Abstract
Prader–Willi syndrome (PWS) is a complex, rare genetic disorder caused by a loss of expression of paternally expressed genes on chromosome 15q11.2-q13. The most common underlying genotypes are paternal deletion (DEL) and maternal uniparental disomy (mUPD). DELs can be subdivided into type 1 (DEL-1) and (smaller) type 2 deletions (DEL-2). Most research has focused on behavioral, cognitive and psychological differences between the different genotypes. However, little is known about physical health problems in relation to genetic subtypes. In this cross-sectional study, we compare physical health problems and other clinical features among adults with PWS caused by DEL (N = 65, 12 DEL-1, 27 DEL-2) and mUPD (N = 65). A meta-analysis, including our own data, showed that BMI was 2.79 kg/m2 higher in adults with a DEL (p = 0.001). There were no significant differences between DEL-1 and DEL-2. Scoliosis was more prevalent among adults with a DEL (80% vs. 58%; p = 0.04). Psychotic episodes were more prevalent among adults with an mUPD (44% vs. 9%; p < 0.001). In conclusion, there were no significant differences in physical health outcomes between the genetic subtypes, apart from scoliosis and BMI. The differences in health problems, therefore, mainly apply to the psychological domain.
1. Introduction
Prader–Willi syndrome (PWS) is a complex, rare genetic disorder with an incidence of 1:16.000–1:20.000 live births [1,2]. It is characterized by intellectual disability, hypotonia, sleep-related disorders and hypothalamic dysfunction [3,4], which in their turn lead to hyperphagia, abnormal pain perception, disturbed thermoregulation and pituitary hormone deficiencies [3,4,5].
PWS is caused by lack of expression of a cluster of paternally expressed genes in the PWS-region on chromosome 15q11.2-q13 [3]. The 15q11.2-q13 region can be subdivided into four distinct sub-regions: a proximal non-imprinted region, the PWS region, the Angelman syndrome (AS) region and a distal non-imprinted region (Figure 1) [3,6]. The genes in the PWS region are maternally imprinted, which means that only the paternally inherited genes are expressed. The PWS region encompasses several genes, including MKRN3, MAGEL2, NDN, NPAP1, SNURF-SNRPN and clusters of small nucleolar RNA genes (snoRNAs) (6). The AS region contains the maternally expressed gene UBE3A [6]. The lack of expression of this maternally expressed gene results in AS.
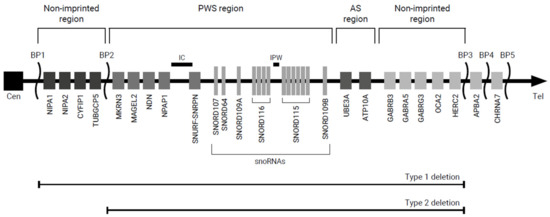
Figure 1.
Overview of the chromosome 15q11.2-q13 region. Abbreviations: AS, Angelman syndrome; BP, breakpoint; Cen, centromere; IC, imprinting center; PWS, Prader–Willi syndrome; Tel, telomere.
The primary genetic mechanisms leading to PWS are paternal deletion (DEL; 65–75%), maternal uniparental disomy (mUPD; 20–30%) and imprinting center defects (ICD; 1–3%) [6]. More rare causes of PWS are balanced translocations (0.1%) and point mutations (<0.1%) [6]. Deletions can be subdivided into type 1 and type 2 deletions (DEL-1, DEL-2). DEL-1 ranges from breakpoint (BP) 1 to BP3, which includes the proximal non-imprinted region, PWS region, AS region and distal non-imprinted region [3,6]. In DEL-2 (BP2 to BP3), the genes in the proximal non-imprinted region are normally expressed [3,6]. Around 8% of the deletions have sizes and/or locations different from DEL-1 and DEL-2 [7].
Several differences between individuals with an mUPD and DEL have been described. Individuals with a DEL tend to have more compulsive behavior [8], better jigsaw skills [9,10], a higher performance intelligence quotient (PIQ) [11] and more hypopigmentation [12,13], whereas individuals with an mUPD are reported to have milder dysmorphic features [12], more psychotic episodes [14], a higher verbal IQ (VIQ) [11,15] and more autism spectrum features [10]. There is no consensus on the differences between individuals with a DEL-1 and DEL-2. Although some studies suggest that individuals with a DEL-1 have more severe compulsive behavior [16,17] and may have delayed speech acquisition [18], other studies did not find any differences between DEL-1 and DEL-2 [19,20]. Although Coupaye et al. [21] showed that adults with a DEL might be more prone to hypothyroidism and might have a higher fat mass (FM) and lean body mass (LBM) than adults with an mUPD, most other studies only focused on differences in behavior, cognitive profile and psychiatric diagnoses and not on somatic differences. Moreover, little is known about the differences in health problems during adulthood.
In this article, we describe the clinical characteristics of 93 adults with PWS according to genotype (mUPD or DEL). Furthermore, we provide a concise overview of the current knowledge on genetic subtype differences in health problems among adults with PWS.
2. Materials and Methods
Approval for this study was waived by the local medical ethics committee of the Erasmus University Medical Center Rotterdam because participants were not subject to procedures or required to follow rules of behavior. In this cross-sectional study, we retrospectively reviewed the medical files of all adults with genetically confirmed PWS who visited the outpatient clinic of the Center for Adults with Complex Rare Genetic Syndromes at the Erasmus University Medical Center, Rotterdam, the Netherlands between January 2015 and June 2021. Adults with multiple genetic syndromes, such as a combination of Klinefelter and Prader–Willi syndrome, were excluded. All patients underwent a systematic health screening as part of regular patient care, including a structured interview, a medical questionnaire, a complete physical examination, biochemical measurements and a review of the medical records, as described previously [22]. The items on physical complaints, symptoms of disease and behavioral challenges were rated on a five-point Likert scale (1 = rarely or never, 2 = not often and/or not severe, 3 = quite often and/or quite severe, 4 = often and/or severe, 5 = very often and/or very severe). An item with a score of 3 or higher was considered present. Patients who had experienced at least one episode of psychotic behavior (diagnosed by a psychologist/psychiatrist) were considered as having a (history of) psychosis. Part of the clinical data of this patient group has been previously published in another context [22,23,24,25,26,27].
2.1. Genetic Diagnosis
Until 2018, the genetic diagnosis was made with a methylation sensitive PCR test, based on allelic methylation differences at the SNRPN locus [28]. In case of hypomethylation, fluorescent in situ hybridization (FISH) was performed with a probe for SNRPN (Prader–Willi/Angelman (SNRPN), Cytocell Ltd., Cambridge, UK) to check for the presence of a DEL as the underlying molecular cause (initially). Later, genomic dosage was assessed using multiplex ligation-dependent probe amplification (MLPA) (MLPA kit ME028, PWS/AS, MRC Holland, Amsterdam, The Netherlands). As of 2018, both methylation status and genomic dosage of the 15q11-q13 region were assessed using methylation specific MLPA.
