
Recent Advances in PROTAC-Based Antiviral Strategies




Abstract
1. Introduction
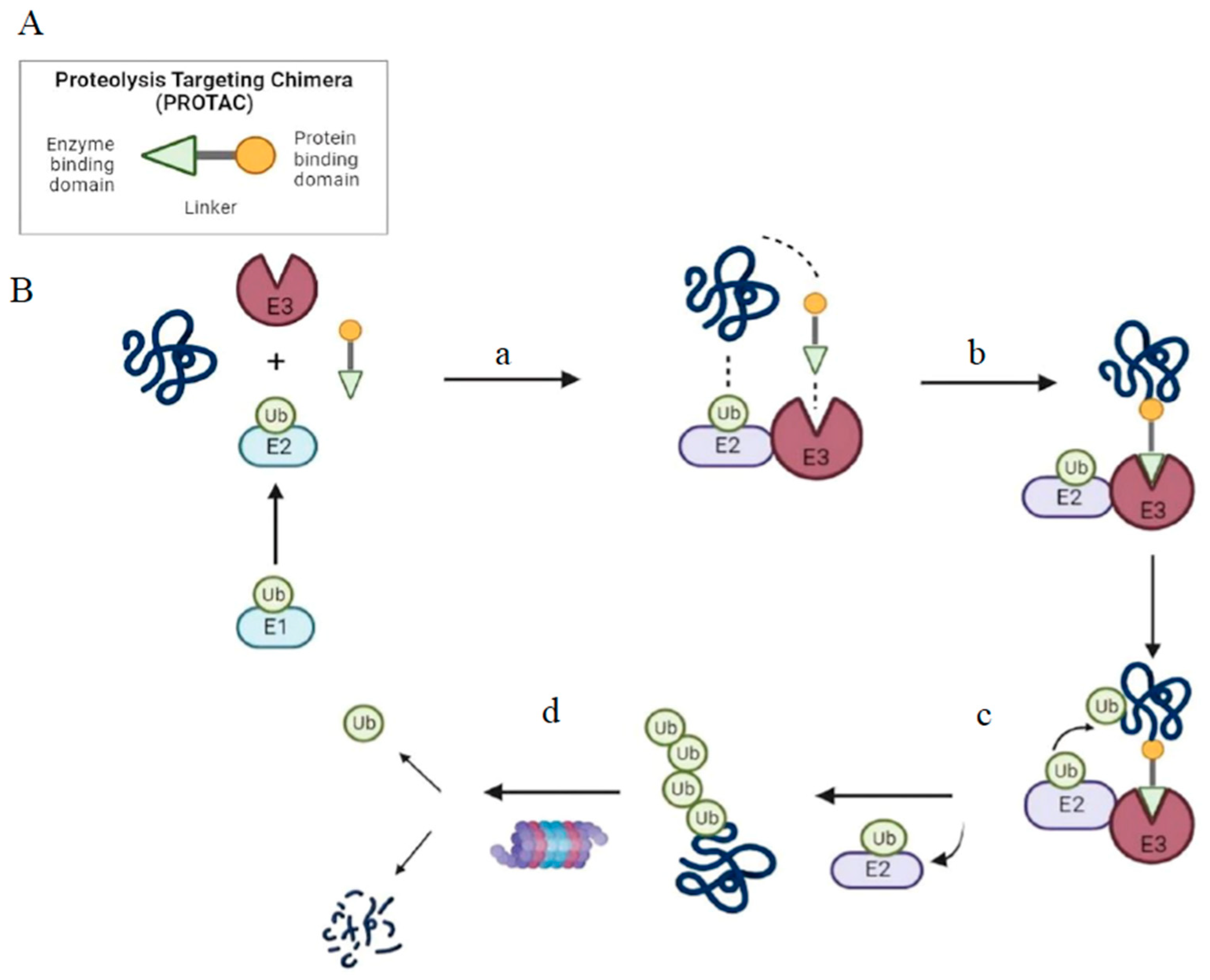
2. PROTAC as a Tool for Targeted Protein Degradation
3. Diversity of PROTACs
4. PROTAC-Based Antiviral Strategies
4.1. PROTAC Virus, a Novel Vaccine Strategy
4.2. Proteases-Targeting PROTAC
4.3. Surface Receptor-Targeting PROTACs
4.4. Host Protein-Targeting PROTAC
4.5. Miscellaneous
5. Advantages of PROTAC-Based Antiviral Strategies
6. Limitations
7. Future Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Radoux, C.J.; Hercules, A.; Ochoa, D.; Dunham, I.; Zalmas, L.-P.; Hessler, G.; Ruf, S.; Shanmugasundaram, V.; Hann, M.M.; et al. The PROTACtable genome. Nat. Rev. Drug Discov. 2021, 20, 789–797. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Khan, S.; Huo, Z.; Lv, D.; Zhang, X.; Liu, X.; Yuan, Y.; Hromas, R.; Xu, M.; Zheng, G.; et al. Proteolysis targeting chimeras (PROTACs) are emerging therapeutics for hematologic malignancies. J. Hematol. Oncol. 2020, 13, 103. [Google Scholar] [CrossRef]
- Kargbo, R.B. PROTAC Degradation of IRAK4 for the Treatment of Neurodegenerative and Cardiovascular Diseases. ACS Med. Chem. Lett. 2019, 10, 1251–1252. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Verma, R.J.; Ransick, A.; Stein, B.; Crews, C.M.; Deshaies, R. Development of Protacs to Target Cancer-promoting Proteins for Ubiquitination and Degradation. Mol. Cell. Proteom. 2003, 2, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Khoo, R.; Peh, K.M.; Teo, J.; Chang, S.C.; Ng, S.; Beilhartz, G.L.; Melnyk, R.A.; Johannes, C.W.; Brown, C.J.; et al. bioPROTACs as versatile modulators of intracellular therapeutic targets including proliferating cell nuclear antigen (PCNA). Proc. Natl. Acad. Sci. USA 2020, 117, 5791–5800. [Google Scholar] [CrossRef]
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2021, 21, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, P.P.; Hamann, L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. [Google Scholar] [CrossRef]
- Lupas, A.; Koster, A.J.; Baumeister, W. Structural Features of 26S and 20S Proteasomes. Enzym. Protein 1993, 47, 252–273. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The Ubiquitin System for Protein Degradation. Annu. Rev. Biochem. 1992, 61, 761–807. [Google Scholar] [CrossRef] [PubMed]
- Kierszenbaum, A.L. The 26S proteasome: Ubiquitin-mediated proteolysis in the tunnel. Mol. Reprod. Dev. 2000, 57, 109–110. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef]
- Ohoka, N.; Okuhira, K.; Ito, M.; Nagai, K.; Shibata, N.; Hattori, T.; Ujikawa, O.; Shimokawa, K.; Sano, O.; Koyama, R.; et al. In Vivo Knockdown of Pathogenic Proteins via Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-dependent Protein Erasers (SNIPERs). J. Biol. Chem. 2017, 292, 4556–4570. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.Z.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Nagai, K.; Morita, Y.; Ujikawa, O.; Ohoka, N.; Hattori, T.; Koyama, R.; Sano, O.; Imaeda, Y.; Nara, H.; et al. Development of Protein Degradation Inducers of Androgen Receptor by Conjugation of Androgen Receptor Ligands and Inhibitor of Apoptosis Protein Ligands. J. Med. Chem. 2018, 61, 543–575. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Peng, Y.; Zhao, Q.; Chen, Y. Small molecule PROTACs: An emerging technology for targeted therapy in drug discovery. RSC Adv. 2019, 9, 16967–16976. [Google Scholar] [CrossRef]
