Abstract
A pathway frequently altered in cancer is glutaminolysis, whereby glutaminase (GA) catalyzes the main step as follows: the deamidation of glutamine to form glutamate and ammonium. There are two types of GA isozymes, named GLS and GLS2, which differ considerably in their expression patterns and can even perform opposing roles in cancer. GLS correlates with tumor growth and proliferation, while GLS2 can function as a context-dependent tumor suppressor. However, both isoenzymes have been described as essential molecules handling oxidant stress because of their involvement in glutathione production. We reviewed the literature to highlight the critical roles of GLS and GLS2 in restraining ROS and regulating both cellular signaling and metabolic stress due to their function as indirect antioxidant enzymes, as well as by modulating both reductive carboxylation and ferroptosis. Blocking GA activity appears to be a potential strategy in the dual activation of ferroptosis and inhibition of cancer cell growth in a ROS-mediated mechanism.
Keywords:
cancer; ferroptosis; glutaminase; glutaminolysis; glutathione; metabolic reprogramming; oxidative stress; ROS 1. Introduction
Glutaminase (GA, EC 3.5.1.2) is the enzyme responsible for catalyzing the conversion of glutamine (Gln) to glutamate (Glu) and represents the first step in glutaminolysis [1]. Gln has essential functions in cancer, providing amino acids, lipids, nucleotides, hexosamines, and polyamines but also rendering metabolic energy in the form of adenosine triphosphate (ATP) or being used as a pleiotropic cell signaling molecule [2]. In addition, Gln is indispensable for synthesizing reduced glutathione (GSH), the most abundant intracellular antioxidant molecule and the main player in the GSH–antioxidant system, formed by GSH, oxidized glutathione (GSSG), glutathione reductase (GR), and glutathione peroxidases (GPXs) [3]. While Glu affords resources for GSH synthesis, the limited oxidation of Gln through glutaminolysis restricts oxidative phosphorylation (OXPHOS) and mitigates reactive oxygen species (ROS) levels that are otherwise harmful to cells [2]. Nonetheless, ROS can cause damage to cancer cells when levels are excessive, but, indeed, they are a needed signaling mechanism for tumor homeostasis [4]; hence, ROS levels are precisely regulated by a set of antioxidant enzymes, including, indirectly, the function of GA isoenzymes [1]. The key role of antioxidant systems and the balance of ROS can be exploited for the design of novel strategies to target cancer [2,3], which is discussed in this review.
2. Glutaminase Isoenzymes in the Control of Cancer Redox Homeostasis
The metabolic alterations observed in cancer are a consequence of the dysregulation of the expression of metabolic enzymes such as GA isoenzymes [5]. In mammals, including humans, the GLS gene encodes two isoforms, the largest kidney-type glutaminase (KGA) and glutaminase C (GAC), the latter usually being the variant most frequently overexpressed in cancers. Both isoforms can be collectively called GLS isoenzymes [6]. On the other hand, the GLS2 gene encodes two more isoforms, glutaminase B (GAB) and the shorter liver-type glutaminase (LGA), and both can be referred to as GLS2 isoenzymes [7]. The secondary functions of these metabolic enzymes are linked with several molecular mechanisms affecting cancer, being sharply different for GLS and GLS2. Therefore, GAs are related to both cancer progression or antitumoral effects by altering the signaling, regulation, and oxidative status of cancer cells (Table 1 and Table 2).

Table 1.
GLS in the control of redox status in cancer.
Table 2.
GLS2 in the control of redox status in cancer.
GLS2 has been related to both tumor suppression properties and prooncogenic effects. Thus, its role in cancer is considered to be context-dependent and has been thoroughly reviewed recently [42]. However, its function as an oncogenic factor or a tumor suppressor has been related, at least partially, to its ability to modulate antioxidant capacity [1]. In the nucleus, GLS2 also participates in redox regulation and the growth-arrest program, interacting with key factors [5,7]. Although human GLS2 has structural determinants for mitochondrial targeting, and it does not possess a typified nuclear localization signal, its nuclear translocation has been reported and it can work as an indirect transcription factor regulating gene expression and modulating other nuclear proteins [43]. In fact, a mechanism implying its tumor suppressor ability is related to p53 and was first characterized in 2010 [44,45]. GLS2 expression was found to be induced by p53 and evoked oxygen consumption, mitochondrial respiration, and ATP generation, increasing Glu, GSH, and nicotinamide adenine dinucleotide (NADH) levels, reducing the amounts of ROS to protect cells from H2O2-induced apoptosis [44]. GLS2 protected cells from DNA oxidation by preventing the generation of 8-hydroxy-2’-deoxyguanosine, the main source of oxidation-associated mutagenesis [45]. In various cell models, GLS2 knockout works as an anticancer factor, correlated with increased levels of ROS in several cancer types, i.e., neuroblastoma [46], cervical cancer [47], and triple-negative breast cancer (TNBC) [48]. Although GLS2 increased GSH levels in neuroblastoma, its expression correlated with the proliferation and aggressiveness of neuroblastoma by an N-MYC-dependent mechanism [46]. In cervical cancer, following radiotherapy, tissues of radiosensitive patients showed diminished GLS2 levels and their lower amounts of GLS2 were concomitant to decreased values of GSH, NADH, and nicotinamide adenine dinucleotide phosphate (NADPH), as well as higher oxidative stress. Hence, GLS2 was associated with the radioresistance of cervical cancer [47]. Of note, when GLS was either pharmacologically inhibited by CB-839 or genetically silenced, GLS2 mimicked its metabolic function, as well as its impact on redox balance, in a long number of TNBC cell lines [48]. Remarkably, after analyzing data from breast cancer tissues available from The Cancer Genome Atlas (TCGA), these authors found a correlation between higher GLS2 expression, increased epithelial-to-mesenchymal transition (EMT), and worse prognosis and mortality [48]. However, in a more in-depth study including 1075 patient samples from the TCGA database, higher GLS/GLS2 ratios were correlated with increased EMT in breast cancer patients and worse survival [49].
3. Mitochondrial Metabolism of Glutamine in Cancer: Redox Balance
Metabolic reprogramming is one of the hallmarks of cancer [50]. Metabolic adaptations are essential for providing the hugely increased bioenergetics and biosynthetic demands to sustain tumor growth [51] but also for maintaining an adequate oxidative–reductive equilibrium for cancer cells’ survival and proliferation [52]. Maximizing OXPHOS is among the metabolic traits most commonly altered in tumor cells [53]. In this metabolic shuffling, Gln is one of the most valuable playing cards [54]. Although Gln is not an essential amino acid, cancer cells usually promote its import and/or additional biosynthesis from other sources [55] through several oncogenes that activate glutaminolysis, i.e., c-MYC [56], KRAS [57], hypoxia-inducible factor 1 (HIF1) [58], isocitrate dehydrogenase 1 and 2 (IDH1/2) [59], epidermal growth factor receptor (EGFR) [60] or even tumor suppressor p53 [44]. Gln is the most abundant amino acid in circulating blood and in muscle [43]. Tumor cells can use Gln for biosynthetic purposes through the tricarboxylic acid cycle (TCA cycle) [61], allowing cancer cells to sustain TCA cycle activity and produce reductive equivalents such as NADPH during proliferation [62]. Additionally, it may be used to produce NADH, flavin adenine dinucleotide (FADH2), and ATP [63]. Oxidation of Gln equally sustains redox homeostasis by a reduction of NADP+ to NADPH by malic enzyme (ME1) due to the transformation of Gln-derived metabolites to pyruvate [54]. On the other hand, Gln is required for the synthesis of the main nonenzymatic antioxidant in the cell, the tripeptide GSH [64], which is synthesized from Glu, cysteine (Cys), and glycine (Gly) [5]. Glutamate production from glutamine is essential for GSH biosynthesis, since it not only provides one of the three amino acids needed but because produced glutamate may also serve to import cysteine to the cell through the xCT transport system; thus, GLS and GLS2 become key enzymes in cancer [3], contributing to the maintenance of redox homeostasis and the balance of ROS levels [65]. Additionally, the TCA cycle’s rewiring causes changes in the redox homeostasis, connecting the cancer metabolisms of amino acids and the redox biologies of tumor cells [66].
