

NCOA4 Regulates Iron Recycling and Responds to Hepcidin Activity and Lipopolysaccharide in Macrophages


Abstract
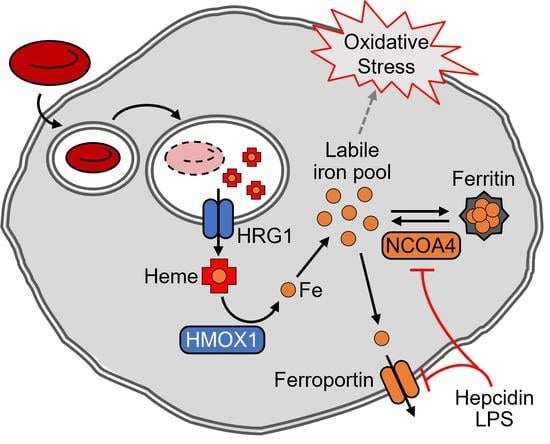
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Iron Treatments
2.2. In Vitro Erythrophagocytosis
2.3. Hepcidin Mimic (PR73) and Endotoxin Treatments
2.4. Gene Silencing by siRNA Transfection
2.5. RNA Isolation, Reverse Transcription, and qPCR
2.6. Protein Extraction and Western Blot Analysis
2.7. Cell Viability Assay
2.8. Cellular Mineral and Heme Analyses
2.9. RNA-seq and Bioinformatic Analysis of Transcriptome Data
2.10. Statistical Analyses
3. Results
3.1. Post-Transcriptional Regulation of NCOA4 by Iron and NCOA4-Dependent Ferritin Turnover in J774 Macrophages
3.2. NCOA4-Dependent Ferritin Turnover and Survival of Iron-Deficient J774 Macrophages
3.3. Cellular Heme and Non-Heme Iron Contents in J774 Macrophages after Erythrophagocytosis
3.4. Facilitated Ferritin Turnover by NCOA4 after Erythrophagocytosis
3.5. Repression of NCOA4 by Hepcidin Activity in J774 Mouse Macrophages
3.6. Enrichment of Genes in Inflammatory Pathways by NCOA4-Responsive Genes in J774 Mouse Macrophages
3.7. NCOA4 Repression in Mouse Macrophages and Spleen by LPS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Papanikolaou, G.; Pantopoulos, K. Iron Metabolism and Toxicity. Toxicol. Appl. Pharmacol. 2005, 202, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Systemic Iron Homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef] [PubMed]
- Knutson, M.; Wessling-Resnick, M. Iron Metabolism in the Reticuloendothelial System. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 61–88. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Dzieciatkowska, M.; Nemkov, T.; Hansen, K.C. Red Blood Cell Proteomics Update: Is There More to Discover? Blood Transfus. 2017, 15, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.M.; Arosio, P. The Ferritins: Molecular Properties, Iron Storage Function and Cellular Regulation. Biochim. Biophys. Acta 1996, 1275, 161–203. [Google Scholar] [CrossRef]
- Kidane, T.Z.; Sauble, E.; Linder, M.C. Release of Iron from Ferritin Requires Lysosomal Activity. Am. J. Physiol. Cell Physiol. 2006, 291, C445–C455. [Google Scholar] [CrossRef]
- Asano, T.; Komatsu, M.; Yamaguchi-Iwai, Y.; Ishikawa, F.; Mizushima, N.; Iwai, K. Distinct Mechanisms of Ferritin Delivery to Lysosomes in Iron-Depleted and Iron-Replete Cells. Mol. Cell. Biol. 2011, 31, 2040–2052. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative Proteomics Identifies NCOA4 as the Cargo Receptor Mediating Ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 Inhibitor Blocks Autophagy and Uncovers a Role for NCOA4 in Ferritin Degradation and Iron Homeostasis in Vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef]
- Knutson, M.D.; Vafa, M.R.; Haile, D.J.; Wessling-Resnick, M. Iron Loading and Erythrophagocytosis Increase Ferroportin 1 (FPN1) Expression in J774 Macrophages. Blood 2003, 102, 4191–4197. [Google Scholar] [CrossRef]
- Bellelli, R.; Federico, G.; Matte’, A.; Colecchia, D.; Iolascon, A.; Chiariello, M.; Santoro, M.; De Franceschi, L.; Carlomagno, F. NCOA4 Deficiency Impairs Systemic Iron Homeostasis. Cell Rep. 2016, 14, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Preza, G.C.; Ruchala, P.; Pinon, R.; Ramos, E.; Qiao, B.; Peralta, M.A.; Sharma, S.; Waring, A.; Ganz, T.; Nemeth, E. Minihepcidins Are Rationally Designed Small Peptides That Mimic Hepcidin Activity in Mice and May Be Useful for the Treatment of Iron Overload. J. Clin. Investig. 2011, 121, 4880–4888. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Guo, H.; Chen, X. Pentraxin 3 Regulates MiR-21 Expression and Secretion in Brown Adipocytes During Lipopolysaccharide-Induced Inflammation. Obesity 2020, 28, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Ryu, M.