
Redox Active Antimicrobial Peptides in Controlling Growth of Microorganisms at Body Barriers

 , , , , and
, , , , and
Abstract
1. Introduction
2. Redox Sensitive AMPs in Skin and Intestine and Mechanisms of Their Redox-Regulated Activities against Microbes

{kind=link}
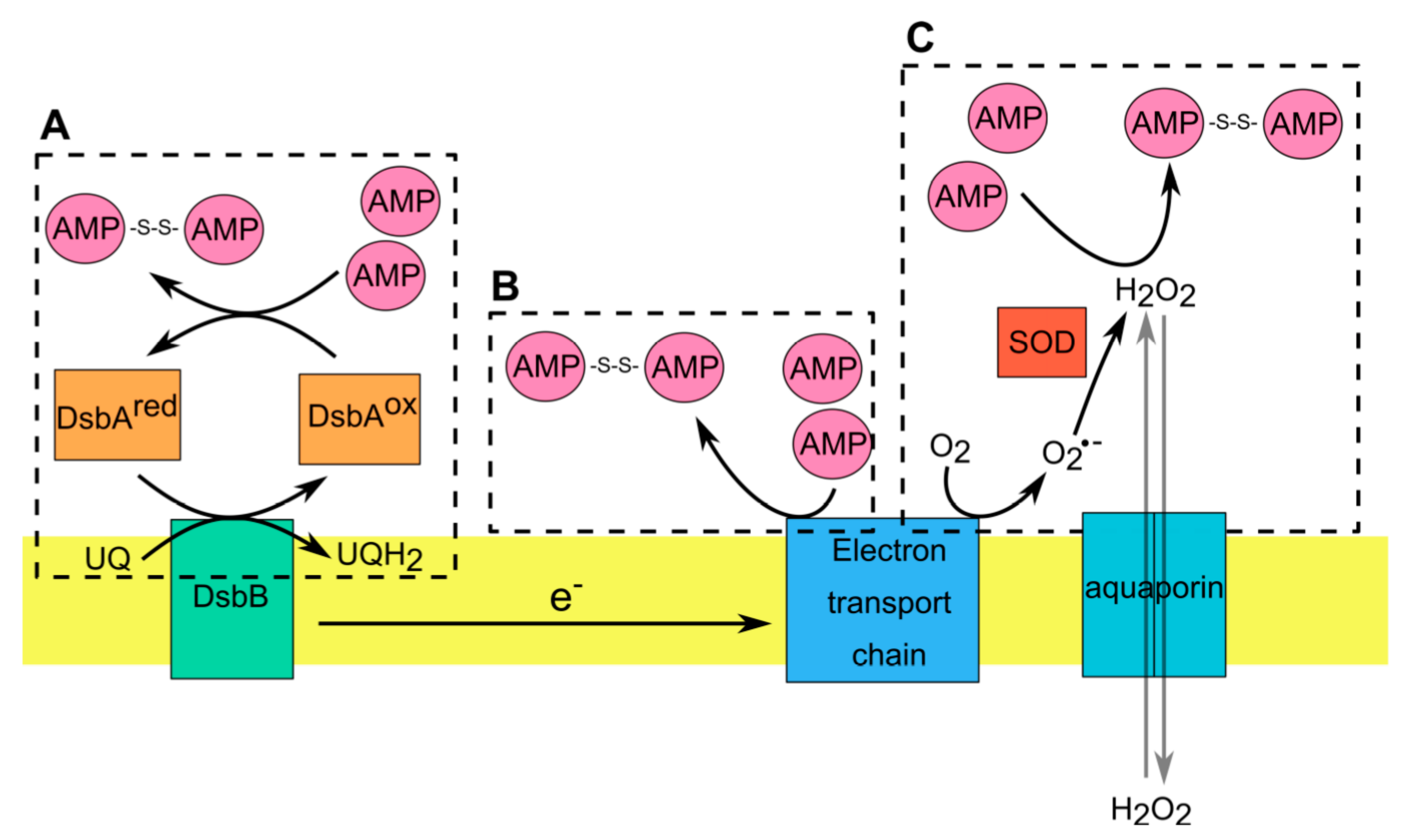{kind=link}
{kind=link}
| AMP | Gene | Expression Sites | Killing Mechanism | Oxidized Form | Reduced Form | Ref |
|---|---|---|---|---|---|---|
| hBD1 | DEFB1 | Keratinocytes; enterocytes, colonocytes (constitutive) | membrane lysis, forms oligomeric structure in reduced form | High activity against E. coli low against B. adolescentis, B. breve, L. acidophilus | High activity against C. albicans, B. adolescentis, B. breve, L. acidophilus; | [3,4,10] |
| hBD2 | DEFB4A | Keratinocytes; enterocytes, colonocytes Skin; respiratory tract (all induced) | membrane lysis No known oligomerization-related activity | High activity, Gram-negative bacteria (i.e., A. baumannii, P. aeruginosa, E. coli, K. pneumoniae, P. mirabilis) Gram-positive bacteria (i.e., E. faecalis, E. faecium, S. aureus) | Low activity | [21] |
| hBD3 | DEFB10 3B | Keratinocytes; enterocytes, colonocytes (all induced) | membrane lysis, inhibition of cell wall synthesis (target: lipid II) | High activity (B. breve); | No significant change in antimicrobial activity, diminished chemotactic activity | [8,10,22] |
| HD6 | DEFA6 | Paneth cells (small intestine) (constitutive) | reduced form: changes in bacterial cell envelope and disintegration of cytoplasmic structures, forms oligomeric structures | Low/no activity (against E.coli, L. acidophilus) | High activity (B. adolescentis, B. breve, L. acidophilus, S. thermophilus) | [3,23] |
| HNP1-3 | DEFA1-DEFA3 | Neutrophils, primary granules, (constitutive) | HNP1: membrane lysis, inhibition of cell wall synthesis (target: lipid II) HNP2: membrane disruption, aggregation and fusion of vesicles HNP3: membrane disruption, pore formation | High activity (E. coli, S. salivarius) | Low activity (E. coli, S. salivarius) | [23,24] |
| HNP4 | DEFA4 | Neutrophils, primary granules, constitutive | alters membrane permeabilization | Low activity (B. adolescentis, L. acidophilus) | High activity (B. adolescentis, L. acidophilus) | [24,25] |
| S100 calcium binding protein A7 (Psoriasin) | S100A7 | Keratinocytes (constitutive at low level);proximal digestive tract (constitutive and induced) | membrane permeabilization (pH dependent), Zn2+ sequestration | High activity against E.coli; lower activity against P. aeruginosa, S. aureus, S. epidermidis and fungi (T. rubrum) | Broad spectrum antifungal activity (i.e., T. rubrum, A. fumigatus, T. mentagrophytes). High activity against E.coli | [2,20,26,27,28] |
| Chemerin (p4) | RARRES2 | Keratinocytes (constitutive); liver; adipose tissue (constitutive) | rapid damage and degradation of cell membrane; targets bacterial electron transport chain | High activity (homodimers) (E. coli, S. aureus) | Low activity (monomer) (E. coli, S. aureus) | [29,30] |