From a strict metabolic point of view, the catabolism of Gln enables cells to sustain enough anaplerotic flux to utilize a large number of TCA cycle intermediates as precursors for macromolecular biosynthesis [67]. Of note, Gln’s main role in many routes of intermediary metabolism that produce Glu and alpha-ketoglutarate (AKG) makes this amino acid an essential source of carbon for the TCA cycle [68]. Gln can be transformed to Glu by mitochondrial GA, which delivers Gln’s amide group as cationic-free ammonium (Figure 1). Glu can be turned to AKG by Glu dehydrogenase (GDH), which releases the amino group as a free ammonium ion, or by mitochondrial aminotransferases such as alanine aminotransferase (ALT) and aspartate aminotransferase (AST), which transfers the amino group of Glu to pyruvate or to oxaloacetate to form alanine or aspartate, respectively [5]. AKG enters the TCA cycle and through sequential reactions generates oxaloacetate (OAA) [67]. Then, Gln can be a substrate for the synthesis of both OAA or, subsequently, lactate, through glutaminolysis, which accumulates reductive capacity as NADH and NADPH [68]. Glutaminases are required by many cells to achieve maximal growth both in vitro and in vivo [69]. However, even Gln-addicted cells can reprogram their carbon metabolism from glucose to synthesize building blocks normally supplied by Gln [70]. The enzyme responsible for this form of glucose-dependent anaplerosis is pyruvate carboxylase (PC), fundamental for cell survival and proliferation of cells adapted to growth in low-Gln concentrations [58]. PC replenishes OAA and generates citrate through reductive carboxylation, facilitating an alternative anaplerotic pathway in Gln-independent tumor cells [71]. Silencing GLS in tumor cells increases PC activity to compensate for reduced anaplerosis from Gln, affecting cancer growth [72]. Gln-dependent anaplerosis is energetically advantageous as PC requires ATP to generate OAA, while GLS is not ATP-dependent [70]. Gln catabolism through the TCA cycle forms reducing equivalents (NADH) for OXPHOS [72]. Cancer cells can utilize the bidirectional metabolism from Gln-derived AKG to synthesize citrate and generate reducing equivalents, which is advantageous for adapting to nutrient availability [71].
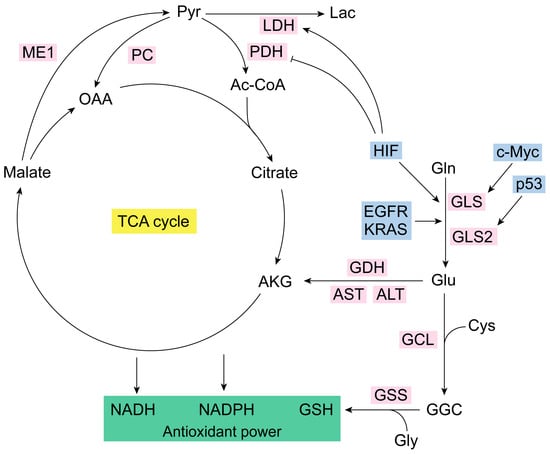
Figure 1.
Glutaminolysis and antioxidant capacity. Glutaminase isoenzymes generate Glu from Gln [7]. GLS is regulated by the oncogene c-Myc, and GLS2 is modulated by tumor-suppressor p53 [44,45,56]. Glu together with Cys (via GCL) and Gly (via GSS) form, respectively, GGC and GSH (the most important antioxidant molecule in the cell) [1]. Glutaminolysis is accurately regulated [3,34]. IDH, KRAS, EGFR, and HIF are some of its key modulators [57,58,59,60]. HIF also controls LDH and PDH [70,71,72,73,74]. NADPH generation does not occur in the TCA cycle but in the IDH1/2 reaction that converts isocitrate to AKG [58]. Ac-CoA, acetyl coenzyme A; AKG, alpha-ketoglutarate; ALT, alanine aminotransferase; AST, aspartate aminotransferase; Cys, cysteine; EGFR, epidermal growth factor receptor; GCL, glutamate-cysteine ligase; GDH, glutamate dehydrogenase; GGC, gamma-glutamylcysteine synthetase; Gln, glutamine; GLS, glutaminase isoenzyme 1; GLS2, glutaminase isoenzyme 2; Glu, glutamate; Gly, glycine; GSH, glutathione; GSS, glutathione synthetase; HIF, hypoxia-inducible factor; Lac, lactate; LDH, lactate dehydrogenase; ME1, malic enzyme 1; NADH, nicotinamide adenine dinucleotide; NADPH, nicotinamide adenine dinucleotide phosphate; OAA, oxaloacetate; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase.
Glutaminase can Trigger Reductive Carboxylation in Cancer
The IDH1 isoform is located in the cytoplasm, whereas the IDH2 and IDH3 isoforms are found in mitochondria. IDH1 and -2 catalyze the decarboxylation of isocitrate into AKG, rendering NADPH, while IDH3 forms AKG, and NADH and CO2 form isocitrate [73]. The noncanonical process of the formation of isocitrate from AKG and CO2 is named reductive carboxylation, and it is fundamental in hypoxia conditions or when a high level of the synthesis of lipids is required [74]. Reductive carboxylation is catalyzed by IDH1/2 through the consumption of NADPH. Of note, NADPH and NADP+ cannot cross the mitochondrial membrane, a fact that implicates IDH1/2 in the balance of the oxidative status in cytosol and mitochondria [63,73,74,75]. IDH1 is related to the scavenging of ROS in the cytoplasm by the generation of NADPH in this cellular compartment [76]. In addition, the generation of cytosolic ROS mediates the induction of reductive carboxylation through IDH1 by negatively regulating a protein tyrosine phosphatase, which results in the increased phosphorylation of IDH1 and, hence, its activation [75].