In the absence of a DEL, the DNA of the index and the parents was tested with a set of highly polymorphic chromosome 15q markers to demonstrate the presence of an mUPD. As of 2018, this marker-test was replaced by SNP array (Infinium Global Screening Array-24 with Multi Disease add-on; Illumina, San Diego, CA, USA). If no mUPD was identified as an underlying molecular cause, we concluded that hypomethylation was caused by an imprinting defect [29].
2.2. Literature Search
Literature was searched in the databases Embase, MEDLINE (via ALL OVID), Web of Science Core Collection, Cochrane Central Register of Controlled Trials and Google Scholar. The full search strategy is included in the Supplementary Materials (Table S1). The literature search, completed in June 2020, only included original research articles comparing the phenotype of PWS adults with different genotypes (mUPD or DEL). We excluded reviews, conference abstracts, data from unpublished research, non-human research, articles with less than 10 subjects and non-English articles. We also excluded articles with incomplete phenotypic or genotypic data, articles describing pediatric patients (mean/median age below 18 years), articles reporting only prenatal characteristics and articles reporting genetic defects outside the chromosome 15q11.2-q13 region, including chromosome 15 duplication syndrome, trisomy 15 syndrome, AS and/or the occurrence of multiple syndromes within one individual. There were no restrictions on period. All articles were independently screened by three reviewers, first by title and abstract (CW and JTG), then by full text (CW, JTG and AR). Data were independently extracted by two reviewers (CW and AR). In case of disagreement, a fourth reviewer (LdG) was consulted.
2.3. Data Analysis
Data were analyzed with IBM Statistical Package for the Social Sciences Statistics version 25.0 and R version 3.6.0. Descriptive statistics are presented as median and interquartile range (IQR) for continuous data and as count and percentage (%) for categorical data. The chi-square test (for categorical variables) and Wilcoxon rank sum test (for continuous variables) were used to detect any differences in clinical features between the genotypes (mUPD vs. DEL and DEL-1 vs. DEL-2). Tests were only performed when at least 10 people had the genetic subtype. Logistic regression analyses were performed to detect differences in clinical features between mUPD and DEL, independent of (past or current) growth hormone (GH) treatment. Additionally, a stratified analysis for differences in health problems between the genetic subtypes was performed to visualize the impact of GH treatment. Further, a meta-analysis for BMI was performed. The result is shown as the pooled mean difference with a 95% confidence interval (CI). Data were pooled with inverse variance weighting on the number of subjects, mean and standard deviation of the studies. A fixed effect model was used when heterogeneity (assessed with I2 statistic) was <50%. p-values < 0.05 were considered statistically significant.
3. Results
A total of 111 patients (51 male and 60 female) had a genetically confirmed diagnosis of PWS. The median age was 28.5 (IQR: 21.1–38.7) years and median body mass index (BMI) was 29.1 (IQR: 26.3–37.3) kg/m2. Of the patients, 28 had an mUPD, 65 a DEL (12 DEL-1, 27 DEL-2) and 3 an ICD. In 15 patients, genetic material from the parents was not available, making it impossible to distinguish between an mUPD and ICD. These patients were excluded from further analyses. The patient characteristics are described in Table 1.

Table 1.
Characteristics of adults with a genetically confirmed diagnosis of Prader–Willi syndrome.
3.1. Paternal Deletion Compared to Maternal Uniparental Disomy
Gender, age and BMI did not differ significantly between adults with a DEL or mUPD (Table 1). The proportion of patients with current or past GH treatment was similar for the DEL and mUPD group. Differences in health problems, physical complaints, symptoms of disease and behavioral challenges according to genotype are described in Table 2.
Table 2.
Differences in health problems, physical complaints, symptoms of disease and behavioral challenges between adults with a deletion and maternal uniparental disomy.
Scoliosis was more often present in adults with a DEL (N = 51 out of 64, 80%) than in adults with an mUPD (N = 15 out of 26, 58%, p = 0.03), even after correcting for GH treatment (p = 0.04). Osteopenia was present in 47% of adults with a DEL (N = 21 out of 45) and 33% of adults with an mUPD (N = 7 out of 21), but this did not reach statistical significance (p > 0.05). Likewise, hypertension was twice as prevalent among adults with an mUPD (N = 7 out of 26, 27%) than among adults with a DEL (N = 8 out of 63, 13%), but this was not significant either. There were no differences in diagnoses of osteoporosis, epilepsy, hypercholesterolemia, diabetes mellitus type 2 (DM2) or hypothyroidism between the genotypes.
Physical complaints and behavioral challenges reported by patients and/or caregivers did not differ significantly between adults with an mUPD and DEL. In both groups, complaints of constipation and daytime sleepiness were frequent. Although not significant, abdominal pain and fatigue were present approximately three times more often in adults with a DEL. Abdominal pain was present in 17% of patients with a DEL (N = 9 out of 53) versus 5% of adults with an mUPD (N = 1 out of 22). Fatigue was present in 26% of patients with a DEL (N = 13 out of 50) versus 10% of adults with an mUPD (N = 2 out of 21). Complaints of the feet, urinary incontinence, cold intolerance and swollen legs, on the other hand, were more often reported among adults with an mUPD compared to DEL (foot complaints: 39% vs. 18%; urinary incontinence: 14% vs. 4%; cold intolerance: 30% vs. 19%; swollen legs: 30% vs. 17%). However, these differences were not statistically significant (p > 0.05 for all).
Twelve (44%) out of 27 adults with an mUPD had a psychotic episode compared to 6 (9%) out of 65 adults with a DEL (p < 0.001). Temper outbursts, food-seeking behavior and skin picking also seemed slightly more prevalent in adults with an mUPD than in adults with a DEL (temper outbursts: 43% vs. 30%; food-seeking behavior: 37% vs. 28%; skin picking: 56% vs. 42%). However, these differences were not significant. Psychotropic drugs were significantly more often used by adults with an mUPD than by adults with a DEL (p = 0.002; Table 1). Thirteen (50%, data missing in two adults) adults with an mUPD used psychotropic drugs, of which nine had a (history of) psychosis. Of adults with a DEL, only twelve (18%) adults used psychotropic drugs, of which four had a (history of) psychosis.
The stratified analysis on GH treatment shows that osteoporosis/osteopenia, hypercholesterolemia, DM2, obesity and hypertension rates are higher in adults who never received GH treatment (Table 3).
Table 3.
Differences in health problems between adults with a deletion and maternal uniparental disomy according to growth hormone treatment.
Among adults who were treated with GH, there were no differences in health problems between DEL and mUPD (apart from the previously mentioned scoliosis and psychotic episodes).
3.2. Deletion Type 1 Compared to Deletion Type 2
Gender, age and BMI did not differ significantly between adults with a DEL-1 or DEL-2 (Table 1). The proportion of patients with current or past GH treatment was similar for the DEL-1 and DEL-2 group. Differences in health problems, physical complaints, symptoms of disease and behavioral challenges between adults with a DEL-1 and DEL-2 are described in Table 4.