3. Redox Ecosystems of the Skin and Intestine in the Context of AMP Bactericidal Activity
4. Redox Pathways Involved in Reciprocal Interactions between Bacteria and AMPs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kwiecien, K.; Zegar, A.; Jung, J.; Brzoza, P.; Kwitniewski, M.; Godlewska, U.; Grygier, B.; Kwiecinska, P.; Morytko, A.; Cichy, J. Architecture of antimicrobial skin defense. Cytokine Growth Factor Rev. 2019, 49, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Hein, K.Z.; Takahashi, H.; Tsumori, T.; Yasui, Y.; Nanjoh, Y.; Toga, T.; Wu, Z.; Grötzinger, J.; Jung, S.; Wehkamp, J.; et al. Disulphide-reduced psoriasin is a human apoptosis-inducing broad-spectrum fungicide. Proc. Natl. Acad. Sci. USA 2015, 112, 13039–13044. [Google Scholar] [CrossRef]
- Raschig, J.; Mailänder-Sánchez, D.; Berscheid, A.; Berger, J.; Strömstedt, A.A.; Courth, L.F.; Malek, N.P.; Brötz-Oesterhelt, H.; Wehkamp, J. Ubiquitously expressed Human Beta Defensin 1 (hBD1) forms bacteria-entrapping nets in a redox dependent mode of action. PLoS Pathog. 2017, 13, e1006261. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O.; Wu, Z.; Nuding, S.; Groscurth, S.; Marcinowski, M.; Beisner, J.; Buchner, J.; Schaller, M.; Stange, E.F.; Wehkamp, J. Reduction of disulphide bonds unmasks potent antimicrobial activity of human β-defensin 1. Nat. Cell Biol. 2011, 469, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef]
- Wang, G. Structures of Human Host Defense Cathelicidin LL-37 and Its Smallest Antimicrobial Peptide KR-12 in Lipid Micelles. J. Biol. Chem. 2008, 283, 32637–32643. [Google Scholar] [CrossRef]
- Schibli, D.J.; Hunter, H.N.; Aseyev, V.; Starner, T.D.; Wiencek, J.M.; McCray, P.B., Jr.; Tack, B.F.; Vogel, H.J. The Solution Structures of the Human β-Defensins Lead to a Better Understanding of the Potent Bactericidal Activity of HBD3 against Staphylococcus aureus. J. Biol. Chem. 2002, 277, 8279–8289. [Google Scholar] [CrossRef] [PubMed]
- Uteng, M.; Hauge, H.H.; Markwick, P.R.L.; Fimland, G.; Mantzilas, D.; Nissen-Meyer, J.; Muhle-Goll, C. Three-Dimensional Structure in Lipid Micelles of the Pediocin-like Antimicrobial Peptide Sakacin P and a Sakacin P Variant That Is Structurally Stabilized by an Inserted C-Terminal Disulfide Bridge †. Biochemestry 2003, 42, 11417–11426. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.; Barran, P.E.; Dorin, J.R. Structure-activity relationships in β-defensin peptides. Biopolymers 2007, 90, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Malmsten, M. Interactions of Antimicrobial Peptides with Bacterial Membranes and Membrane Components. Curr. Top. Med. Chem. 2015, 16, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y.; Hauger, R.L.; Grigoriadis, D.E.; Dallman, M.F.; Plotsky, P.M.; Vale, W.W.; Dautzenberg, F.M. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Mavri, J.; Vogel, H.J. Ion pair formation of phosphorylated amino acids and lysine and arginine side chains: A theoretical study. Proteins 1996, 24, 495–501. [Google Scholar] [CrossRef]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of Hydrophobicity and Charge Distribution of Cationic Antimicrobial Peptides in Peptide-Membrane Interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef]
