus Arteriosus Communis" Journal of Clinical Medicine 13, no. 15: 4465. https://doi.org/10.3390/jcm13154465
APA StyleWolter, A., Haessig, A., Kurkevych, A., Weichert, J., Bosselmann, S., Mielke, G., Bedei, I. A., Schenk, J., Widriani, E., & Axt-Fliedner, R. (2024). Prenatal Diagnosis, Course and Outcome of Patients with Truncus Arteriosus Communis. Journal of Clinical Medicine, 13(15), 4465. https://doi.org/10.3390/jcm13154465