Table 4.
Differences in health problems, physical complaints, symptoms of disease and behavioral challenges between adults with a deletion type 1 and deletion type 2.
None of the health problems differed significantly between adults with a DEL-1 and DEL-2. Scoliosis was present in more than 80% of the adults with a DEL-1 or DEL-2. Osteopenia seemed slightly more prevalent among adults with a DEL-2 (N = 13 out of 23; 57%) compared to DEL-1 (N = 2 out of 9, 22%, p = 0.18). There were no differences in diagnoses of osteoporosis, psychotic episodes, epilepsy, hypercholesterolemia, DM2, hypertension and hypothyroidism between the genetic subgroups.
Physical complaints and behavioral challenges reported by the patients and/or caregivers did not differ significantly between adults with DEL-1 and DEL-2. At least 30% of the adults with a DEL-1 or DEL-2 experienced daytime sleepiness and/or temper outbursts. Although not significant, constipation and skin picking seemed more prevalent among adults with a DEL-2 compared to DEL-1 (constipation: 40% vs. 25%; skin picking: 39% vs. 20%). On the other hand, fatigue, cold intolerance, foot complaints, swollen legs and food-seeking behavior were more often reported in adults with a DEL-1 compared to DEL-2 (fatigue: 40% vs. 19%; cold intolerance: 30% vs. 14%; foot complaints: 20% vs. 5%; swollen legs: 33% vs. 10%; food-seeking behavior: 33% vs. 22%), but these differences did not reach statistical significance. There was also no significant difference in the use of psychotropic drugs between the two groups.
The stratified analysis on GH treatment shows that osteoporosis/osteopenia and obesity rates seem higher in adults who never received GH treatment (Table 5). However, there do not seem to be any differences in health problems between DEL and mUPD within the strata.
Table 5.
Differences in health problems between adults with a deletion type 1 and deletion type 2 according to growth hormone treatment.
3.3. Literature Review
The original search yielded 7762 articles. After deduplication, the titles and abstracts of 4174 articles were screened. Of 444 articles, we assessed the full text article, after which we included 30 articles in the literature review (Figure 2). The study characteristics and findings of the included articles are described in Table 6 and Table 7 and visualized in Figure 3. Most studies focused on behavior (10 studies), cognition (9 studies), psychopathology (5 studies) and/or hyperphagia (6 studies). Three studies investigated general somatic health problems [21,30,31].
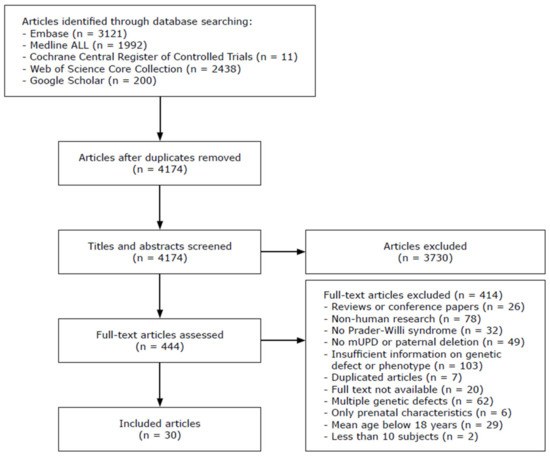
Figure 2.
Flowchart of included studies. Abbreviations: mUPD, maternal uniparental disomy.
Table 6.
Study characteristics of previous literature.
Table 7.
Differences between maternal uniparental disomy and paternal deletion according to previous literature.
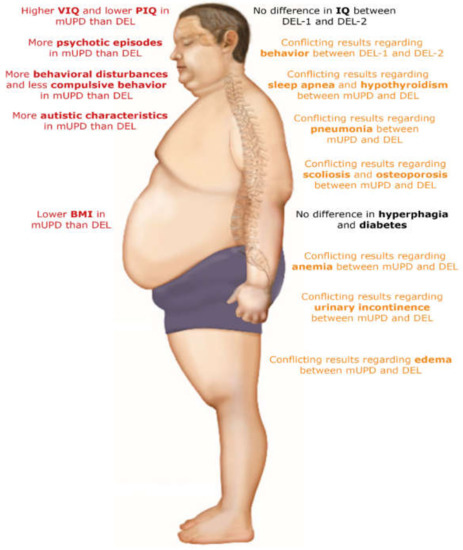
Figure 3.
Overview of differences between adults with a maternal uniparental disomy and paternal deletion. Abbreviations: BMI, body mass index; DEL, paternal deletion; DEL-1, paternal deletion type 1; DEL-2, paternal deletion type 2; IQ, intelligence quotient; mUPD, maternal uniparental disomy; PIQ, performance intelligence quotient; VIQ, verbal intelligence quotient. The figure was adapted from Zorgatlas Groeihormoon, Esculaap Media bv, 2017 [53]. Red represents differences between groups, orange represents conflicting results regarding differences in health problems between groups and black represents no differences between groups.
There was no difference in hyperphagia between adults with an mUPD and DEL [19,21,34,37,43], nor between DEL-1 and DEL-2 [19,36]. There was no difference in the presence of scoliosis and diabetes, but hypothyroidism, sleep apnea, osteoporosis and edema might be more prevalent in patients with a DEL than in mUPD patients [21,30,31]. Moreover, FM and LBM were higher in adults with a DEL [21]. Pneumonia, anemia and urinary incontinence might be more prevalent among patients with an mUPD compared to DEL [31].
Most studies found non-significantly higher BMI in adults with a DEL compared to mUPD. Therefore, we conducted a meta-analysis on BMI including our own data. We found that BMI was 2.79 kg/m2 higher in adults with a DEL compared to adults with an mUPD (95% CI: 1.10–4.47 kg/m2, p = 0.001; Figure 4).
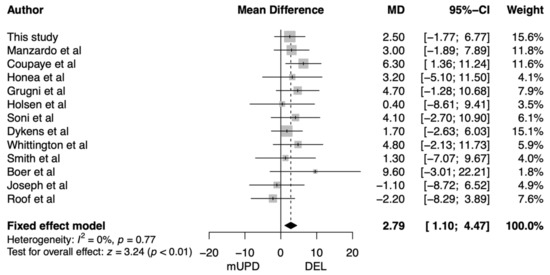
Figure 4.
Forest plot of the difference in body mass index between adults with a paternal deletion and maternal uniparental disomy. Abbreviations: CI, confidence interval; DEL, paternal deletion; MD, mean difference; mUPD, maternal uniparental disomy.