There are tumor-associated IDH1/2 mutant versions that consume NADPH to transform AKG to the (R) enantiomer of 2-hydroxyglutarate, (R)-2HG, which accrues in cancer cells [73]. These (R)-2HG-producing mutants of IDH1/2 are commonly found in a subtype of gliomas [77]. (R)-2HG is considered an oncometabolite capable of inhibiting the branched-chain amino acid aminotransferases 1 and 2 (BCAT1/2), which use AKG and branched-chain amino acids as substrates to produce Glu and the corresponding branched-chain alpha-ketoacid, in a reversible amino group transfer reaction [78]. Hence, in this molecular context, the generation of Glu by transamination is compromised and becomes limiting for the biosynthesis of GSH [79], therefore impacting redox homeostasis (Figure 2). The activation of reductive carboxylation reprograms Gln metabolism, increasing GSH biosynthesis in a global compensatory rewiring [66]. This mechanism follows TCA cycle inhibition or impairment of the mitochondrial thioredoxin reductase (TrxR) antioxidant system [68]. Thus, to normalize antioxidant power, reductive carboxylation of Gln-derived AKG takes place [80]. The exchanging of glucose to Gln in the TCA cycle toward reductive Gln metabolism is conducted by HIFs in a mechanism regulated by the Warburg effect, also known as fermentative glycolysis [81]. For example, in normoxic 3D spheroids lung cancer cells, reductive citrate, produced by IDH1 in the cytosol from AKG derived from Gln is used to maintain redox homeostasis. The citrate generated in the cytosol was subsequently imported into the mitochondria, where an oxidative metabolism occurs via IDH2, forming NADPH, which can be used by various antioxidant systems to detoxify mitochondrial ROS [82]. Similarly, in H460 lung cancer cells, AKG’s reductive carboxylation allowed for the flow of citrate from the cytosol to the mitochondria, resulting in oxidative stress mitigation and faster growth rates [80]. Eventually, the increased consumption of Gln by reductive carboxylation of Gln-derived AKG supports anabolic reactions, such as lipid synthesis, to activate cell proliferation [83]. Reductive carboxylation of Gln-derived AKG was equally found in hepatoma cells and mouse xenograft models to support de novo lipogenesis and the cellular antioxidant system by increasing the levels of NADPH and GSH [84]. On the other hand, Gln-derived metabolites in the TCA cycle may serve for synthesizing aspartate, an essential metabolite for de novo nucleotide biosynthesis. In renal cell carcinoma, GLS inhibitor BPTES impaired the reductive transformation of Gln-derived carbons to aspartate and subsequently minimized the de novo synthesis of pyrimidines [85,86]. Accordingly, in three GBM cell lines treated with the more potent GLS inhibitor CB-839, a sharp drop in aspartate together with diminishments in both the oxidative metabolism in the TCA cycle and reductive carboxylation of Gln-derived AKG were found. These findings correlated with lower levels of metabolites from de novo purine and pyrimidine biosynthesis pathways [87]. In NSCLC, reductive carboxylation of AKG increased cytosolic ROS by a mechanism that includes the upregulation of NADPH oxidase 2 (NOX2) [75]. On the other hand, GLS inhibition by BPTES also evoked higher intracellular ROS levels by affecting GSH formation [85].
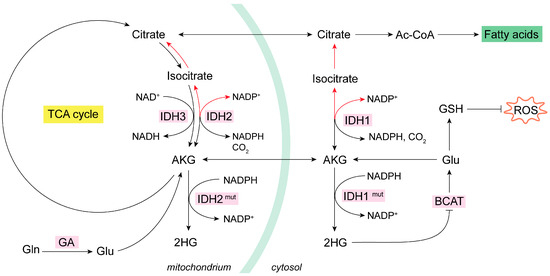
Figure 2.
Mitochondrial oxidative and reductive metabolism of Gln. IDH isoenzymes modulate the oxidative or reductive metabolism of Gln in NADP-dependent reactions [74]. In cancer cells, the increase in glycolysis and glutaminolysis boost ROS levels [55]. In some cancer types, mutant IDHs produce oncometabolite 2HG, which inhibits BCAT isoenzymes and lowers GSH [77,78,79]. Many types of tumor cells activate the reductive metabolism (red arrows) of AKG to generate Ac-CoA for de novo lipogenesis as a response to their biosynthetic need for rapid proliferation [80,81,82,83,84]. Reductive carboxylation by IDH1 in the cytosol consumes NADPH to generate isocitrate and then citrate; subsequently, the citrate is imported into the mitochondria and there follows oxidative metabolism (black arrows) and decarboxylation via IDH2 to generate mitochondrial NADPH [73,74,75]. Ac-CoA, acetyl coenzyme A; AKG, alpha-ketoglutarate; BCAT, branched-chain amino acid aminotransferase; GA, glutaminase; GDH, glutamate dehydrogenase; GLS, glutaminase isoenzyme 1; GLS2, glutaminase isoenzyme 2; Gln, glutamine; Glu, glutamate; GSH, glutathione; HG, hydroxyglutarate; HIF, hypoxia-inducible factor; NADH, nicotinamide adenine dinucleotide; NADPH, nicotinamide adenine dinucleotide phosphate; OAA, oxaloacetate; ROS, reactive oxygen species.
4. Glutaminases, Ferroptosis, and ROS
Ferroptosis is an iron-dependent specific and regulated type of cell death. The mechanism is mainly characterized by high levels of intracellular iron, which through the Fenton reaction leads to the production of hydroxyl radicals, subsequently provoking polyunsaturated fatty acid peroxidation, lastly resulting in membrane disruption and cell death [88]. Glutamate, which is produced by GLS and GLS2, is essential for the synthesis of GSH, which reduces lipid hydroperoxides via glutathione peroxidase 4 (GPX4) [89]. Thus, a decrease in GSH levels can lead to an increase in the lipid peroxidation process, and, therefore, the ferroptosis phenomena, because GSH is the most important nonenzymatic antioxidant and serves as a fundamental eliminator of ROS [88]. GPX4 may act as an oncogene, as it works as the major inhibitor of ferroptosis, also being related to the regulation of the electron transfer chain (ETC), OXPHOS, and other metabolic pathways in cancer [89]. Noteworthy, erastin, an inhibitor of the xCT cystine-Glu antiporter, is another fundamental regulator of ferroptosis (Figure 3). An induction of ferroptosis by erastin evokes GSH depletion by limiting the cysteine supply [90]. Erastin-dependent induction of ferroptosis is impacted by the mitochondrial metabolism of Gln, as it has been found that the reductive carboxylation of Gln-derived metabolites favors resistance in NSCLC cells via a redox mechanism involving the GSH antioxidant system [91]. GA isoenzymes have a profound link with ferroptosis, since they are fundamental enzymes for achieving a high glutaminolytic flux in cancer cells [81]. Hence, ferroptosis is modulated by the TrxR system upon erastin action, as well as by glutaminolysis [90]. Thus, a connection between Gln metabolism and ferroptosis via the GLS2-p53 axis has been demonstrated [92,93]. Mechanistically, p53 inhibited the transcription of the cystine/Glu antiporter SLC7A11 (system xCT) [94]. Additionally, in ccRCC, GLS2 was upregulated during ferroptosis induced by erastin or RSL3. GLS2 is related to higher amounts of GSH in the cell and is considered a potential ferroptosis suppressor in ccRCC [95]. Ferroptosis was equally activated in human TNBC cells in a mechanism dependent on the action of the activating transcription factor 4 (ATF4) and ChaC glutathione-specific gamma-glutamylcyclotransferase 1 (CHAC1), which degrades GSH and induces cystine deprivation [96]. Mechanistically, this might result in the relocation of reducing equivalents from cystine to thioredoxin (via TrxR1) and thereby avoids ROS augmentation [97]. Although GSH prevents the interaction of iron, oxygen, and polyunsaturated fatty acids from triggering ferroptosis, the exact role of total GSH (tGSH) in ferroptosis and whether there exist differences between cytosolic and mitochondrial GSH for the induction of ferroptosis are still to be clarified [98].