- Harris, F.; Dennison, S.R.; Phoenix, D.A. Anionic Antimicrobial Peptides from Eukaryotic Organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Levine, R.L. Protein Oxidation. Ann. N. Y. Acad. Sci. 2006, 899, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; Duran, M.; Loupatty, F.J. Enzymology of the branched-chain amino acid oxidation disorders: The valine pathway. J. Inherit. Metab. Dis. 2010, 35, 5–12. [Google Scholar] [CrossRef]
- Janssen-Heininger, Y.M.; Mossman, B.T.; Heintz, N.H.; Forman, H.J.; Kalyanaraman, B.; Finkel, T.; Stamler, J.S.; Rhee, S.G.; van der Vliet, A. Redox-based regulation of signal transduction: Principles, pitfalls, and promises. Free. Radic. Biol. Med. 2008, 45, 1–17. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free. Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef]
- Lee, R.L.E.K.C.; Eckert, R.L. S100A7 (Psoriasin)—Mechanism of Antibacterial Action in Wounds. J. Investig. Dermatol. 2007, 127, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Routsias, J.G.; Karagounis, P.; Parvulesku, G.; Legakis, N.J.; Tsakris, A. In vitro bactericidal activity of human β-defensin 2 against nosocomial strains. Pept. 2010, 31, 1654–1660. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Hoover, D.M.; Yang, D.; Boulègue, C.; Santamaria, F.; Oppenheim, J.J.; Lubkowski, J.; Lu, W. Engineering disulfide bridges to dissect antimicrobial and chemotactic activities of human -defensin 3. Proc. Natl. Acad. Sci. USA 2003, 100, 8880–8885. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Ehmann, D.; Precht, J.C.; Castillo, P.A.; Küchler, R.; Berger, J.; Schaller, M.; Stange, E.F.; Wehkamp, J. Paneth cell α-defensin 6 (HD-6) is an antimicrobial peptide. Mucosal Immunol. 2015, 8, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Ericksen, B.; Wu, Z.; Lu, W.; Lehrer, R.I. Antibacterial activity and specificity of the six human {alpha}-defensins. Antimicrob. Agents Chemother. 2005, 49, 269–275. [Google Scholar] [CrossRef]
- Szyk, A.; Wu, Z.; Tucker, K.; Yang, D.; Lu, W.; Lubkowski, J. Crystal structures of human α-defensins HNP4, HD5, and HD6. Protein Sci. 2006, 15, 2749–2760. [Google Scholar] [CrossRef]
- Cunden, L.S.; Brophy, M.B.; Rodriguez, G.E.; Flaxman, H.A.; Nolan, E.M. Biochemical and Functional Evaluation of the Intramolecular Disulfide Bonds in the Zinc-Chelating Antimicrobial Protein Human S100A7 (Psoriasin). Biochemestry 2017, 56, 5726–5738. [Google Scholar] [CrossRef]