Regarding behavioral challenges, there was no consensus on the differences between the genetic subtypes. However, it seems that adults with an mUPD might have more challenging behavior in general, but less compulsive behavior than adults with a DEL [8,17,19,34,36,38]. Although one study found more severe compulsive behavior in DEL-2 [8] and another study found less adaptive behavior in DEL-1 [17], other studies reported no differences in overall behavior between DEL-1 and DEL-2 [19,36]. However, some small differences may exist [17]. For cognition, most studies reported lower VIQ [11,15,16] and higher PIQ [11,42,48] in adults with a DEL than in adults with an mUPD. While total IQ was similar in most individual studies [15,16,42,45,46,48], a meta-analysis by Yang et al. [11] showed lower total IQ for DEL compared to mUPD. No differences in VIQ, PIQ and total IQ between DEL-1 and DEL-2 were reported [16,47]. Psychosis and bipolar disorder were more common in adults with an mUPD than DEL [11,39,44,51]. Additionally, adults with a DEL-1 might have more psychiatric diagnoses than adults with a DEL-2 [33].
4. Discussion
We studied genetic subtype differences in relation to health problems among 93 adults with PWS due to an mUPD or DEL. Supported by our literature overview, we conclude that differences between the genetic subtypes seem mostly present in the psychological domain. Psychotic episodes, especially, were more frequent in adults with an mUPD than in in adults with DEL. Apart from an increased frequency of scoliosis in adults with a DEL, there were no significant differences in physical health outcomes between the genetic subtypes.
In our cohort, 68% of the adults with a genetically confirmed diagnoses of PWS had a DEL, 29% an mUPD and 3% an ICD, which is similar to the previously reported distribution of genetic subtypes in PWS [6]. The size of the deletion was determined in 45 adults (69%), of whom 27% had a DEL-1, 60% a DEL-2 and 13% had an atypical microdeletion. The proportion of adults with a DEL-1 was slightly lower than in most previously reported studies [8,16,17,33,38,47]. A possible explanation for this discrepancy is that when analyzing the size of the deletion, sometimes, only the proximal BP is determined instead of both the proximal and distal BPs [7]. This can lead to inaccurate classification of deletion types and, therefore, a higher proportion of DEL-1 in previous literature. In our study, we found that 13% of the adults had an atypical microdeletion, which is higher than previously reported [6] and supports the hypothesis that some deletions classified as DEL-1 might actually be atypical microdeletions.
4.1. Physical Health Problems
We found that scoliosis was more prevalent in adults with a DEL compared to mUPD, even after correcting for previous GH treatment, which, in the past, has been suggested to increase the chance of scoliosis [54]. This finding was not reported in previous literature. Part of our population lived in specialized PWS group homes (20%), of which 75% had a DEL and 25% an mUPD. As caregivers in these homes have extensive PWS experience, this might have had an influence on the prevalence of scoliosis among adults with a DEL due to increased awareness and detection. However, a difference in the presence of scoliosis due to the different genetic mechanisms underlying PWS may also exist.
BMI was slightly higher in adults with a DEL compared to mUPD. Coupaye et al. [21] suggested that this difference might be due to increased adipocyte expansion in individuals with a DEL. Another explanation might be a difference in lifestyle between the two groups. More research is needed to unravel the difference in BMI between the genetic subtypes.
Smith et al. [49] reported that mortality was higher in individuals with an mUPD than in individuals with DELs. In our cohort, four patients had died, of which two had an mUPD (7%) and two had a DEL (3%; p = 0.38). The cause of death was unknown in one patient and of cardiovascular origin in the other patients.
4.2. Psychopathology, Cognition and Behavior
Similar to previous literature, we found that psychotic episodes were more prevalent in adults with an mUPD [11,51]. In our cohort, psychotic episodes were present in 44% of the adults with an mUPD. This is in agreement with previous studies, where psychotic features were present in approximately 30–60% of the adults with an mUPD [33,39,44]. A likely explanation for the increased prevalence of psychosis in adults with mUPD is overexpression of the (in the brain) paternally imprinted gene UBE3A. The expression of UBE3A is higher in persons with an mUPD than in DEL and higher than in persons without PWS [55]. UBE3A plays a role in the development and function of the nervous system and may indirectly alter the GABA and glutamatergic system [56,57]. Overexpression of UBE3A can disrupt these systems, which disturbs the excitatory–inhibitory balance in the brain, possibly leading to psychosis [57]. The hypothesis that overexpression of UBE3A can result in psychosis is supported by studies about patients with a 15q11-q13 duplication [58,59]. Isles et al. found that maternal duplications of this region are associated with psychosis [58]. Additionally, Noor et al. reported symptoms of psychosis in patients with maternal microduplications of UBE3A only [59]. Notably, both studies also reported symptoms of autism, which suggests that overexpression of UBE3A might also contribute to the increased prevalence of autistic characteristics in patients with an mUPD [60,61]. Other genes and snoRNAs that might play a role in the onset of psychosis are GABRG3, NDN, CYFIP1 and SNORD115 [57]. The most likely hypothesis is that those genes and snoRNAs are involved in a two-hit model, as previously suggested [44]. This model is based on the idea that several genetic and neural alterations are needed for the onset of psychosis [57]. Loss of expression of GABRG3, NDN, CYFIP1 and the snoRNA cluster SNORD115 may act as a first hit [57]. Overexpression of UBE3A may be the second hit, ultimately leading to psychosis in adults with an mUPD.
Several studies reported better verbal capacities and lower PIQ in adults with an mUPD compared to DEL [11,15,16,17,42,48]. We hypothesize that overexpression of UBE3A may contribute to the decreased performance capacities in adults with an mUPD. This is supported by a study of Baker et al. who showed that higher UBE3A mRNA levels were associated with lower PIQ scores [62]. However, the maternally imprinted genes may also play a role. When it comes to VIQ, not only the imprinted genes but also the non-imprinted genes in the chromosome 15q11.2-q13 region might be involved. Individuals with a DEL are hemizygous for these genes, whereas individuals with an mUPD are suspected to have normal expression. This might influence the verbal capacities in adults with a DEL. Indeed, studies on 15q11.2 BP1-BP2 microdeletions, in which the non-imprinted genes NIPA1, NIPA2, CYFIP1 and TUBGCP5 are deleted, showed that verbal IQ was impaired in these patients [63]. This also suggests that there might be a difference in VIQ between individuals with a DEL-1 and DEL-2, which was confirmed by Milner et al., who found higher VIQ scores in children with a DEL-2 [20]. However, other studies, among adults, did not find a difference in VIQ between the DEL subtypes [16,17].
As a post-hoc analysis, we studied the occurrence of mutism or other speech/language abnormalities in relation to the genetic subtype in our cohort. In total, five adults (four mUPD, one atypical microdeletions) had impaired speech, of which three had complete mutism. What is remarkable is that all adults with complete mutism had an mUPD. The patient with an atypical microdeletion encompassing the genes between TUBGCP5 and SNRPN had a remarkable speech, speaking fluently but placing the words in the sentences in a random order. Language disorders in individuals with PWS were previously described by Dimitropoulos et al. [64]. They showed that core language ability was impaired in PWS and that both expressive and receptive language abilities were lower than VIQ. In addition, they reported that individuals with an mUPD had expressive language abilities exceeding receptive language abilities, whereas expressive and receptive language abilities did not differ within individuals with a DEL. More research is needed to elucidate the effect of the genetic subtypes on language abilities.