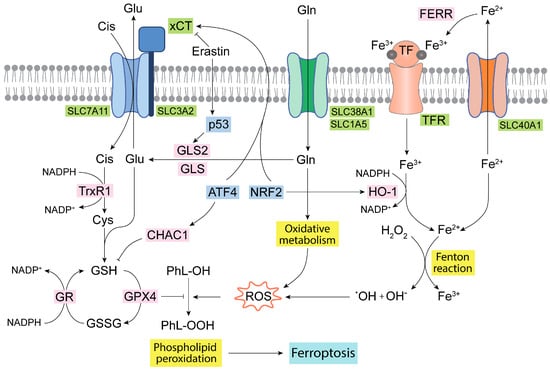
Figure 3.
Ferroptosis and Gln homeostasis. Iron metabolism in the mitochondria generates oxidative molecules and radicals that together with the oxidative metabolism of Gln produce a large amount of ROS, leading to lipid peroxidation and ferroptosis [41,98]. The glutathione antioxidant system formed by GR, GPX, and TrxR isoenzymes, as well as by GSH, is the main antioxidant network to detoxify ROS [90,97]. Some transcription factors, such as ATF4 and NRF2, regulate and connect the GSH antioxidant system, Glu transport, and iron metabolism [29,96]. ATF4, activating transcription factor 4; CHAC1, ChaC glutathione specific gamma-glutamylcyclotransferase 1; Cis, cystine; Cys, cysteine; FERR, ferritin; GLS, glutaminase isoenzyme 1; GLS2, glutaminase isoenzyme 2; Gln, glutamine; Glu, glutamate; GPX4, glutathione peroxidase 4; GR, glutathione reductase; GSH, reduced glutathione; GSSG, oxidized glutathione; HO-1, heme oxygenase 1; NADPH, nicotinamide adenine dinucleotide phosphate; NRF2, nuclear erythroid factor 2-related factor 2; PhL-OOH, peroxidized phospholipids; PhL-OH, phospholipids; ROS, reactive oxygen species; TF, transferrin; TFR, transferrin receptor; TrxR1, thioredoxin 1; xCT, cystine/glutamate antiporter.
Ferroptosis modulation is essential in the chemoresistance event in TNBC [99], whereby tumor cells are characterized as commonly being Gln-dependent via glutaminolysis, which favors drug resistance [100]. To defeat these cancer cells, a dual combination strategy has been suggested by targeting both GLS and ferroptosis [99]. Gallbladder cancer cells, which also need GLS for growth and proliferation, triggered ferroptosis following GLS inhibition and a drop in antioxidant capacity (GSH- and NADPH-dependent), which evoked oxidative stress [101]. Ovarian cancer cells are enriched in iron and rewire their Gln metabolism through HIF-mediated metabolic reprogramming [102]. However, ovarian cancer cells are resistant to ferroptosis through NRF2-mediated activation of additional antioxidant mechanisms, as those elicited by ferritin and heme oxygenase-1 (HO-1) decrease free iron amounts, modulating iron metabolism and ROS levels [103]. In these conditions, therapeutic strategies that either block the xCT system or target specific metabolic vulnerabilities may be an option to overcome ferroptosis resistance [102]. In several types of aggressive and metastatic cancers a combination treatment including the natural product beta-elemene, in a codelivery strategy, was an efficacious drug for tumor patients reversing multidrug resistance and activating ferroptosis [104]. In KRAS-mutant CRC cells, a combination of beta-elemene and cetuximab was successful, both in vitro and in vivo, and resulted in the induction of ferroptosis via the downregulation of GLS, xCT, GPX4, ferritin, and ferroportin (SLC40A1), which exports intracellular iron out of the cell [105].
5. Future Perspectives
As reported above, the GLS and GLS2 isoenzymes are metabolic enzymes [42,43,54,63] whose roles may imply key antioxidant functions [64,65,91,95], which might lead to therapeutic implications for cancer therapy [15,16,26,35]. Of interest, several GLS inhibitors can block cancer progression by promoting oxidative stress [106]. Thus, a novel derivative of lonidamine, named HYL001, greatly inhibited cancer stem cells (CSCs) by hampering Gln metabolism, with 380-fold and 340-fold lower IC50 against breast CSCs and hepatocellular carcinoma (HCC) stem cells, respectively, as compared to lonidamine, while having minimum toxic effects toward nontumor cells and immune-competent mice [13]. Although lonidamine increased ROS and promoted cell death by reduction in the pentose phosphate pathway (which lowered NADPH and GSH formation) [107], it is not approved in the clinic due to its hepatotoxicity, which is caused by its low solubility and pharmacokinetic characteristics [108]. However, HYL001 constitutes a promising derivative and is confirmed to be a good candidate for clinical trials, such as through reduced cancer cell proliferation on fresh tumor tissues from HCC patients by diminishing GSH, increasing ROS levels, and inducing apoptosis [13]. Similarly, Gln metabolism and, particularly, GLS activity were found to be essential for androgen-receptor antagonist-resistant prostate cancer cells, xenografts, patient-derived organoids, patient-derived explants, and tumor samples by increasing antioxidant capacity [106].
Cancer is extremely adaptive with compensatory metabolic pathways that inevitably ease resistance to Gln shortages, specifically with monotherapy [109]. Blocking GLS by CB-839 or genetic silencing made resistant cancer cells vulnerable in an antioxidant-dependent mechanism, which included GSH action [106]. However, because of the plasticity of tumors and heterogeneity in cancer, it is not simple to target their vulnerabilities, as normal and malignant cells share many essential enzymes, metabolic network pathways, and antioxidant systems [52]. Additional efforts are vital to delimitate how to modulate the redox status against every type of cancer and optimize combinations for synergistic therapy, hopefully to achieve personalized successful treatments. For example, because poly (ADP-ribose) polymerase (PARP) inhibitors synergize with GLS ones [85], a phase II clinical trial was started to explore the consequences of the association of CB-839 with a PARP inhibitor in metastatic cancer prostate cells (NCT04824937). Ferredoxin 1 (FDX1) is another targetable antioxidant system because of its key function in the mitochondrial membrane of tumor cells with an impaired ETC [110]. Resistant pancreas cancer cells were also sensitive to an FDX1-selective drug, which increased ROS levels and decreased cell viability [106]. A compensatory dual strategy targeting the GLS-associated antioxidant program might be reinforced by a ferredoxin inhibition and/or a ferroptosis activation to provide novel therapeutic tools [91]. Thus, targeting the xCT system, ferroptosis and GLS in a combination therapy, consisting of erastin + doxorubicin + CB-839, has shown promising results in TNBC, tumor cells very sensitive to CB-839 treatment, because its high reliance on Gln and elevated GLS expression [100]. Other efforts to synergistically cooperate with both GLS silencing and ROS enhancement might span MYC, and the PTEN/PI3K/mTOR axis [109], as well as xCT, GPX, and glucose transporters (GLUTs) [102]. Interestingly, disrupting both cellular redox balance and Gln availability by erastin and CB-839 has produced a positive synergistic effect in cisplatin therapy, sensitizing chemo-resistant TNBC cells [100]. These findings offer promising clinical relevance and deserve additional in-depth studies.