- Gläser, R.; Harder, J.; Lange, H.; Bartels, J.; Christophers, E.; Schröder, J.-M. Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat. Immunol. 2004, 6, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Michalek, M.; Gelhaus, C.; Hecht, O.; Podschun, R.; Schröder, J.M.; Leippe, M.; Grötzinger, J. The human antimicrobial protein psoriasin acts by permeabilization of bacterial membranes. Dev. Comp. Immunol. 2009, 33, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Banas, M.; Zabieglo, K.; Kasetty, G.; Kapinska-Mrowiecka, M.; Borowczyk, J.; Drukala, J.; Murzyn, K.; Zabel, B.A.; Butcher, E.C.; Schroeder, J.M.; et al. Chemerin Is an Antimicrobial Agent in Human Epidermis. PLoS ONE 2013, 8, e58709. [Google Scholar] [CrossRef]
- Godlewska, U.; Bilska, B.; Zegar, A.; Brzoza, P.; Borek, A.; Murzyn, K.; Bochenska, O.; Morytko, A.; Kuleta, P.; Kozik, A.; et al. The antimicrobial activity of chemerin-derived peptide p4 requires oxidative conditions. J. Biol. Chem. 2019, 294, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Banas, M.; Zegar, A.; Kwitniewski, M.; Zabieglo, K.; Marczynska, J.; Kapinska-Mrowiecka, M.; LaJevic, M.; Zabel, B.A.; Cichy, J. The Expression and Regulation of Chemerin in the Epidermis. PLoS ONE 2015, 10, e0117830. [Google Scholar] [CrossRef]
- Kulig, P.; Kantyka, T.; Zabel, B.A.; Banaś, M.; Chyra, A.; Stefańska, A.; Tu, H.; Allen, S.J.; Handel, T.M.; Kozik, A.; et al. Regulation of Chemerin Chemoattractant and Antibacterial Activity by Human Cysteine Cathepsins. J. Immunol. 2011, 187, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Pazgier, M.; Jung, G.; Nuccio, S.-P.; Castillo, P.A.; De Jong, M.F.; Winter, M.G.; Winter, S.E.; Wehkamp, J.; Shen, B.; et al. Human -Defensin 6 Promotes Mucosal Innate Immunity Through Self-Assembled Peptide Nanonets. Science 2012, 337, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.; Schröder, J.-M. RNase 7, a Novel Innate Immune Defense Antimicrobial Protein of Healthy Human Skin. J. Biol. Chem. 2002, 277, 46779–46784. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-C.; Lin, Y.-M.; Chang, T.-W.; Wu, S.-J.; Lee, Y.-S.; Chang, M.D.-T.; Chen, C.; Liao, Y.-D. The Flexible and Clustered Lysine Residues of Human Ribonuclease 7 Are Critical for Membrane Permeability and Antimicrobial Activity. J. Biol. Chem. 2007, 282, 4626–4633. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.D.; Schwaderer, A.L.; Eichler, T.; Wang, H.; Kline, J.; Justice, S.S.; Cohen, D.M.; Hains, D.S. An endogenous ribonuclease inhibitor regulates the antimicrobial activity of ribonuclease 7 in the human urinary tract. Kidney Int. 2014, 85, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Abtin, A.; Eckhart, L.; Mildner, M.; Ghannadan, M.; Harder, J.; Schröder, J.M.; Tschachler, E. Degradation by Stratum Corneum Proteases Prevents Endogenous RNase Inhibitor from Blocking Antimicrobial Activities of RNase 5 and RNase 7. J. Investig. Dermatol. 2009, 129, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- Dressel, S.; Harder, J.; Cordes, J.; Wittersheim, M.; Sunderkötter, C.; Gläser, R.; Meyer-Hoffert, U. Differential expression of antimicrobial peptides in margins of chronic wounds. Exp. Dermatol. 2010, 19, 628–632. [Google Scholar] [CrossRef]