- An, S.; Fu, L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. Ebiomedicine 2018, 36, 553–562. [Google Scholar] [CrossRef]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25, 67–77. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.-Y.; Liu, Z.; Wang, M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511. [Google Scholar] [CrossRef]
- Potjewyd, F.; Turner, A.-M.W.; Beri, J.; Rectenwald, J.M.; Norris-Drouin, J.L.; Cholensky, S.H.; Margolis, D.M.; Pearce, K.H.; Herring, L.E.; James, L.I. Degradation of Polycomb Repressive Complex 2 with an EED-Targeted Bivalent Chemical Degrader. Cell Chem. Biol. 2020, 27, 47–56. [Google Scholar] [CrossRef]
- McCoull, W.; Cheung, T.; Anderson, E.; Barton, P.; Burgess, J.; Byth, K.; Cao, Q.; Castaldi, M.P.; Chen, H.; Chiarparin, E.; et al. Development of a Novel B-Cell Lymphoma 6 (BCL6) PROTAC To Provide Insight into Small Molecule Targeting of BCL6. ACS Chem. Biol. 2018, 13, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- De Dominici, M.; Porazzi, P.; Xiao, Y.; Chao, A.; Tang, H.-Y.; Kumar, G.; Fortina, P.; Spinelli, O.; Rambaldi, A.; Peterson, L.F.; et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood 2020, 135, 1560–1573. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.; Jiang, B.; Bauer, S.; Donovan, K.A.; Liang, Y.; Wang, E.S.; Nowak, R.P.; Yuan, J.C.; Zhang, T.; Kwiatkowski, N.; et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem. Biol. 2019, 26, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-H.; Zengerle, M.; Testa, A.; Ciulli, A. Impact of Target Warhead and Linkage Vector on Inducing Protein Degradation: Comparison of Bromodomain and Extra-Terminal (BET) Degraders Derived from Triazolodiazepine (JQ1) and Tetrahydroquinoline (I-BET726) BET Inhibitor Scaffolds. J. Med. Chem. 2018, 61, 504–513. [Google Scholar] [CrossRef]
- Kong, X.; Pan, P.; Sun, H.; Xia, H.; Wang, X.; Li, Y.; Hou, T. Drug Discovery Targeting Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2019, 62, 10927–10954. [Google Scholar] [CrossRef]
- Nalawansha, D.A.; Crews, C.M. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, Y.; Aguilar, A.; Bernard, D.; Yang, C.-Y. Targeting the MDM2–p53 Protein–Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a026245. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lan, T.; Su, S.; Rao, Y. Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem. Commun. 2019, 55, 369–372. [Google Scholar] [CrossRef]
- Hines, J.; Lartigue, S.; Dong, H.; Qian, Y.; Crews, C.M. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Res. 2019, 79, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhao, X.; Ding, N.; Gao, H.; Wu, Y.; Yang, Y.; Zhao, M.; Hwang, J.; Song, Y.; Liu, W.; et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018, 28, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Burgess, K. TrkC-Targeted Kinase Inhibitors and PROTACs. Mol. Pharm. 2019, 16, 4313–4318. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Tu, G.; Hu, Y.; Jiang, Q.; Liu, J.; Lin, S.; Yu, Z.; Li, G.; Wu, X.; Tang, Y.; et al. Discovery of BP3 as an efficacious proteolysis targeting chimera (PROTAC) degrader of HSP90 for treating breast cancer. Eur. J. Med. Chem. 2022, 228, 114013. [Google Scholar] [CrossRef]
- Naito, M.; Ohoka, N.; Shibata, N. SNIPERs—Hijacking IAP activity to induce protein degradation. Drug Discov. Today Technol. 2019, 31, 35–42. [Google Scholar] [CrossRef]