Author Contributions
Conceptualization, J.D.l.S.-J., J.A.C.-S. and J.M.M.; methodology, J.D.l.S.-J., J.A.C.-S. and J.M.M.; resources, J.M. and J.M.M.; writing—original draft preparation, J.D.l.S.-J., J.A.C.-S. and J.M.M.; writing—review and editing, J.D.l.S.-J., J.A.C.-S., F.J.A., J.M. and J.M.M.; project administration, J.M. and J.M.M.; funding acquisition, J.M. and J.M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministerio de Ciencia e Innovación of Spain, grant number: PID2022-140388OB-I00 (to J.M.M.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
AKG, alpha-ketoglutarate; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ATF4, activating transcription factor 4; ATP, adenosine triphosphate; BCAT1/2, branched chain amino acid aminotransferases 1 and 2; ccRCC, clear cell renal cell carcinoma; CHAC1, ChaC glutathione specific gamma-glutamylcyclotransferase 1; CSC, cancer stem cell; Cys, cysteine; EGFR, epidermal growth factor receptor; EMT, epithelial–mesenchymal transition; ESCC, esophageal squamous cell carcinoma; ETC, electron transfer chain; FADH2, flavin adenine dinucleotide; FDX1, ferredoxin 1; GA, glutaminase; GAB, longer GLS2 transcript and isoform; GAC, shorter GLS transcript and isoform; GBM, glioblastoma; GDH, glutamate dehydrogenase; Gln, glutamine; GLS, glutaminase isoenzyme; GLS2, glutaminase isoenzyme 2; Glu, glutamate; GLUT, glucose transporter; Gly, glycine; GR, glutathione reductase; GSH, reduced glutathione; GSSG; oxidized glutathione; GPX4, glutathione peroxidase 4; HCC, hepatocellular carcinoma; HIF1, hypoxia-inducible factor 1; HNSCC, head and neck cancer squamous cell carcinoma; HO-1, heme oxygenase-1; IDH1, isocitrate dehydrogenase 1; KGA, longer GLS transcript and isoform; LGA, shorter GLS2 transcript and isoform; mTOR, mammalian target of rapamycin; NADH, nicotinamide adenine dinucleotide; NADPH, nicotinamide adenine dinucleotide phosphate; NOX2, NADPH oxidase 2; NRF2, nuclear erythroid factor 2-related factor 2; NSCLC, non-small-cell lung carcinoma; OAA, oxaloacetate; OXPHOS, oxidative phosphorylation; PARP, poly(ADP-ribose) polymerase; PC, pyruvate carboxylase; PDAC, pancreatic ductal adenocarcinoma; PI3K, phosphatidylinositol-3-kinase; PTEN, phosphatase and tensin homolog; ROS, reactive oxygen species; TCA, tricarboxylic acid; TCGA, The Cancer Genome Atlas; tGSH, total glutathione; TNBC, triple-negative breast cancer.
References
- Matés, J.M.; Campos-Sandoval, J.A.; De Los Santos-Jiménez, J.; Márquez, J. Glutaminases regulate glutathione and oxidative stress in cancer. Arch. Toxicol. 2020, 94, 2603–2623. [Google Scholar] [CrossRef]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef]
- Matés, J.M.; Di Paola, F.J.; Campos-Sandoval, J.A.; Mazurek, S.; Márquez, J. Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. Semin. Cell Dev. Biol. 2020, 98, 34–43. [Google Scholar] [CrossRef]
- Palma, F.R.; Gantner, B.N.; Sakiyama, M.J.; Kayzuka, C.; Shukla, S.; Lacchini, R.; Cunniff, B.; Bonini, M.G. ROS production by mitochondria: Function or dysfunction? Oncogene 2024, 43, 295–303. [Google Scholar] [CrossRef]
- Matés, J.M.; Campos-Sandoval, J.A.; Santos-Jiménez, J.L.; Márquez, J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 2019, 467, 29–39. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Katt, W.P.; Cerione, R.A. Alone and together: Current approaches to targeting glutaminase enzymes as part of anti-cancer therapies. Future Drug Discov. 2023, 4, FDD79. [Google Scholar] [CrossRef]
- Matés, J.M.; Campos-Sandoval, J.A.; Márquez, J. Glutaminase isoenzymes in the metabolic therapy of cancer. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 158–164. [Google Scholar] [CrossRef]
- Fox, D.B.; Garcia, N.M.G.; McKinney, B.J.; Lupo, R.; Noteware, L.C.; Newcomb, R.; Liu, J.; Locasale, J.W.; Hirschey, M.D.; Alvarez, J.V. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat. Metab. 2020, 2, 318–334. [Google Scholar] [CrossRef]
- Singh, B.; Tai, K.; Madan, S.; Raythatha, M.R.; Cady, A.M.; Braunlin, M.; Irving, L.R.; Bajaj, A.; Lucci, A. Selection of metastatic breast cancer cells based on adaptability of their metabolic state. PLoS ONE 2012, 7, e36510. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Safi, R.; Liu, X.; Baldi, R.; Liu, W.; Liu, J.; Locasale, J.W.; Chang, C.Y.; McDonnell, D.P. Inhibition of ERRalpha Prevents Mitochondrial Pyruvate Uptake Exposing NADPH-Generating Pathways as Targetable Vulnerabilities in Breast Cancer. Cell Rep. 2019, 27, 3587–3601.e4. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.; Zhang, Y.; Wang, J.; Chen, Z.; Wen, T. Zeolitic imidazolate framework-based nanoparticles for the cascade enhancement of cancer chemodynamic therapy by targeting glutamine metabolism. Nanoscale 2022, 14, 8727–8743. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Owada, S.; Inagaki, Y.; Shida, Y.; Tatemichi, M. Metabolic reprogramming sustains cancer cell survival following extracellular matrix detachment. Redox Biol. 2020, 36, 101643. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, S.; Xu, C.; Hua, A.; Wang, C.; Xiong, Y.; Deng, Q.; Chen, X.; Yang, T.; Wan, J.; et al. A novel lonidamine derivative targeting mitochondria to eliminate cancer stem cells by blocking glutamine metabolism. Pharmacol. Res. 2023, 190, 106740. [Google Scholar] [CrossRef] [PubMed]
- Ornelas, A.; McCullough, C.R.; Lu, Z.; Zacharias, N.M.; Kelderhouse, L.E.; Gray, J.; Yang, H.; Engel, B.J.; Wang, Y.; Mao, W.; et al. Induction of autophagy by ARHI (DIRAS3) alters fundamental metabolic pathways in ovarian cancer models. BMC Cancer 2016, 16, 824. [Google Scholar] [CrossRef] [PubMed]
- Dorai, T.; Shah, A.; Summers, F.; Mathew, R.; Huang, J.; Hsieh, T.C.; Wu, J.M. NRH:quinone oxidoreductase 2 (NQO2) and glutaminase (GLS) both play a role in large extracellular vesicles (LEV) formation in preclinical LNCaP-C4-2B prostate cancer model of progressive metastasis. Prostate 2018, 78, 1181–1195. [Google Scholar] [CrossRef] [PubMed]
- Mukha, A.; Kahya, U.; Linge, A.; Chen, O.; Löck, S.; Lukiyanchuk, V.; Richter, S.; Alves, T.C.; Peitzsch, M.; Telychko, V.; et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. Theranostics 2021, 11, 7844–7868. [Google Scholar] [CrossRef] [PubMed]