- Moenner, M.; Vosoghi, M.; Ryazantsev, S.; Glitz, D.G. Ribonuclease Inhibitor Protein of Human Erythrocytes: Characterization, Loss of Activity in Response to Oxidative Stress, and Association with Heinz Bodies. Blood Cells Mol. Dis. 1998, 24, 149–164. [Google Scholar] [CrossRef]
- Blázquez, M.; Fominaya, J.M.; Hofsteenge, J. Oxidation of Sulfhydryl Groups of Ribonuclease Inhibitor in Epithelial Cells Is Sufficient for Its Intracellular Degradation. J. Biol. Chem. 1996, 271, 18638–18642. [Google Scholar] [CrossRef]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968–983.e24. [Google Scholar] [CrossRef]
- Evans, N.; Naylor, P. The systemic oxygen supply to the surface of human skin. Respir. Physiol. 1967, 3, 21–37. [Google Scholar] [CrossRef]
- Stewart, F.A.; Denekamp, J.; Randhawa, V.S. Skin sensitization by misonidazole: A demonstration of uniform mild hypoxia. Br. J. Cancer 1982, 45, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Stücker, M.; Struk, A.; Altmeyer, P.; Herde, M.; Baumgärtl, H.; Lübbers, D.W. The cutaneous uptake of atmospheric oxygen contributes significantly to the oxygen supply of human dermis and epidermis. J. Physiol. 2002, 538, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.M.; Schrlau, A.E.; Chalian, A.A.; Zhang, P.; Koch, C.J. Oxygen Levels in Normal and Previously Irradiated Human Skin as Assessed by EF5 Binding. J. Investig. Dermatol. 2006, 126, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Shirshin, E.A.; Gurfinkel, Y.I.; Priezzhev, A.V.; Fadeev, V.V.; Lademann, J.; Darvin, M.E. Two-photon autofluorescence lifetime imaging of human skin papillary dermis in vivo: Assessment of blood capillaries and structural proteins localization. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Boutin, A.T.; Zinkernagel, A.S.; Datta, V.; Nizet, V.; Johnson, R.S. Critical Role of HIF-1α in Keratinocyte Defense against Bacterial Infection. J. Investig. Dermatol. 2008, 128, 1964–1968. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Ahuir, A.; Manzanares-Estreder, S.; Proft, M. Pro- and Antioxidant Functions of the Peroxisome-Mitochondria Connection and Its Impact on Aging and Disease. Oxidative Med. Cell. Longev. 2017, 2017, 1–17. [Google Scholar] [CrossRef]
- Albrecht, S.; Elpelt, A.; Kasim, C.; Reble, C.; Mundhenk, L.; Pischon, H.; Hedtrich, S.; Witzel, C.; Lademann, J.; Zastrow, L.; et al. Quantification and characterization of radical production in human, animal and 3D skin models during sun irradiation measured by EPR spectroscopy. Free. Radic. Biol. Med. 2019, 131, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Bossi, O.; Gartsbein, M.; Leitges, M.; Kuroki, T.; Grossman, S.; Tennenbaum, T. UV irradiation increases ROS production via PKCδ signaling in primary murine fibroblasts. J. Cell. Biochem. 2008, 105, 194–207. [Google Scholar] [CrossRef]