The studies on behavior were inconclusive. Most studies agree that compulsive behavior is more prevalent in adults with a DEL than in mUPD [8,17], but the difference between DEL-1 and DEL-2 is less clear. One study found more severe compulsive behavior in adults with a DEL-2 compared to DEL-1 [8], whereas other studies suggest that compulsive behavior might be more prevalent in DEL-1 [16,17]. Although Butler et al. [17] showed that skin picking is not associated with compulsive behavior, several parents and caregivers of PWS adults who attended our clinic do report a relation between the two (personal communication), with an increase in both skin picking and other compulsive behavior during stressful episodes. Some studies showed that skin picking, like compulsive behavior, also seems more prevalent in adults with a DEL compared to mUPD [17,19]. In our cohort, we found no significant difference in skin picking between the genetic subtypes, but small differences may exist. We hypothesize that the genes in the non-imprinted regions may contribute to the compulsive behavior in adults with a DEL. Since compulsive behavior may be more severe in adults with a DEL-1, the genes in the proximal non-imprinted region (NIPA1, NIPA2, CYFIP1, TUBGCP5) might play a significant role. In addition, Bittel et al. [65] showed that NIPA2 and CYFIP1 mRNA levels especially were associated with several sub-items of compulsive behavior. Moreover, variants in TUBGCP5 are known to be related to obsessive-compulsive disorders [66]. Contrary to compulsive behavior and skin picking, other challenging behavioral phenotypes seem more prevalent in adults with an mUPD [34,36,38]. When looking at the DEL subtypes, one study reported less adaptive behavior in DEL-1 than in DEL-2 and more maladaptive behavior in DEL-2 than in mUPD [17], whereas another study reported no differences in overall behavioral profile between DEL-1 and DEL-2 [36]. In our cohort, we also found that temper outbursts tended to be more prevalent among adults with an mUPD (43%) compared to DEL (30%). However, this was not significant. We found no difference in temper outbursts between DEL-1 and DEL-2. We hypothesize that the reported challenging behavior in patients with an mUPD might be partly due to overexpression of UBE3A, but also other genes within the chromosome 15q11.2-q13 region are probably involved.
4.3. Growth Hormone Treatment
Nowadays, most people with PWS receive GH treatment during childhood, which is often continued into adulthood. Previous research showed that the positive effects of GH treatment are seen in both adults with a DEL and mUPD [67]. Coupaye et al. [21] found that the effects of previous GH treatment on reduction in body fat and adipocyte volume might be greater in people with a DEL. As expected, we noticed in our cohort that adults who had never received GH treatment had higher rates of several health problems, such as obesity. In our stratified analysis, we also show that among adults who had ever been treated with GH, scoliosis was more prevalent for DEL and psychosis was more prevalent for mUPD. No differences were detected among adults who had never received GH treatment. While this might be due to differences in effect of GH treatment between the genetic subtypes, it is most likely caused by a lack of power due to small sample sizes within the strata. However, more research is needed to elucidate the possible genetic subtype specific effects of GH treatment on health problems in adults with PWS.
4.4. Strengths and Limitations
Like every study, our study has strengths and limitations. A strength of our study is the relatively large cohort of adults with a confirmed genetic diagnosis of PWS. Furthermore, we used a systematic health screening to evaluate the health of all adults with PWS attending our clinic, which improved the quality of our data. Moreover, we provide a thorough overview of the current knowledge, including a meta-analysis, and relate the previous findings to our own results. However, some limitations remain. Fifteen patients were excluded from analyses because it was impossible to distinguish between mUPD and ICD due to unavailable parental genetic material. Since this group generally resembles an older population with less involved parents, this might have influenced the results. However, the age difference between adults with an mUPD and DEL is small, which suggests that the groups are still comparable. Furthermore, since additional tests for osteoporosis were only performed on indication, we had, relatively, many missing values for osteoporosis/osteopenia. We had no data on the chronicity of the psychosis, i.e., whether it was a single episode of psychotic behavior or a recurrent form of psychosis. Furthermore, the questionnaire on physical complaints, symptoms of disease and behavioral challenges was not filled out by all participants, which reduced our statistical power. Therefore, more (small) differences may exist between the genetic subtypes, and our results should be interpreted with caution.
4.5. Future Research
To further explore the differences in health problems between adults with an mUPD, DEL-1 and DEL-2, more research is needed. Further research should not only focus on the differences in behavior and cognition, but also on physical aspects, such as BMI and scoliosis. Moreover, differences in language abilities between the genetic subtypes should be studied. In addition, more research on atypical microdeletions is needed to further unravel the influence of the individual genes in the chromosome 15q11.2-q13 region on specific components of the PWS phenotype.
5. Conclusions
Differences in health problems between adults with an mUPD and DEL seem mostly present in the psychological domain. Especially, psychotic episodes were more frequent in adults with an mUPD. Apart from scoliosis and the meta-analysis on BMI, there were no significant differences in physical health outcomes between the genetic subtypes. More research is needed to further explore the differences in health problems between adults with different genetic subtypes. In addition, more data on individuals with an atypical microdeletion are needed to unravel the influence of the genes in the chromosome 15q11.2-q13 region on the PWS phenotype.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jcm11144033/s1, Table S1: Literature search. Table S2: Differences between maternal uniparental disomy and paternal deletion according to previous literature.
Author Contributions
Conceptualization, A.G.W.R. and L.C.G.D.G.; methodology, A.G.W.R. and L.C.G.D.G.; formal analysis, A.G.W.R.; investigation, A.G.W.R.; resources, L.C.G.D.G.; data curation, A.G.W.R., C.M.W., J.M.T.G., K.P. and L.C.G.D.G.; writing—original draft preparation, A.G.W.R.; writing—review and editing, A.G.W.R., C.M.W., J.M.T.G., K.P., D.H.V.A., K.D., L.J.C.M.V.Z., H.T.B., J.L.R., A.J.V.d.L. and L.C.G.D.G.; visualization, A.G.W.R.; supervision, L.C.G.D.G. and A.J.V.d.L.; project administration, L.C.G.D.G.; funding acquisition, L.C.G.D.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki. Ethical review and approval were waived for this study by the Institutional Review Board (or Ethics Committee) of the Erasmus University Medical Center (protocol code MEC-2018-1389, 24 September 2018), as this is not applicable for retrospective data collection from patient records.
Informed Consent Statement
Informed consent was obtained from subjects involved in the study or anonymized patient data was collected.