- Aurora, A.B.; Khivansara, V.; Leach, A.; Gill, J.G.; Martin-Sandoval, M.; Yang, C.; Kasitinon, S.Y.; Bezwada, D.; Tasdogan, A.; Gu, W.; et al. Loss of glucose 6-phosphate dehydrogenase function increases oxidative stress and glutaminolysis in metastasizing melanoma cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2120617119. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Ju, R.; Gao, H.; Huang, Y.; Guo, L.; Zhang, D. Targeting glutamine utilization to block metabolic adaptation of tumor cells under the stress of carboxyamidotriazole-induced nutrients unavailability. Acta Pharm. Sin. B 2022, 12, 759–773. [Google Scholar] [CrossRef]
- Hamada, S.; Matsumoto, R.; Tanaka, Y.; Taguchi, K.; Yamamoto, M.; Masamune, A. Nrf2 Activation Sensitizes K-Ras Mutant Pancreatic Cancer Cells to Glutaminase Inhibition. Int. J. Mol. Sci. 2021, 22, 1870. [Google Scholar] [CrossRef]
- Tong, Y.; Guo, D.; Lin, S.H.; Liang, J.; Yang, D.; Ma, C.; Shao, F.; Li, M.; Yu, Q.; Jiang, Y.; et al. SUCLA2-coupled regulation of GLS succinylation and activity counteracts oxidative stress in tumor cells. Mol. Cell 2021, 81, 2303–2316.e8. [Google Scholar] [CrossRef]
- Zhang, J.; Han, Z.Q.; Wang, Y.; He, Q.Y. Alteration of mitochondrial protein succinylation against cellular oxidative stress in cancer. Mil. Med. Res. 2022, 9, 6. [Google Scholar] [CrossRef]
- Ammar, N.; Hildebrandt, M.; Geismann, C.; Röder, C.; Gemoll, T.; Sebens, S.; Trauzold, A.; Schäfer, H. Monocarboxylate Transporter-1 (MCT1)-Mediated Lactate Uptake Protects Pancreatic Adenocarcinoma Cells from Oxidative Stress during Glutamine Scarcity Thereby Promoting Resistance against Inhibitors of Glutamine Metabolism. Antioxidants 2023, 12, 1818. [Google Scholar] [CrossRef] [PubMed]
- Abu Aboud, O.; Habib, S.L.; Trott, J.; Stewart, B.; Liang, S.; Chaudhari, A.J.; Sutcliffe, J.; Weiss, R.H. Glutamine Addiction in Kidney Cancer Suppresses Oxidative Stress and Can Be Exploited for Real-Time Imaging. Cancer Res. 2017, 77, 6746–6758. [Google Scholar] [CrossRef] [PubMed]
- Teng, R.; Liu, Z.; Tang, H.; Zhang, W.; Chen, Y.; Xu, R.; Chen, L.; Song, J.; Liu, X.; Deng, H. HSP60 silencing promotes Warburg-like phenotypes and switches the mitochondrial function from ATP production to biosynthesis in ccRCC cells. Redox Biol. 2019, 24, 101218. [Google Scholar] [CrossRef]
- Tronci, L.; Caria, P.; Frau, D.V.; Liggi, S.; Piras, C.; Murgia, F.; Santoru, M.L.; Pibiri, M.; Deiana, M.; Griffin, J.L.; et al. Crosstalk between Metabolic Alterations and Altered Redox Balance in PTC-Derived Cell Lines. Metabolites 2019, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, R.; Jayachandran, K.; Zhang, J.; Menon, V.; Muhammad, N.; Zahner, M.; Ruiz, F.; Zhang, S.; Cho, K.; Wang, Y.; et al. Glutaminase Inhibitors Induce Thiol-Mediated Oxidative Stress and Radiosensitization in Treatment-Resistant Cervical Cancers. Mol. Cancer Ther. 2020, 19, 2465–2475. [Google Scholar] [CrossRef]
- Wicker, C.A.; Hunt, B.G.; Krishnan, S.; Aziz, K.; Parajuli, S.; Palackdharry, S.; Elaban, W.R.; Wise-Draper, T.M.; Mills, G.B.; Waltz, S.E.; et al. Glutaminase inhibition with telaglenastat (CB-839) improves treatment response in combination with ionizing radiation in head and neck squamous cell carcinoma models. Cancer Lett. 2021, 502, 180–188. [Google Scholar] [CrossRef]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef]
- LeBoeuf, S.E.; Wu, W.L.; Karakousi, T.R.; Karadal, B.; Jackson, S.R.; Davidson, S.M.; Wong, K.K.; Koralov, S.B.; Sayin, V.I.; Papagiannakopoulos, T. Activation of Oxidative Stress Response in Cancer Generates a Druggable Dependency on Exogenous Non-essential Amino Acids. Cell Metab. 2020, 31, 339–350.e4. [Google Scholar] [CrossRef]
- Bruntz, R.C.; Belshoff, A.C.; Zhang, Y.; Macedo, J.K.A.; Higashi, R.M.; Lane, A.N.; Fan, T.W. Inhibition of Anaplerotic Glutaminolysis Underlies Selenite Toxicity in Human Lung Cancer. Proteomics 2019, 19, e1800486. [Google Scholar] [CrossRef]
- Ulanet, D.B.; Couto, K.; Jha, A.; Choe, S.; Wang, A.; Woo, H.K.; Steadman, M.; DeLaBarre, B.; Gross, S.; Driggers, E.; et al. Mesenchymal phenotype predisposes lung cancer cells to impaired proliferation and redox stress in response to glutaminase inhibition. PLoS ONE 2014, 9, e115144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Nabel, C.S.; Li, D.; O’Connor, R.Í.; Crosby, C.R.; Chang, S.M.; Hao, Y.; Stanley, R.; Sahu, S.; Levin, D.S.; et al. Histone Deacetylase 6 Inhibition Exploits Selective Metabolic Vulnerabilities in LKB1 Mutant, KRAS Driven NSCLC. J. Thorac. Oncol. 2023, 18, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, D.; Yang, L.; Wang, N.; Shen, J.; Wang, J.; Zhang, L.; Chen, L.; Gao, S.; Zong, W.X.; et al. Activation of polyamine catabolism promotes glutamine metabolism and creates a targetable vulnerability in lung cancer. Proc. Natl. Acad. Sci. USA 2024, 121, e2319429121. [Google Scholar] [CrossRef] [PubMed]
- Moreira Franco, Y.E.; Alves, M.J.; Uno, M.; Moretti, I.F.; Trombetta-Lima, M.; de Siqueira Santos, S.; Dos Santos, A.F.; Arini, G.S.; Baptista, M.S.; Lerario, A.M.; et al. Glutaminolysis dynamics during astrocytoma progression correlates with tumor aggressiveness. Cancer Metab. 2021, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Lee, G.; Oh, T.I.; Kim, B.M.; Shim, D.W.; Lee, K.H.; Kim, Y.J.; Lim, B.O.; Lim, J.H. Inhibition of glutamine utilization sensitizes lung cancer cells to apigenin-induced apoptosis resulting from metabolic and oxidative stress. Int. J. Oncol. 2016, 48, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Martín-Rufián, M.; Nascimento-Gomes, R.; Higuero, A.; Crisma, A.R.; Campos-Sandoval, J.A.; Gómez-García, M.C.; Cardona, C.; Cheng, T.; Lobo, C.; Segura, J.A.; et al. Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells. J. Mol. Med. 2014, 92, 277–290. [Google Scholar] [CrossRef] [PubMed]
- de Los Santos-Jiménez, J.; Campos-Sandoval, J.A.; Márquez-Torres, C.; Urbano-Polo, N.; Brøndegaard, D.; Martín-Rufián, M.; Lobo, C.; Peñalver, A.; Gómez-García, M.C.; Martín-Campos, J.; et al. Glutaminase isoforms expression switches microRNA levels and oxidative status in glioblastoma cells. J. Biomed. Sci. 2021, 28, 14. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Bai, R.; Liu, Y.; Bi, H.; Shi, X.; Qu, C. Long Non-Coding RNA ATXN8OS Promotes Ferroptosis and Inhibits the Temozolomide-Resistance of Gliomas through the ADAR/GLS2 Pathway. Brain Res. Bull. 2022, 186, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Markert, E.K.; Rufini, A.; Antonov, A.V.; Sayan, B.S.; Tucci, P.; Agostini, M.; Mineo, T.C.; Levine, A.J.; Melino, G. p73 regulates serine biosynthesis in cancer. Oncogene 2014, 33, 5039–5046. [Google Scholar] [CrossRef]