- Mena, S.; Ortega, A.; Estrela, J.M. Oxidative stress in environmental-induced carcinogenesis. Mutat. Res. Toxicol. Environ. Mutagen. 2009, 674, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Tanaka, R.; Okuda, Y.; Ikota, N.; Yamamoto, H.; Urano, S.; Ozawa, T.; Anzai, K. Quantitative Measurements of Oxidative Stress in Mouse Skin Induced by X-Ray Irradiation. Chem. Pharm. Bull. 2005, 53, 1411–1415. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grange, P.A.; Chéreau, C.; Raingeaud, J.; Nicco, C.; Weill, B.; Dupin, N.; Batteux, F. Production of Superoxide Anions by Keratinocytes Initiates P. acnes-Induced Inflammation of the Skin. PLoS Pathog. 2009, 5, e1000527. [Google Scholar] [CrossRef]
- Thiele, J.; Schroeter, C.; Hsieh, S.; Podda, M.; Packer, L. The Antioxidant Network of the Stratum corneum. Sex. Transm. Dis. Adv. Diagn. Treat. 2000, 29, 26–42. [Google Scholar] [CrossRef]
- Shindo, Y.; Witt, E.; Han, D.; Epstein, W.; Packer, L. Enzymic and Non-Enzymic Antioxidants in Epidermis and Dermis of Human Skin. J. Investig. Dermatol. 1994, 102, 122–124. [Google Scholar] [CrossRef]
- Hellemans, L.; Corstjens, H.; Neven, A.; Declercq, L.; Maes, D. Antioxidant Enzyme Activity in Human Stratum Corneum Shows Seasonal Variation with an Age-Dependent Recovery. J. Investig. Dermatol. 2003, 120, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S.J.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. JBIC J. Biol. Inorg. Chem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Van Der Flier, L.G.; Clevers, H. Stem Cells, Self-Renewal, and Differentiation in the Intestinal Epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef]
- Bevins, C.L.; Salzman, N.H. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Genet. 2011, 9, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.C.; Bevins, C.L. Paneth Cells: Maestros of the Small Intestinal Crypts. Annu. Rev. Physiol. 2013, 75, 289–311. [Google Scholar] [CrossRef]
- Willson, M. Microbial Inhabitants of Humans: Their Ecology and Role in Health and Disease; Cambridge University Press: Cambridge, UK, 2005; p. 476. [Google Scholar]
- Aw, T.Y. Biliary glutathione promotes the mucosal metabolism of luminal peroxidized lipids by rat small intestine in vivo. J. Clin. Investig. 1994, 94, 1218–1225. [Google Scholar] [CrossRef]
- Dahm, L.J.; Jones, D.P. Rat Jejunum Controls Luminal Thiol-Disulfide Redox. J. Nutr. 2000, 130, 2739–2745. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Redox biology of the intestine. Free. Radic. Res. 2011, 45, 1245–1266. [Google Scholar] [CrossRef]
- Takaishi, S.; Sawada, M.; Seno, H.; Kayahara, T.; Morita-Fujisawa, Y.; Fukuzawa, H.; Chiba, T. Growth promoting effect of thioredoxin on intestinal epithelial cells. Dig. Dis. Sci. 2003, 48, 379–385. [Google Scholar] [CrossRef]