Data Availability Statement
Some or all datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
Acknowledgments
The authors wish to thank Maarten F.M. Engel from the Erasmus MC Medical Library for developing the search strategy.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lionti, T.; Reid, S.M.; White, S.M.; Rowell, M.M. A population-based profile of 160 Australians with Prader-Willi syndrome: Trends in diagnosis, birth prevalence and birth characteristics. Am. J. Med. Genet. Part A 2015, 167, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Bar, C.; Diene, G.; Molinas, C.; Bieth, E.; Casper, C.; Tauber, M. Early diagnosis and care is achieved but should be improved in infants with Prader-Willi syndrome. Orphanet. J. Rare Dis. 2017, 12, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [Green Version]
- Swaab, D.F. Prader-Willi syndrome and the hypothalamus. Acta Paediatr. Suppl. 1997, 423, 50–54. [Google Scholar] [CrossRef] [Green Version]
- Holm, V.A.; Cassidy, S.B.; Butler, M.G.; Hanchett, J.M.; Greenswag, L.R.; Whitman, B.Y.; Greenberg, F. Prader-Willi syndrome: Consensus diagnostic criteria. Pediatrics 1993, 91, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Cheon, C.K. Genetics of Prader-Willi syndrome and Prader-Will-Like syndrome. Ann. Pediatr. Endocrinol. Metab. 2016, 21, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Miller, J.L.; Kuipers, P.J.; German, J.R.; Beaudet, A.L.; Sahoo, T.; Driscoll, D.J. Unique and atypical deletions in Prader-Willi syndrome reveal distinct phenotypes. Eur. J. Hum. Genet. 2012, 20, 283–290. [Google Scholar] [CrossRef]
- Novell-Alsina, R.; Esteba-Castillo, S.; Caixas, A.; Gabau, E.; Gimenez-Palop, O.; Pujol, J.; Deus, J.; Torrents-Rodas, D. Compulsions in Prader-Willi syndrome: Occurrence and severity as a function of genetic subtype. Actas Esp. Psiquiatr. 2019, 47, 79–87. [Google Scholar]
- Dykens, E.M. Are jigsaw puzzle skills ‘spared’ in persons with Prader-Willi syndrome? J. Child Psychol. Psychiatry 2002, 43, 343–352. [Google Scholar] [CrossRef]
- Veltman, M.W.; Thompson, R.J.; Roberts, S.E.; Thomas, N.S.; Whittington, J.; Bolton, P.F. Prader-Willi syndrome—A study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. Eur. Child Adolesc. Psychiatry 2004, 13, 42–50. [Google Scholar] [CrossRef]
- Yang, L.; Zhan, G.D.; Ding, J.J.; Wang, H.J.; Ma, D.; Huang, G.Y.; Zhou, W.H. Psychiatric illness and intellectual disability in the Prader-Willi syndrome with different molecular defects—A meta analysis. PLoS ONE 2013, 8, e72640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassidy, S.B.; Forsythe, M.; Heeger, S.; Nicholls, R.D.; Schork, N.; Benn, P.; Schwartz, S. Comparison of phenotype between patients with Prader-Willi syndrome due to deletion 15q and uniparental disomy 15. Am. J. Med. Genet. 1997, 68, 433–440. [Google Scholar] [CrossRef]
- Torrado, M.; Araoz, V.; Baialardo, E.; Abraldes, K.; Mazza, C.; Krochik, G.; Ozuna, B.; Leske, V.; Caino, S.; Fano, V.; et al. Clinical-etiologic correlation in children with Prader-Willi syndrome (PWS): An interdisciplinary study. Am. J. Med. Genet. Part A 2007, 143, 460–468. [Google Scholar] [CrossRef]
- Soni, S.; Whittington, J.; Holland, A.J.; Webb, T.; Maina, E.; Boer, H.; Clarke, D. The course and outcome of psychiatric illness in people with Prader-Willi syndrome: Implications for management and treatment. J. Intellect. Disabil. Res. 2007, 51 Pt 1, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Roof, E.; Stone, W.; MacLean, W.; Feurer, I.D.; Thompson, T.; Butler, M.G. Intellectual characteristics of Prader-Willi syndrome: Comparison of genetic subtypes. J. Intellect. Disabil. Res. 2000, 44 Pt 1, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Zarcone, J.; Napolitano, D.; Peterson, C.; Breidbord, J.; Ferraioli, S.; Caruso-Anderson, M.; Holsen, L.; Butler, M.G.; Thompson, T. The relationship between compulsive behaviour and academic achievement across the three genetic subtypes of Prader-Willi syndrome. J. Intellect. Disabil. Res. 2007, 51 Pt 6, 478–487. [Google Scholar] [CrossRef]
- Butler, M.G.; Bittel, D.C.; Kibiryeva, N.; Talebizadeh, Z.; Thompson, T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics 2004, 113 Pt 1, 565–573. [Google Scholar] [CrossRef]
- Varela, M.C.; Kok, F.; Setian, N.; Kim, C.A.; Koiffmann, C.P. Impact of molecular mechanisms, including deletion size, on Prader-Willi syndrome phenotype: Study of 75 patients. Clin. Genet. 2005, 67, 47–52. [Google Scholar] [CrossRef]
- Dykens, E.M.; Roof, E. Behavior in Prader-Willi syndrome: Relationship to genetic subtypes and age. J. Child Psychol. Psychiatry 2008, 49, 1001–1008. [Google Scholar] [CrossRef]
- Milner, K.M.; Craig, E.E.; Thompson, R.J.; Veltman, M.W.; Thomas, N.S.; Roberts, S.; Bellamy, M.; Curran, S.R.; Sporikou, C.M.; Bolton, P.F. Prader-Willi syndrome: Intellectual abilities and behavioural features by genetic subtype. J. Child Psychol. Psychiatry 2005, 46, 1089–1096. [Google Scholar] [CrossRef]
- Coupaye, M.; Tauber, M.; Cuisset, L.; Laurier, V.; Bieth, E.; Lacorte, J.M.; Oppert, J.M.; Clement, K.; Poitou, C. Effect of Genotype and Previous GH Treatment on Adiposity in Adults with Prader-Willi Syndrome. J. Clin. Endocrinol. Metab. 2016, 101, 4895–4903. [Google Scholar] [CrossRef] [PubMed]
- Pellikaan, K.; Rosenberg, A.G.W.; Kattentidt-Mouravieva, A.A.; Kersseboom, R.; Bos-Roubos, A.G.; Veen-Roelofs, J.M.C.; van Wieringen, N.; Hoekstra, F.M.E.; van den Berg, S.A.A.; van der Lely, A.J.; et al. Missed Diagnoses and Health Problems in Adults with Prader-Willi Syndrome: Recommendations for Screening and Treatment. J. Clin. Endocrinol. Metab. 2020, 105, e4671–e4687. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.G.W.; Pellikaan, K.; Poitou, C.; Goldstone, A.P.; Hoybye, C.; Markovic, T.; Grugni, G.; Crino, A.; Caixas, A.; Coupaye, M.; et al. Central Adrenal Insufficiency Is Rare in Adults with Prader-Willi Syndrome. J. Clin. Endocrinol. Metab. 2020, 105, e2563–e2571. [Google Scholar] [CrossRef] [PubMed]