- Chen, G.Y.; Chao, H.C.; Liao, H.W.; Tsai, I.L.; Kuo, C.H. Rapid quantification of glutaminase 2 (GLS2)-related metabolites by HILIC-MS/MS. Anal. Biochem. 2017, 539, 39–44. [Google Scholar] [CrossRef]
- Suzuki, S.; Venkatesh, D.; Kanda, H.; Nakayama, A.; Hosokawa, H.; Lee, E.; Miki, T.; Stockwell, B.R.; Yokote, K.; Tanaka, T.; et al. GLS2 Is a Tumor Suppressor and a Regulator of Ferroptosis in Hepatocellular Carcinoma. Cancer Res. 2022, 82, 3209–3222. [Google Scholar] [CrossRef]
- Buczkowska, J.; Szeliga, M. Two Faces of Glutaminase GLS2 in Carcinogenesis. Cancers 2023, 15, 5566. [Google Scholar] [CrossRef]
- Márquez, J.; Matés, J.M.; Alonso, F.J.; Martín-Rufián, M.; Lobo, C.; Campos-Sandoval, J.A. Canceromis studies unravel tumor’s glutamine addiction after metabolic reprogramming. In Tumor Cell Metabolism: Pathways, Regulation and Biology; Mazurek, S., Shoshan, M., Eds.; Springer Verlag: Wien, Austria, 2015; pp. 257–286. [Google Scholar]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a Novel P53 Target Gene Regulating Energy Metabolism and Antioxidant Function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-Activated Glutaminase (GLS2), a P53-Inducible Regulator of Glutamine Metabolism and Reactive Oxygen Species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef]
- Xiao, D.; Ren, P.; Su, H.; Yue, M.; Xiu, R.; Hu, Y.; Liu, H.; Qing, G. Myc Promotes Glutaminolysis in Human Neuroblastoma through Direct Activation of Glutaminase 2. Oncotarget 2015, 6, 40655–40666. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Xie, G.; Liu, C.; Zhou, J.; Chen, J.; Yu, S.; Li, J.; Pang, X.; Shi, H.; Liang, H. Knock-down of Glutaminase 2 Expression Decreases Glutathione, NADH, and Sensitizes Cervical Cancer to Ionizing Radiation. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2013, 1833, 2996–3005. [Google Scholar] [CrossRef]
- Dias, M.M.; Adamoski, D.; dos Reis, L.M.; Ascenção, C.F.R.; de Oliveira, K.R.S.; Mafra, A.C.P.; da Silva Bastos, A.C.; Quintero, M.; Cassago, C.d.G.; Ferreira, I.M.; et al. GLS2 Is Protumorigenic in Breast Cancers. Oncogene 2020, 39, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Peña, E.; Arnold, J.; Shivakumar, V.; Joseph, R.; Vidhya Vijay, G.; den Hollander, P.; Bhangre, N.; Allegakoen, P.; Prasad, R.; Conley, Z.; et al. The Epithelial to Mesenchymal Transition Promotes Glutamine Independence by Suppressing GLS2 Expression. Cancers 2019, 11, 1610. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed]
- Emadi, A.; Jun, S.A.; Tsukamoto, T.; Fathi, A.T.; Minden, M.D.; Dang, C.V. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp. Hematol. 2014, 42, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Qie, S.; Chu, C.; Li, W.; Wang, C.; Sang, N. ErbB2 activation upregulates glutaminase 1 expression which promotes breast cancer cell proliferation. J. Cell. Biochem. 2014, 115, 498–509. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Deberardinis, R.J.; Sayed, N.; Ditsworth, D.; Thompson, C.B. Brick by brick: Metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 2008, 18, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M.; Pérez-Gómez, C.; Núñez de Castro, I.; Asenjo, M.; Márquez, J. Glutamine and its relationship with intracellular redox status, oxidative stress and cell proliferation/death. Int. J. Biochem. Cell Biol. 2002, 34, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M.; Segura, J.A.; Campos-Sandoval, J.A.; Lobo, C.; Alonso, L.; Alonso, F.J.; Márquez, J. Glutamine homeostasis and mitochondrial dynamics. Int. J. Biochem. Cell Biol. 2009, 41, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.G.; Yang, M.; Prag, H.A.; Blanco, G.R.; Nikitopoulou, E.; Segarra-Mondejar, M.; Powell, C.A.; Young, T.; Burger, N.; Miljkovic, J.L.; et al. Disruption of the TCA cycle reveals an ATF4-dependent integration of redox and amino acid metabolism. Elife 2021, 10, e72593. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Alberghina, L.; Gaglio, D. Redox control of glutamine utilization in cancer. Cell Death Dis. 2014, 5, e1561. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Deberardinis, R.J. Cancer metabolism: When more is less. Nature 2012, 489, 511–512. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Sudderth, J.; Yang, C.; Mullen, A.R.; Jin, E.S.; Matés, J.M.; DeBerardinis, R.J. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8674–8679. [Google Scholar] [CrossRef]
- Mullen, A.R.; Hu, Z.; Shi, X.; Jiang, L.; Boroughs, L.K.; Kovacs, Z.; Boriack, R.; Rakheja, D.; Sullivan, L.B.; Linehan, W.M.; et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014, 7, 1679–1690. [Google Scholar] [CrossRef]
- Sellers, K.; Fox, M.P.; Bousamra, M., 2nd; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Chen, J.; Xie, Z.; Chen, Z. Wild-Type Isocitrate Dehydrogenase-Dependent Oxidative Decarboxylation and Reductive Carboxylation in Cancer and Their Clinical Significance. Cancers 2022, 14, 5779. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chen, D.; Fan, H.; Wu, R.; Tu, J.; Zhang, F.Q.; Wang, M.; Zheng, H.; Qu, C.K.; Elf, S.E.; et al. Cellular signals converge at the NOX2-SHP-2 axis to induce reductive carboxylation in cancer cells. Cell Chem. Biol. 2022, 29, 1200–1208.e6. [Google Scholar] [CrossRef]
- Itsumi, M.; Inoue, S.; Elia, A.J.; Murakami, K.; Sasaki, M.; Lind, E.F.; Brenner, D.; Harris, I.S.; Chio, I.I.C.; Afzal, S.; et al. Idh1 protects murine hepatocytes from endotoxin-induced oxidative stress by regulating the intracellular NADP(+)/NADPH ratio. Cell Death Differ. 2015, 22, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Kaelin, W.G., Jr. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e25. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Wang, Z.; Wang, G.; Wang, Q.A.; DeBerardinis, R.J.; Jiang, L. FASN deficiency induces a cytosol-to-mitochondria citrate flux to mitigate detachment-induced oxidative stress. Cell Rep. 2023, 42, 112971. [Google Scholar] [CrossRef]