- Demizu, Y.; Okuhira, K.; Motoi, H.; Ohno, A.; Shoda, T.; Fukuhara, K.; Okuda, H.; Naito, M.; Kurihara, M. Design and synthesis of estrogen receptor degradation inducer based on a protein knockdown strategy. Bioorg. Med. Chem. Lett. 2012, 22, 1793–1796. [Google Scholar] [CrossRef]
- Demizu, Y.; Shibata, N.; Hattori, T.; Ohoka, N.; Motoi, H.; Misawa, T.; Shoda, T.; Naito, M.; Kurihara, M. Development of BCR-ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorg. Med. Chem. Lett. 2016, 26, 4865–4869. [Google Scholar] [CrossRef]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. 2016, 55, 807–810. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25, 78–87. [Google Scholar] [CrossRef]
- Schneekloth, J.S., Jr.; Fonseca, F.N.; Koldobskiy, M.; Mandal, A.; Deshaies, R.; Sakamoto, K.; Crews, C.M. Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. J. Am. Chem. Soc. 2004, 126, 3748–3754. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Maniaci, C.; Hughes, S.J.; Testa, A.; Chen, W.; Lamont, D.J.; Rocha, S.; Alessi, D.R.; Romeo, R.; Ciulli, A. Homo-PROTACs: Bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun. 2017, 8, 830. [Google Scholar] [CrossRef] [PubMed]
- Steinebach, C.; Lindner, S.; Udeshi, N.D.; Mani, D.C.; Kehm, H.; Köpff, S.; Carr, S.A.; Gütschow, M.; Krönke, J. Homo-PROTACs for the Chemical Knockdown of Cereblon. ACS Chem. Biol. 2018, 13, 2771–2782. [Google Scholar] [CrossRef] [PubMed]
- Girardini, M.; Maniaci, C.; Hughes, S.J.; Testa, A.; Ciulli, A. Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs. Bioorg. Med. Chem. 2019, 27, 2466–2479. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.E.; Du, G.; Bushman, J.W.; He, Z.; Zhang, T.; Fischer, E.S.; Gray, N.S. Selective degradation-inducing probes for studying cereblon (CRBN) biology. RSC Med. Chem. 2021, 12, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, J.; Aguilar, A.; McEachern, D.; Przybranowski, S.; Liu, L.; Yang, C.-Y.; Wang, M.; Han, X.; Wang, S. Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J. Med. Chem. 2019, 62, 448–466. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, J.; Tandon, I.; Wu, S.; Teng, P.; Liao, J.; Tang, W. Development of MDM2 degraders based on ligands derived from Ugi reactions: Lessons and discoveries. Eur. J. Med. Chem. 2021, 219, 113425. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolicb, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, e3. [Google Scholar] [CrossRef]
- Zheng, M.; Huo, J.; Gu, X.; Wang, Y.; Wu, C.; Zhang, Q.; Wang, W.; Liu, Y.; Liu, Y.; Zhou, X.; et al. Rational Design and Synthesis of Novel Dual PROTACs for Simultaneous Degradation of EGFR and PARP. J. Med. Chem. 2021, 64, 7839–7852. [Google Scholar] [CrossRef]
- Gabizon, R.; London, N. The rise of covalent proteolysis targeting chimeras. Curr. Opin. Chem. Biol. 2021, 62, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Kiely-Collins, H.; Winter, G.E.; Bernardes, G.J. The role of reversible and irreversible covalent chemistry in targeted protein degradation. Cell Chem. Biol. 2021, 28, 952–968. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R. Dual targeting strategies with bispecific antibodies. Mabs 2012, 4, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.B.; Wells, J.A. Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598. [Google Scholar] [CrossRef]
- Meyer, H.; Ehmann, R.; Smith, G.L. Smallpox in the Post-Eradication Era. Viruses 2020, 12, 138. [Google Scholar] [CrossRef] [PubMed]