- Motohashi, K.; Romano, P.G.N.; Hisabori, T. Identification of Thioredoxin Targeted Proteins Using Thioredoxin Single-Cysteine Mutant-Immobilized Resin. Methods Mol. Biol. 2009, 479, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.; Go, Y.-M.; Jones, D.P. Nonequilibrium thermodynamics of thiol/disulfide redox systems: A perspective on redox systems biology. Free. Radic. Biol. Med. 2008, 44, 921–937. [Google Scholar] [CrossRef]
- Gasdaska, J.R.; Gasdaska, P.Y.; Gallegos, A.; Powis, G. Human Thioredoxin Reductase Gene Localization to Chromosomal Position 12q23–q24.1 and mRNA Distribution in Human Tissue. Genome 1996, 37, 257–259. [Google Scholar] [CrossRef]
- Jaeger, S.U.; Schroeder, B.O.; Meyer-Hoffert, U.; Courth, L.; Fehr, S.N.; Gersemann, M.; Stange, E.F.; Wehkamp, J. Cell-mediated reduction of human β-defensin 1: A major role for mucosal thioredoxin. Mucosal Immunol. 2013, 6, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Kelly, C.J.; Colgan, S.P. Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Physiol. 2015, 309, C350–C360. [Google Scholar] [CrossRef] [PubMed]
- Schaible, B.; Schaffer, K.; Taylor, C.T. Hypoxia, innate immunity and infection in the lung. Respir. Physiol. Neurobiol. 2010, 174, 235–243. [Google Scholar] [CrossRef]
- Kelly, C.J.; Glover, L.E.; Campbell, E.L.; Kominsky, D.J.; Ehrentraut, S.F.; Bowers, B.E.; Bayless, A.J.; Saeedi, B.J.; Colgan, S.P. Fundamental role for HIF-1α in constitutive expression of human β defensin-1. Mucosal Immunol. 2013, 6, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Cook, G.M.; Greening, C.; Hards, K.; Berney, M. Energetics of Pathogenic Bacteria and Opportunities for Drug Development. Adv. Microb. Physiol. 2014, 65, 1–62. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Genet. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Godlewska, U.; Bilska, B.; Majewski, P.; Pyza, E.; Zabel, B.A.; Cichy, J. Bacteria Modify Their Sensitivity to Chemerin-Derived Peptides by Hindering Peptide Association with the Cell Surface and Peptide Oxidation. Front. Microbiol. 2020, 11, 1819. [Google Scholar] [CrossRef]
- Engstrom, G.; Rajagukguk, R.; Saunders, A.J.; Patel, C.N.; Rajagukguk, S.; Merbitz-Zahradnik, T.; Xiao, K.; Pielak, G.J.; Trumpower, B.; Yu, C.-A.; et al. Design of a Ruthenium-Labeled CytochromecDerivative to Study Electron Transfer with the Cytochromebc1Complex †. Biochemestry 2003, 42, 2816–2824. [Google Scholar] [CrossRef]
- Sarewicz, M.; Borek, A.; Daldal, F.; Froncisz, W.; Osyczka, A. Demonstration of Short-lived Complexes of Cytochrome c with Cytochrome bc1 by EPR Spectroscopy. J. Biol. Chem. 2008, 283, 24826–24836. [Google Scholar] [CrossRef]
- Unden, G.; Bongaerts, J. Alternative respiratory pathways of Escherichia coli: Energetics and transcriptional regulation in response to electron acceptors. Biochim. Biophys. Acta (BBA) Bioenerg. 1997, 1320, 217–234. [Google Scholar] [CrossRef]
- Ezraty, B.; Gennaris, A.; Barras, F.; Collet, J.-F. Oxidative stress, protein damage and repair in bacteria. Nat. Rev. Microbiol. 2017, 15, 385–396. [Google Scholar] [CrossRef]