-S.; Zhang, D.; Protchenko, O.; Shakoury-Elizeh, M.; Philpott, C.C. PCBP1 and NCOA4 Regulate Erythroid Iron Storage and Heme Biosynthesis. J. Clin. Investig. 2017, 127, 1786–1797. [Google Scholar] [CrossRef]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 Is Essential for Heme Transport from the Phagolysosome of Macrophages during Erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A Tool to Design Target-Specific Primers for Polymerase Chain Reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Yook, J.-S.; You, M.; Kim, J.; Toney, A.M.; Fan, R.; Puniya, B.L.; Helikar, T.; Vaulont, S.; Deschemin, J.-C.; Okla, M.; et al. Essential Role of Systemic Iron Mobilization and Redistribution for Adaptive Thermogenesis through HIF2-α/Hepcidin Axis. Proc. Natl. Acad. Sci. USA 2021, 118, e2109186118. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. G:Profiler: A Web Server for Functional Enrichment Analysis and Conversions of Gene Lists (2019 Update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and Interpreting Genome Data at the Network Level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Bernuzzi, F.; Recalcati, S. A Precious Metal: Iron, an Essential Nutrient for All Cells. Genes Nutr. 2006, 1, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef] [PubMed]
- Winn, N.C.; Volk, K.M.; Hasty, A.H. Regulation of Tissue Iron Homeostasis: The Macrophage “Ferrostat”. JCI Insight 2020, 5, e132964. [Google Scholar] [CrossRef]
- Fung, E.; Chua, K.; Ganz, T.; Nemeth, E.; Ruchala, P. Thiol-Derivatized Minihepcidins Retain Biological Activity. Bioorg. Med. Chem. Lett. 2015, 25, 763–766. [Google Scholar] [CrossRef]
- Corna, G.; Campana, L.; Pignatti, E.; Castiglioni, A.; Tagliafico, E.; Bosurgi, L.; Campanella, A.; Brunelli, S.; Manfredi, A.A.; Apostoli, P.; et al. Polarization Dictates Iron Handling by Inflammatory and Alternatively Activated Macrophages. Haematologica 2010, 95, 1814–1822. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Ornatowska, M.; Azim, A.C.; Wang, X.; Christman, J.W.; Xiao, L.; Joo, M.; Sadikot, R.T. Functional Genomics of Silencing TREM-1 on TLR4 Signaling in Macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1377–L1384. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Iron Homeostasis in Host Defence and Inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef]
- Ryu, M.-S.; Duck, K.A.; Philpott, C.C. Ferritin Iron Regulators, PCBP1 and NCOA4, Respond to Cellular Iron Status in Developing Red Cells. Blood Cells. Mol. Dis. 2018, 69, 75–81. [Google Scholar] [CrossRef]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 Is Required for Erythropoiesis and Is Regulated by Iron Dependent HERC2-Mediated Proteolysis. eLife 2015, 4, e10308. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, Y.; Truman, M.; Cohen, L.A.; Leichtmann-Bardoogo, Y.; Meyron-Holtz, E.G. Endoplasmic Reticulum Anchored Heme-Oxygenase 1 Faces the Cytosol. Haematologica 2012, 97, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Gableske, S.; del Rey, M.Q.; Małachowska, B.; Jedrychowski, M.P.; Biancur, D.E.; Schmidt, P.J.; Fleming, M.D.; Fendler, W.; Harper, J.W.; et al. NCOA4 Maintains Murine Erythropoiesis via Cell Autonomous and Non-Autonomous Mechanisms. Haematologica 2019, 104, 1342–1354. [Google Scholar] [CrossRef] [PubMed]
- Nai, A.; Lidonnici, M.R.; Federico, G.; Pettinato, M.; Olivari, V.; Carrillo, F.; Geninatti Crich, S.; Ferrari, G.; Camaschella, C.; Silvestri, L.; et al. NCOA4-Mediated Ferritinophagy in Macrophages Is Crucial to Sustain Erythropoiesis in Mice. Haematologica 2020, 106, 795–805. [Google Scholar] [CrossRef]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A Cytosolic Iron Chaperone That Delivers Iron to Ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef]