- Trilla, A.; Trilla, G.; Daer, C. The 1918 “Spanish Flu” in Spain. Clin. Infect. Dis. 2008, 47, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Su, S.; Yang, H.; Jiang, S. Antivirals with common targets against highly pathogenic viruses. Cell 2021, 184, 1604–1620. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519–530. [Google Scholar] [CrossRef]
- Morens, D.M.; Fauci, A.S. Emerging Pandemic Diseases: How We Got to COVID-19. Cell 2020, 182, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Huang, T.; Song, L.; Xu, S.; Cheng, Y.; Cherukupalli, S.; Kang, D.; Zhao, T.; Sun, L.; Zhang, J.; et al. Medicinal chemistry strategies towards the development of effective SARS-CoV-2 inhibitors. Acta Pharm. Sin. B 2022, 12, 581–599. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Fifty Years in Search of Selective Antiviral Drugs. J. Med. Chem. 2019, 62, 7322–7339. [Google Scholar] [CrossRef]
- Zhan, P.; Pannecouque, C.; De Clercq, E.; Liu, X. Anti-HIV Drug Discovery and Development: Current Innovations and Future Trends. J. Med. Chem. 2016, 59, 2849–2878. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Frutos-Beltrán, E.; Kang, D.; Pannecouque, C.; De Clercq, E.; Menéndez-Arias, L.; Liu, X.; Zhan, P. Medicinal chemistry strategies for discovering antivirals effective against drug-resistant viruses. Chem. Soc. Rev. 2021, 50, 4514–4540. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.J. Recent Advances in Vaccine Technologies. Vet. Clin. N. Am. Small Anim. Pract. 2018, 48, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Dane, D.S.; Dick, G.W.A.; Briggs, M.; Nelson, R. Vaccination Against Poliomyelitis with Live Virus Vaccines. 5. Neutralizing anti body levels one year after vaccination. Br. Med. J. 1958, 2, 1187–1188. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.I.; Cordeiro, M.; Sevilla, E.; Liu, J. Comparison of egg and high yielding MDCK cell-derived live attenuated influenza virus for commercial production of trivalent influenza vaccine: In vitro cell susceptibility and influenza virus replication kinetics in permissive and semi-permissive cells. Vaccine 2010, 28, 3848–3855. [Google Scholar] [CrossRef]
- Pica, N.; Palese, P. Toward a Universal Influenza Virus Vaccine: Prospects and Challenges. Annu. Rev. Med. 2013, 64, 189–202. [Google Scholar] [CrossRef]
- Si, L.; Shen, Q.; Li, J.; Chen, L.; Shen, J.; Xiao, X.; Bai, H.; Feng, T.; Ye, A.Y.; Le Li, L.; et al. Generation of a live attenuated influenza A vaccine by proteolysis targeting. Nat. Biotechnol. 2022, 40, 1370–1377. [Google Scholar] [CrossRef]
- De Wispelaere, M.; Du, G.; Donovan, K.A.; Zhang, T.; Eleuteri, N.A.; Yuan, J.C.; Kalabathula, J.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat. Commun. 2019, 10, 3468. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Banerjee, S.; Ghosh, K.; Gayen, S.; Jha, T. Protease targeted COVID-19 drug discovery and its challenges: Insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorg. Med. Chem. 2021, 29, 115860. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, C.; Xin, L.; Ren, X.; Tian, L.; Ju, X.; Li, H.; Wang, Y.; Zhao, Q.; Liu, H.; et al. The development of Coronavirus 3C-Like protease (3CLpro) inhibitors from 2010 to 2020. Eur. J. Med. Chem. 2020, 206, 112711. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y. Sialobiology of Influenza: Molecular Mechanism of Host Range Variation of Influenza Viruses. Biol. Pharm. Bull. 2005, 28, 399–408. [Google Scholar] [CrossRef]