- Delgir, S.; Bastami, M.; Ilkhani, K.; Safi, A.; Seif, F.; Alivand, M.R. The pathways related to glutamine metabolism, glutamine inhibitors and their implication for improving the efficiency of chemotherapy in triple-negative breast cancer. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108366. [Google Scholar] [CrossRef]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Wang, Z.; Wang, Q.A.; Chan, D.; Jiang, L. Metabolic reprogramming in the OPA1-deficient cells. Cell. Mol. Life Sci. 2022, 79, 517. [Google Scholar] [CrossRef]
- Dai, W.; Xu, L.; Yu, X.; Zhang, G.; Guo, H.; Liu, H.; Song, G.; Weng, S.; Dong, L.; Zhu, J.; et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J. Hepatol. 2020, 72, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, A.; Gameiro, P.A.; Christodoulou, D.; Laviollette, L.; Schneider, M.; Chaves, F.; Stemmer-Rachamimov, A.; Yazinski, S.A.; Lee, R.; Stephanopoulos, G.; et al. Glutaminase and poly(ADP-ribose) polymerase inhibitors suppress pyrimidine synthesis and VHL-deficient renal cancers. J. Clin. Investig. 2017, 127, 1631–1645. [Google Scholar] [CrossRef]
- Sigoillot, F.D.; Berkowski, J.A.; Sigoillot, S.M.; Kotsis, D.H.; Guy, H.I. Cell cycle-dependent regulation of pyrimidine biosynthesis. J. Biol. Chem. 2003, 278, 3403–3409. [Google Scholar] [CrossRef]
- De Los Santos-Jiménez, J.; Rosales, T.; Ko, B.; Campos-Sandoval, J.A.; Alonso, F.J.; Márquez, J.; DeBerardinis, R.J.; Matés, J.M. Metabolic Adjustments following Glutaminase Inhibition by CB-839 in Glioblastoma Cell Lines. Cancers 2023, 15, 531. [Google Scholar] [CrossRef] [PubMed]
- Motooka, Y.; Toyokuni, S. Ferroptosis As Ultimate Target of Cancer Therapy. Antioxid. Redox Signal. 2023, 39, 206–223. [Google Scholar] [CrossRef]
- Zhang, X.; Sui, S.; Wang, L.; Li, H.; Zhang, L.; Xu, S.; Zheng, X. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J. Cell. Physiol. 2020, 235, 3425–3437. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sun, S.; Xu, W.; Zhang, Y.; Yang, R.; Ma, K.; Zhang, J.; Xu, J. Piperlongumine Inhibits Thioredoxin Reductase 1 by Targeting Selenocysteine Residues and Sensitizes Cancer Cells to Erastin. Antioxidants 2022, 11, 710. [Google Scholar] [CrossRef]
- Hu, Q.; Dai, J.; Zhang, Z.; Yu, H.; Zhang, J.; Zhu, X.; Qin, Y.; Zhang, L.; Zhang, P. ASS1-Mediated Reductive Carboxylation of Cytosolic Glutamine Confers Ferroptosis Resistance in Cancer Cells. Cancer Res. 2023, 83, 1646–1665. [Google Scholar] [CrossRef]
- Jennis, M.; Kung, C.P.; Basu, S.; Budina-Kolomets, A.; Leu, J.I.; Khaku, S.; Scott, J.P.; Cai, K.Q.; Campbell, M.R.; Porter, D.K.; et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016, 30, 918–930. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.Y.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Zheng, J.; Liang, Q.; Liu, Y.; Yang, Y.; Wang, R.; Wang, M.; Zhang, Q.; Xuan, Z.; Sun, H.; et al. Identification and Validation of a Novel Ferroptotic Prognostic Genes-Based Signature of Clear Cell Renal Cell Carcinoma. Cancers 2022, 14, 4690. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Wang, S.F.; Hsu, C.Y.; Yin, P.H.; Yeh, T.S.; Lee, H.C.; Tseng, L.M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget 2017, 8, 114588. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; Morrow, J.D.; Burk, R.F. Thioredoxin reductase reduces lipid hydroperoxides and spares alpha-tocopherol. Biochem. Biophys. Res. Commun. 2002, 292, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef]
- Choi, H.; Gupta, M.; Hensley, C.; Lee, H.; Lu, Y.T.; Pantel, A.; Mankoff, D.; Zhou, R. Disruption of redox balance in glutaminolytic triple negative breast cancer by inhibition of glutamate export and glutaminase. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zeng, H.; Fan, J.; Wang, F.; Xu, C.; Li, Y.; Tu, J.; Nephew, K.P.; Long, X. Glutamine metabolism in breast cancer and possible therapeutic targets. Biochem. Pharmacol. 2023, 210, 115464. [Google Scholar] [CrossRef]
- Li, W.; Wang, Z.; Lin, R.; Huang, S.; Miao, H.; Zou, L.; Liu, K.; Cui, X.; Wang, Z.; Zhang, Y.; et al. Lithocholic acid inhibits gallbladder cancer proliferation through interfering glutaminase-mediated glutamine metabolism. Biochem. Pharmacol. 2022, 205, 115253. [Google Scholar] [CrossRef]
- Kobayashi, H.; Yoshimoto, C.; Matsubara, S.; Shigetomi, H.; Imanaka, S. A comprehensive overview of recent developments on the mechanisms and pathways of ferroptosis in cancer: The potential implications for therapeutic strategies in ovarian cancer. Cancer Drug Resist. 2023, 6, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H. Recent advances in understanding the metabolic plasticity of ovarian cancer: A systematic review. Heliyon 2022, 8, e11487. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Zhao, F.; Qi, Q.R.; Yue, B.S.; Zhai, B.T. Targeted drug delivery systems for elemene in cancer therapy: The story thus far. Biomed. Pharmacother. 2023, 166, 115331. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, X.; Zhang, R.; Liu, S.; Xiang, Y.; Zhang, M.; Chen, X.; Pan, T.; Yan, L.; Feng, J.; et al. Combinative treatment of beta-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics 2020, 10, 5107–5119. [Google Scholar] [CrossRef] [PubMed]
- Blatt, E.B.; Parra, K.; Neeb, A.; Buroni, L.; Bogdan, D.; Yuan, W.; Gao, Y.; Gilbreath, C.; Paschalis, A.; Carreira, S.; et al. Critical role of antioxidant programs in enzalutamide-resistant prostate cancer. Oncogene 2023, 42, 2347–2359. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Lee, S.-C.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.P.; et al. Mechanism of antineoplastic activity of lonidamine. BBA Rev. Cancer 2016, 1866, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Erez, I.; Issacson, C.; Lavi, Y.; Shaco-Levy, R.; Milam, J.; Laster, B.; Gheber, L.A.; Rapaport, H. Antitumor Effect of Lonidamine-Polypeptide-Peptide Nanoparticles in Breast Cancer Models. ACS Appl. Mater. Interfaces 2019, 11, 32670–32678. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.; Posadas, E.; Ellis, L.; Freedland, S.J.; Vizio, D.D.; Freeman, M.R.; Theodorescu, D.; Figlin, R.; Gong, J. Targeting Glutamine Metabolism in Prostate Cancer. Front. Biosci. 2023, 15, 2. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, Y.; Guo, X.; Du, Y.; Zhu, Q.; Wang, X.; Duan, C. FDX1 can Impact the Prognosis and Mediate the Metabolism of Lung Adenocarcinoma. Front. Pharmacol. 2021, 12, 749134. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).