- Pellikaan, K.; Ben Brahim, Y.; Rosenberg, A.G.W.; Davidse, K.; Poitou, C.; Coupaye, M.; Goldstone, A.P.; Høybye, C.; Markovic, T.P.; Grugni, G.; et al. Hypogonadism in Adult Males with Prader-Willi Syndrome-Clinical Recommendations Based on a Dutch Cohort Study, Review of the Literature and an International Expert Panel Discussion. J. Clin. Med. 2021, 10, 4361. [Google Scholar] [CrossRef]
- Pellikaan, K.; Ben Brahim, Y.; Rosenberg, A.G.W.; Davidse, K.; Poitou, C.; Coupaye, M.; Goldstone, A.P.; Høybye, C.; Markovic, T.P.; Grugni, G.; et al. Hypogonadism in Women with Prader-Willi Syndrome-Clinical Recommendations Based on a Dutch Cohort Study, Review of the Literature and an International Expert Panel Discussion. J. Clin. Med. 2021, 10, 5781. [Google Scholar] [CrossRef]
- Pellikaan, K.; Snijders, F.; Rosenberg, A.G.W.; Davidse, K.; van den Berg, S.A.A.; Visser, W.E.; van der Lely, A.J.; de Graaff, L.C.G. Thyroid Function in Adults with Prader-Willi Syndrome, a Cohort Study and Literature Review. J. Clin. Med. 2021, 10, 3804. [Google Scholar] [CrossRef]
- Pellikaan, K.; Rosenberg, A.G.W.; Davidse, K.; Kattentidt-Mouravieva, A.A.; Kersseboom, R.; Bos-Roubos, A.G.; Grootjen, L.N.; Damen, L.; van den Berg, S.A.A.; van der Lely, A.J.; et al. Effects of Childhood Multidisciplinary Care and Growth Hormone Treatment on Health Problems in Adults with Prader-Willi Syndrome. J. Clin. Med. 2021, 10, 3250. [Google Scholar] [CrossRef]
- Zeschnigk, M.; Lich, C.; Buiting, K.; Doerfler, W.; Horsthemke, B. A single-tube PCR test for the diagnosis of Angelman and Prader-Willi syndrome based on allelic methylation differences at the SNRPN locus. Eur. J. Hum. Genet. 1997, 5, 94–98. [Google Scholar] [CrossRef]
- Beygo, J.; Buiting, K.; Ramsden, S.C.; Ellis, R.; Clayton-Smith, J.; Kanber, D. Update of the EMQN/ACGS best practice guidelines for molecular analysis of Prader-Willi and Angelman syndromes. Eur. J. Hum. Genet. 2019, 27, 1326–1340. [Google Scholar] [CrossRef] [Green Version]
- Laurier, V.; Lapeyrade, A.; Copet, P.; Demeer, G.; Silvie, M.; Bieth, E.; Coupaye, M.; Poitou, C.; Lorenzini, F.; Labrousse, F.; et al. Medical, psychological and social features in a large cohort of adults with Prader-Willi syndrome: Experience from a dedicated centre in France. J. Intellect. Disabil. Res. 2015, 59, 411–421. [Google Scholar] [CrossRef]
- Sinnema, M.; Maaskant, M.A.; van Schrojenstein Lantman-de Valk, H.M.; van Nieuwpoort, I.C.; Drent, M.L.; Curfs, L.M.; Schrander-Stumpel, C.T. Physical health problems in adults with Prader-Willi syndrome. Am. J. Med. Genet. Part A 2011, 155, 2112–2124. [Google Scholar] [CrossRef] [PubMed]
- Debladis, J.; Valette, M.; Strenilkov, K.; Mantoulan, C.; Thuilleaux, D.; Laurier, V.; Molinas, C.; Barone, P.; Tauber, M. Face processing and exploration of social signals in Prader-Willi syndrome: A genetic signature. Orphanet. J. Rare Dis. 2019, 14, 262. [Google Scholar] [CrossRef] [PubMed]
- Manzardo, A.M.; Weisensel, N.; Ayala, S.; Hossain, W.; Butler, M.G. Prader-Willi syndrome genetic subtypes and clinical neuropsychiatric diagnoses in residential care adults. Clin. Genet. 2018, 93, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Ihara, H.; Ogata, H.; Sayama, M.; Gito, M.; Murakami, N.; Ayabe, T.; Oto, Y.; Takahashi, A.; Nagai, T. Autistic, Aberrant, and Food-Related Behaviors in Adolescents and Young Adults with Prader-Willi Syndrome: The Effects of Age and Genotype. Behav. Neurol. 2017, 2017, 4615451. [Google Scholar] [CrossRef] [PubMed]
- Key, A.P.; Jones, D.; Dykens, E.M. Social and emotional processing in Prader-Willi syndrome: Genetic subtype differences. J. Neurodev. Disord. 2013, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Jauregi, J.; Laurier, V.; Copet, P.; Tauber, M.; Thuilleaux, D. Behavioral profile of adults with Prader-Willi syndrome: Correlations with individual and environmental variables. J. Neurodev. Disord. 2013, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Honea, R.A.; Holsen, L.M.; Lepping, R.J.; Perea, R.; Butler, M.G.; Brooks, W.M.; Savage, C.R. The neuroanatomy of genetic subtype differences in Prader-Willi syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Sinnema, M.; Einfeld, S.L.; Schrander-Stumpel, C.T.; Maaskant, M.A.; Boer, H.; Curfs, L.M. Behavioral phenotype in adults with Prader-Willi syndrome. Res. Dev. Disabil. 2011, 32, 604–612. [Google Scholar] [CrossRef] [Green Version]
- Sinnema, M.; Boer, H.; Collin, P.; Maaskant, M.A.; van Roozendaal, K.E.; Schrander-Stumpel, C.T.; Curfs, L.M. Psychiatric illness in a cohort of adults with Prader-Willi syndrome. Res. Dev. Disabil. 2011, 32, 1729–1735. [Google Scholar] [CrossRef]
- Grugni, G.; Giardino, D.; Crinò, A.; Malvestiti, F.; Ballarati, L.; Di Giorgio, G.; Marzullo, P. Growth hormone secretion among adult patients with Prader-Willi syndrome due to different genetic subtypes. J. Endocrinol. Investig. 2011, 34, 493–497. [Google Scholar]
- Maas, A.P.; Sinnema, M.; Didden, R.; Maaskant, M.A.; Smits, M.G.; Schrander-Stumpel, C.T.; Curfs, L.M. Sleep disturbances and behavioural problems in adults with Prader-Willi syndrome. J. Intellect. Disabil. Res. 2010, 54, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Copet, P.; Jauregi, J.; Laurier, V.; Ehlinger, V.; Arnaud, C.; Cobo, A.M.; Molinas, C.; Tauber, M.; Thuilleaux, D. Cognitive profile in a large French cohort of adults with Prader-Willi syndrome: Differences between genotypes. J. Intellect. Disabil. Res. 2010, 54, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Holsen, L.M.; Zarcone, J.R.; Chambers, R.; Butler, M.G.; Bittel, D.C.; Brooks, W.M.; Thompson, T.I.; Savage, C.R. Genetic subtype differences in neural circuitry of food motivation in Prader-Willi syndrome. Int. J. Obes. 2009, 33, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, S.; Whittington, J.; Holland, A.J.; Webb, T.; Maina, E.N.; Boer, H.; Clarke, D. The phenomenology and diagnosis of psychiatric illness in people with Prader-Willi syndrome. Psychol. Med. 2008, 38, 1505–1514. [Google Scholar] [CrossRef]