- Schwartz, A.J.; Das, N.K.; Ramakrishnan, S.K.; Jain, C.; Jurkovic, M.T.; Wu, J.; Nemeth, E.; Lakhal-Littleton, S.; Colacino, J.A.; Shah, Y.M. Hepatic Hepcidin/Intestinal HIF-2α Axis Maintains Iron Absorption during Iron Deficiency and Overload. J. Clin. Investig. 2019, 129, 336–348. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Núñez, G.; Sakamoto, K.; Soares, M.P. Innate Nutritional Immunity. J. Immunol. 2018, 201, 11–18. [Google Scholar] [CrossRef]
- Nairz, M.; Ferring-Appel, D.; Casarrubea, D.; Sonnweber, T.; Viatte, L.; Schroll, A.; Haschka, D.; Fang, F.C.; Hentze, M.W.; Weiss, G.; et al. Iron Regulatory Proteins Mediate Host Resistance to Salmonella Infection. Cell Host Microbe 2015, 18, 254–261. [Google Scholar] [CrossRef]
- Miller, L.L.; Miller, S.C.; Torti, S.V.; Tsuji, Y.; Torti, F.M. Iron-Independent Induction of Ferritin H Chain by Tumor Necrosis Factor. Proc. Natl. Acad. Sci. USA 1991, 88, 4946–4950. [Google Scholar] [CrossRef]
- Mesquita, G.; Silva, T.; Gomes, A.C.; Oliveira, P.F.; Alves, M.G.; Fernandes, R.; Almeida, A.A.; Moreira, A.C.; Gomes, M.S. H-Ferritin Is Essential for Macrophages’ Capacity to Store or Detoxify Exogenously Added Iron. Sci. Rep. 2020, 10, 3061. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Yewdall, S.J.; Harrison, P.; Santambrogio, P.; Cozzi, A.; Rovida, E.; Albertini, A.; Arosio, P. Evidence That H- and L-Chains Have Co-Operative Roles in the Iron-Uptake Mechanism of Human Ferritin. Biochem. J. 1992, 288, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Madu, A.J.; Ughasoro, M.D. Anaemia of Chronic Disease: An In-Depth Review. Med. Princ. Pract. 2017, 26, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.R.; Adams, P.C.; Kowdley, K.V.; Powell, L.W.; Tavill, A.S. Diagnosis and Management of Hemochromatosis: 2011 Practice Guideline by the American Association for the Study of Liver Diseases. Hepatology 2011, 54, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Bilanges, B.; Vanhaesebroeck, B. Cinderella Finds Her Shoe: The First Vps34 Inhibitor Uncovers a New PI3K–AGC Protein Kinase Connection. Biochem. J. 2014, 464, e7–e10. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript | Primer Set | Sequence |
|---|---|---|
| Ncoa4 | Forward | 5’-AGCTAAGGCACCCAAGGCTA-3′ |
| Reverse | 5’-CTTAGGGCCTCCTTTGCACG-3′ | |
| Tfrc | Forward | 5′-TCACTTCCTGTCGCCCTATGT-3′ |
| Reverse | 5′-AGAGTGTGAGAGCCAGAGCC-3′ | |
| Fth1 | Forward | 5′-CCACGTGACCAACTTACGCA-3′ |
| Reverse | 5′-TCTCATCACCGTGTCCCAGG-3′ | |
| Ftl1 | Forward | 5′-GGAGCGTCTCCTCGAGTTTC-3′ |
| Reverse | 5’-GAGATGGCTTCTGCACATCCT-3′ | |
| Ptgs2 | Forward | 5’-GCTCAGCCAGGCAGCAAATC-3′ |
| Reverse | 5’-AGTCCGGGTACAGTCACACTT-3′ | |
| Il6 | Forward | 5’-CTCGGCAAACCTACTGCGTT-3′ |
| Reverse | 5’-TGACCACAGTGAGGAATGTCCA-3′ | |
| Actb | Forward | 5’-AGGAGTACGATGAGTCCGGC-3′ |
| Reverse | 5’-AGCTCAGTAACAGTCCGCCT-3′ | |
| Tbp | Forward | 5’-AGTTGTGCAGAAGTTGGGCT-3′ |
| Reverse | 5’-TACTGAACTGCTGGTGGGTCA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guggisberg, C.A.; Kim, J.; Lee, J.; Chen, X.; Ryu, M.-S. NCOA4 Regulates Iron Recycling and Responds to Hepcidin Activity and Lipopolysaccharide in Macrophages. Antioxidants 2022, 11, 1926. https://doi.org/10.3390/antiox11101926
Guggisberg CA, Kim J, Lee J, Chen X, Ryu M-S. NCOA4 Regulates Iron Recycling and Responds to Hepcidin Activity and Lipopolysaccharide in Macrophages. Antioxidants. 2022; 11(10):1926. https://doi.org/10.3390/antiox11101926
Chicago/Turabian StyleGuggisberg, Cole A., Juyoung Kim, Jaekwon Lee, Xiaoli Chen, and Moon-Suhn Ryu. 2022. "NCOA4 Regulates Iron Recycling and Responds to Hepcidin Activity and Lipopolysaccharide in Macrophages" Antioxidants 11, no. 10: 1926. https://doi.org/10.3390/antiox11101926
APA StyleGuggisberg, C. A., Kim, J., Lee, J., Chen, X., & Ryu, M.-S. (2022). NCOA4 Regulates Iron Recycling and Responds to Hepcidin Activity and Lipopolysaccharide in Macrophages. Antioxidants, 11(10), 1926. https://doi.org/10.3390/antiox11101926