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Gallop, J.L. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 2005, 438, 590–596. [Google Scholar] [CrossRef]
- Wolf, M.; Garcea, R.L.; Grigorieff, N.; Harrison, S.C. Subunit interactions in bovine papillomavirus. Proc. Natl. Acad. Sci. USA 2010, 107, 6298–6303. [Google Scholar] [CrossRef]
- Xu, Z.; Liu, X.; Ma, X.; Zou, W.; Chen, Q.; Chen, F.; Deng, X.; Liang, J.; Dong, C.; Lan, K.; et al. Discovery of oseltamivir-based novel PROTACs as degraders targeting neuraminidase to combat H1N1 influenza virus. Cell Insight 2022, 1, 100030. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Jr, R.M.W.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Pervushin, K.; Tan, E.; Parthasarathy, K.; Lin, X.; Jiang, F.L.; Yu, D.; Vararattanavech, A.; Soong, T.W.; Liu, D.X.; Torres, J. Structure and Inhibition of the SARS Coronavirus Envelope Protein Ion Channel. PLoS Pathog. 2009, 5, e1000511. [Google Scholar] [CrossRef]
- Ge, X.-Y.; Li, J.-L.; Yang, X.-L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Bansal, P.; Kumar, R.; Singh, J.; Dhanda, S. In silico molecular docking of SARS-CoV-2 surface proteins with microbial non-ribosomal peptides: Identification of potential drugs. J. Proteins Proteom. 2021, 12, 177–184. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Yamada, T.; Komoto, J.; Watanabe, K.; Ohmiya, Y.; Takusagawa, F. Crystal Structure and Possible Catalytic Mechanism of Microsomal Prostaglandin E Synthase Type 2 (mPGES-2). J. Mol. Biol. 2005, 348, 1163–1176. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Terracciano, R.; Preianò, M.; Fregola, A.; Pelaia, C.; Montalcini, T.; Savino, R. Mapping the SARS-CoV-2–Host Protein–Protein Interactome by Affinity Purification Mass Spectrometry and Proximity-Dependent Biotin Labeling: A Rational and Straightforward Route to Discover Host-Directed Anti-SARS-CoV-2 Therapeutics. Int. J. Mol. Sci. 2021, 22, 532. [Google Scholar] [CrossRef]
- Desantis, J.; Mercorelli, B.; Celegato, M.; Croci, F.; Bazzacco, A.; Baroni, M.; Siragusa, L.; Cruciani, G.; Loregian, A.; Goracci, L. Indomethacin-based PROTACs as pan-coronavirus antiviral agents. Eur. J. Med. Chem. 2021, 226, 113814. [Google Scholar] [CrossRef]
- Gutierrez-Chamorro, L.; Felip, E.; Ezeonwumelu, I.J.; Margelí, M.; Ballana, E. Cyclin-dependent Kinases as Emerging Targets for Developing Novel Antiviral Therapeutics. Trends Microbiol. 2021, 29, 836–848. [Google Scholar] [CrossRef]
- Hahn, F.; Hamilton, S.T.; Wangen, C.; Wild, M.; Kicuntod, J.; Brückner, N.; Follett, J.E.L.; Herrmann, L.; Kheimar, A.; Kaufer, B.B.; et al. Development of a PROTAC-Based Targeting Strategy Provides a Mechanistically Unique Mode of Anti-Cytomegalovirus Activity. Int. J. Mol. Sci. 2021, 22, 12858. [Google Scholar] [CrossRef]
- Murakami, S. Hepatitis B Virus X Protein: Structure, Function and Biology. Intervirology 1999, 42, 81–99. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Duan, L.X. Hepatitis B virus X antigen in the pathogenesis of chronic infections and the development of hepatocellular carcinoma. Am. J. Pathol. 1997, 150, 1141–1157. [Google Scholar]
- Montrose, K.; Krissansen, G.W. Design of a PROTAC that antagonizes and destroys the cancer-forming X-protein of the hepatitis B virus. Biochem. Biophys. Res. Commun. 2014, 453, 735–740. [Google Scholar] [CrossRef]