- Descheemaeker, M.J.; Govers, V.; Vermeulen, P.; Fryns, J.P. Pervasive developmental disorders in Prader-Willi syndrome: The Leuven experience in 59 subjects and controls. Am. J. Med. Genet. Part A 2006, 140, 1136–1142. [Google Scholar] [CrossRef]
- Stauder, J.E.; Boer, H.; Gerits, R.H.; Tummers, A.; Whittington, J.; Curfs, L.M. Differences in behavioural phenotype between parental deletion and maternal uniparental disomy in Prader-Willi syndrome: An ERP study. Clin. Neurophysiol. 2005, 116, 1464–1470. [Google Scholar] [CrossRef]
- Hartley, S.L.; Maclean, W.E., Jr.; Butler, M.G.; Zarcone, J.; Thompson, T. Maladaptive behaviors and risk factors among the genetic subtypes of Prader-Willi syndrome. Am. J. Med. Genet. Part A 2005, 136, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Whittington, J.; Holland, A.; Webb, T.; Butler, J.; Clarke, D.; Boer, H. Cognitive abilities and genotype in a population-based sample of people with Prader-Willi syndrome. J. Intellect. Disabil. Res. 2004, 48 Pt 2, 172–187. [Google Scholar] [CrossRef]
- Smith, A.; Loughnan, G.; Steinbeck, K. Death in adults with Prader-Willi syndrome may be correlated with maternal uniparental disomy. J. Med. Genet. 2003, 40, e63. [Google Scholar] [CrossRef] [Green Version]
- Webb, T.; Whittington, J.; Clarke, D.; Boer, H.; Butler, J.; Holland, A. A study of the influence of different genotypes on the physical and behavioral phenotypes of children and adults ascertained clinically as having PWS. Clin. Genet. 2002, 62, 273–281. [Google Scholar] [CrossRef]
- Boer, H.; Holland, A.; Whittington, J.; Butler, J.; Webb, T.; Clarke, D. Psychotic illness in people with Prader Willi syndrome due to chromosome 15 maternal uniparental disomy. Lancet 2002, 359, 135–136. [Google Scholar] [CrossRef]
- Joseph, B.; Egli, M.; Sutcliffe, J.S.; Thompson, T. Possible dosage effect of maternally expressed genes on visual recognition memory in Prader-Willi syndrome. Am. J. Med. Genet. 2001, 105, 71–75. [Google Scholar] [CrossRef]
- De Graaff, L.C.G.; van der Kaay, D.C.M. Zorgatlas Groeihormoon; Esculaap Media: Beuningen, The Netherlands, 2017. [Google Scholar]
- Diene, G.; de Gauzy, J.S.; Tauber, M. Is scoliosis an issue for giving growth hormone to children with Prader-Willi syndrome? Arch. Dis. Child. 2008, 93, 1004–1006. [Google Scholar] [CrossRef]
- Bittel, D.C.; Kibiryeva, N.; Talebizadeh, Z.; Butler, M.G. Microarray analysis of gene/transcript expression in Prader-Willi syndrome: Deletion versus UPD. J. Med. Genet. 2003, 40, 568–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, S.J.; Segal, D.J.; LaSalle, J.M. UBE3A: An E3 Ubiquitin Ligase with Genome-Wide Impact in Neurodevelopmental Disease. Front. Mol. Neurosci. 2019, 11, 476. [Google Scholar] [CrossRef] [Green Version]
- Aman, L.C.S.; Manning, K.E.; Whittington, J.E.; Holland, A.J. Mechanistic insights into the genetics of affective psychosis from Prader-Willi syndrome. Lancet Psychiatry 2018, 5, 370–378. [Google Scholar] [CrossRef]
- Isles, A.R.; Ingason, A.; Lowther, C.; Walters, J.; Gawlick, M.; Stöber, G.; Rees, E.; Martin, J.; Little, R.B.; Potter, H.; et al. Parental Origin of Interstitial Duplications at 15q11.2-q13.3 in Schizophrenia and Neurodevelopmental Disorders. PLoS Genet. 2016, 12, e1005993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noor, A.; Dupuis, L.; Mittal, K.; Lionel, A.C.; Marshall, C.R.; Scherer, S.W.; Stockley, T.; Vincent, J.B.; Mendoza-Londono, R.; Stavropoulos, D.J. 15q11.2 Duplication Encompassing Only the UBE3A Gene Is Associated with Developmental Delay and Neuropsychiatric Phenotypes. Hum. Mutat. 2015, 36, 689–693. [Google Scholar] [CrossRef]
- Bennett, J.A.; Germani, T.; Haqq, A.M.; Zwaigenbaum, L. Autism spectrum disorder in Prader–Willi syndrome: A systematic review. Am. J. Med. Genet. Part A 2015, 167, 2936–2944. [Google Scholar] [CrossRef]
- Hogart, A.; Wu, D.; LaSalle, J.M.; Schanen, N.C. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol. Dis. 2010, 38, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Baker, E.K.; Butler, M.G.; Hartin, S.N.; Ling, L.; Bui, M.; Francis, D.; Rogers, C.; Field, M.J.; Slee, J.; Gamage, D.; et al. Relationships between UBE3A and SNORD116 expression and features of autism in chromosome 15 imprinting disorders. Transl. Psychiatry 2020, 10, 362. [Google Scholar] [CrossRef]
- Cox, D.M.; Butler, M.G. The 15q11.2 BP1-BP2 microdeletion syndrome: A review. Int. J. Mol. Sci. 2015, 16, 4068–4082. [Google Scholar] [CrossRef] [PubMed]
- Dimitropoulos, A.; Ferranti, A.; Lemler, M. Expressive and receptive language in Prader–Willi syndrome: Report on genetic subtype differences. J. Commun. Disord. 2013, 46, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Bittel, D.C.; Kibiryeva, N.; Butler, M.G. Expression of 4 genes between chromosome 15 breakpoints 1 and 2 and behavioral outcomes in Prader-Willi syndrome. Pediatrics 2006, 118, e1276–e1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wolf, V.; Brison, N.; Devriendt, K.; Peeters, H. Genetic counseling for susceptibility loci and neurodevelopmental disorders: The del15q11.2 as an example. Am. J. Med. Genet. Part A 2013, 161, 2846–2854. [Google Scholar] [CrossRef]
- Damen, L.; Donze, S.H.; Kuppens, R.J.; Bakker, N.E.; de Graaff, L.C.G.; van der Velden, J.; Hokken-Koelega, A.C.S. Three years of growth hormone treatment in young adults with Prader-Willi syndrome: Sustained positive effects on body composition. Orphanet. J. Rare Dis. 2020, 15, 163. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).