- Landesman, S.H.; Ginzburg, H.M.; Weiss, S. The AIDS Epidemic. N. Engl. J. Med. 1985, 312, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.G.; Summers, M.F. Structural biology of HIV. J. Mol. Biol. 1999, 285, 1–32. [Google Scholar] [CrossRef]
- Zhang, Z.; Yuan, S.; Xu, S.; Guo, D.; Chen, L.; Hou, W.; Wang, M. Suppression of HIV-1 Integration by Targeting HIV-1 Integrase for Degradation with A Chimeric Ubiquitin Ligase. Virol. Sin. 2021, 36, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef]
- Rana, S.; Bendjennat, M.; Kour, S.; King, H.M.; Kizhake, S.; Zahid, M.; Natarajan, A. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg. Med. Chem. Lett. 2019, 29, 1375–1379. [Google Scholar] [CrossRef]
- Crew, A.P.; Raina, K.; Dong, H.; Qian, Y.; Wang, J.; Vigil, D.; Serebrenik, Y.V.; Hamman, B.D.; Morgan, A.; Ferraro, C.; et al. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J. Med. Chem. 2018, 61, 583–598. [Google Scholar] [CrossRef]
- Zhou, B.; Hu, J.; Xu, F.; Chen, Z.; Bai, L.; Fernandez-Salas, E.; Lin, M.; Liu, L.; Yang, C.-Y.; Zhao, Y.; et al. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem. 2018, 61, 462–481. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Cao, P.; Gao, Y.; Wu, M.; Lin, Y.; Tian, Y.; Yuan, W. Differential expression of p38 MAPK α, β, γ, δ isoforms in nucleus pulposus modulates macrophage polarization in intervertebral disc degeneration. Sci. Rep. 2016, 6, 22182. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Schultz, A.R.; Bondeson, D.P.; Eide, C.A.; Stevens, S.L.S.; Druker, B.J.; Crews, C.M. Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res 2019, 79, 4744–4753. [Google Scholar] [CrossRef]
- Burslem, G.M.; Song, J.; Chen, X.; Hines, J.; Crews, C.M. Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion. J. Am. Chem. Soc. 2018, 140, 16428–16432. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Reits, E.; Neefjes, J. Making sense of mass destruction: Quantitating MHC class I antigen presentation. Nat. Rev. Immunol. 2003, 3, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.; Setz, C.; Wild, J.; Schubert, U. The PTAP Sequence within the p6 Domain of Human Immunodeficiency Virus Type 1 Gag Regulates Its Ubiquitination and MHC Class I Antigen Presentation. J. Immunol. 2011, 186, 5706–5718. [Google Scholar] [CrossRef]
- Martinez-Ortiz, W.; Zhou, M.-M. Could PROTACs Protect Us From COVID-19? Drug Discov. Today 2020, 25, 1894–1896. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.-J.; Crank, M.C.; Shiver, J.; Graham, B.S.; Mascola, J.R.; Nabel, G.J. Next-generation influenza vaccines: Opportunities and challenges. Nat. Rev. Drug Discov. 2020, 19, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ma, J.; Liu, Y.; Xia, J.; Li, Y.; Wang, Z.P.; Wei, W. PROTACs: A novel strategy for cancer therapy. Semin. Cancer Biol. 2020, 67, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Coen, M.; Zhang, A.X.; Pachl, F.; Castaldi, M.P.; Dahl, G.; Boyd, H.; Scott, C.; Newham, P. Proteolysis-targeting chimeras in drug development: A safety perspective. Br. J. Pharmacol. 2020, 177, 1709–1718. [Google Scholar] [CrossRef]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Costales, M.G.; Matsumoto, Y.; Velagapudi, S.P.; Disney, M.D. Small Molecule Targeted Recruitment of a Nuclease to RNA. J. Am. Chem. Soc. 2018, 140, 6741–6744. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Jha, B.K.; Silverman, R.H. New insights into the role of RNase L in innate immunity. J. Interferon Cytokine Res. 2011, 37, 49–57. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Target | Examples | E3 Ligase | Mechanism of Action | Ref. |
|---|---|---|---|---|
| Viral proteases | ||||
| Telaprevir | CRBN | Telaprevir acts as a protein ligand in the ternary complex and may reversibly bind to and inhibit the viral proteases. | [71] | |
| Surface proteins | ||||
| Oseltamivir | VHL/CRBN | Oseltamivir binds to the neuraminidase enzyme in the viral envelope and inhibits it. The PROTAC molecule employs the cellular ligases for the subsequent degradation of neuraminidase. | [79] |
| ||||
| Thalidomide | VHL | Targets envelop protein E and lead to its degradation via the proteasomal degradation pathway. | |
| Host protein | ||||
| Indomethacin | Indomethacin binds to the host protein, PGES-2. PGES2 and NSP-7 form the viral primase complex. Targeting PGES-2 for degradation hinders the formation of the viral polymerase. Alternatively, it interacts with NSPs in the viral primase complex and regulates the PKR pathway to inhibit protein synthesis. | [89] | |
| SNS032 | CRBN/IAP/VHL | CDK-directed PROTACs cause the degradation of other essential CDKs that participate in the formation of the nuclear capsid of the virus. | [91] |
| Target | PROTACs | Conventional Antivirals |
|---|---|---|
| Specificity | PROTACs offer highly specific and precise machinery to degrade the target protein and eliminate its activity. | They may not be highly specific and are also unable to cause complete elimination of the target proteins because they can only inhibit the activity of the protein. |
| Efficiency | They are highly potent and do not depend on the target protein’s high affinity. The formation of a ternary complex is sufficient to induce protein degradation. | They require a very high binding affinity with the target protein and hence are not very efficient in action. |
| Mechanism of action | They exhibit a catalytic mechanism of action because a single molecule can degrade more than one target protein molecule. | They have been known to function in a stoichiometric manner; that is, a single molecule can only inhibit a single molecule of the target protein. |
| Dosage | Since they are highly specific, potent, and efficient, even a nanomolar concentration range can induce targeted protein degradation. | Due to the stoichiometric mechanism of action and weaker efficiency, they are required to be administered in higher doses. |
| Resistance | It is highly unlikely for viruses to develop resistance to PROTACs. | Prolonged exposure can induce drug resistance because of antigenic shifts and drift in the viral genome. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, H.; Zia, B.; Husain, H.; Husain, A. Recent Advances in PROTAC-Based Antiviral Strategies. Vaccines 2023, 11, 270. https://doi.org/10.3390/vaccines11020270
Ahmad H, Zia B, Husain H, Husain A. Recent Advances in PROTAC-Based Antiviral Strategies. Vaccines. 2023; 11(2):270. https://doi.org/10.3390/vaccines11020270
Chicago/Turabian StyleAhmad, Haleema, Bushra Zia, Hashir Husain, and Afzal Husain. 2023. "Recent Advances in PROTAC-Based Antiviral Strategies" Vaccines 11, no. 2: 270. https://doi.org/10.3390/vaccines11020270
APA StyleAhmad, H., Zia, B., Husain, H., & Husain, A. (2023). Recent Advances in PROTAC-Based Antiviral Strategies. Vaccines, 11(2), 270. https://doi.org/10.3390/vaccines11020270