- Imlay, J.A. The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nat. Rev. Genet. 2013, 11, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.; Imlay, J.A. Two sources of endogenous hydrogen peroxide inEscherichia coli. Mol. Microbiol. 2010, 75, 1389–1401. [Google Scholar] [CrossRef] [PubMed]
- McBee, M.E.; Chionh, Y.H.; Sharaf, M.L.; Ho, P.; Cai, M.W.L.; Dedon, P.C. Production of Superoxide in Bacteria Is Stress- and Cell State-Dependent: A Gating-Optimized Flow Cytometry Method that Minimizes ROS Measurement Artifacts with Fluorescent Dyes. Front. Microbiol. 2017, 8, 459. [Google Scholar] [CrossRef] [PubMed]
- Najmuldeen, H.; Alghamdi, R.; Alghofaili, F.; Yesilkaya, H. Functional assessment of microbial superoxide dismutase isozymes suggests a differential role for each isozyme. Free. Radic. Biol. Med. 2019, 134, 215–228. [Google Scholar] [CrossRef]
- Kim, S.Y.; Park, C.; Jang, H.-J.; Kim, B.-O.; Bae, H.-W.; Chung, I.-Y.; Kim, E.S.; Cho, Y.-H. Antibacterial strategies inspired by the oxidative stress and response networks. J. Microbiol. 2019, 57, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Wang, X.; Dong, Y.; Hu, Q.; Zhao, Z.; Zhu, Y.; Dong, L.; Bai, F.; Dong, X. A Streptococcus aquaporin acts as peroxiporin for efflux of cellular hydrogen peroxide and alleviation of oxidative stress. J. Biol. Chem. 2019, 294, 4583–4595. [Google Scholar] [CrossRef] [PubMed]
- Godlewska, U.; Zegar, A.; Cichy, J. Rola chemeryny i jej antybakteryjnych pochodnych w utrzymaniu funkcji barierowych nabłonka. Postępy Biochemii 2020, 66, 1–9. [Google Scholar] [CrossRef]
- Pagacz, J.; Broniec, A.; Wolska, M.; Osyczka, A.; Borek, A. ROS signaling capacity of cytochrome bc1: Opposing effects of adaptive and pathogenic mitochondrial mutations. Free. Radic. Biol. Med. 2021, 163, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.-F.; Cho, S.-H.; Iorga, B.I.; Goemans, C.V. How the assembly and protection of the bacterial cell envelope depend on cysteine residues. J. Biol. Chem. 2020, 295, 11984–11994. [Google Scholar] [CrossRef] [PubMed]
- Arts, I.S.; Gennaris, A.; Collet, J.-F. Reducing systems protecting the bacterial cell envelope from oxidative damage. FEBS Lett. 2015, 589, 1559–1568. [Google Scholar] [CrossRef]
- Messens, J.; Collet, J.-F. Pathways of disulfide bond formation in Escherichia coli. Int. J. Biochem. Cell Biol. 2006, 38, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Wendler, J.; Ehmann, D.; Courth, L.; Schroeder, B.O.; Malek, N.P.; Wehkamp, J. Bacterial Periplasmic Oxidoreductases Control the Activity of Oxidized Human Antimicrobial beta-Defensin 1. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brzoza, P.; Godlewska, U.; Borek, A.; Morytko, A.; Zegar, A.; Kwiecinska, P.; Zabel, B.A.; Osyczka, A.; Kwitniewski, M.; Cichy, J. Redox Active Antimicrobial Peptides in Controlling Growth of Microorganisms at Body Barriers. Antioxidants 2021, 10, 446. https://doi.org/10.3390/antiox10030446
Brzoza P, Godlewska U, Borek A, Morytko A, Zegar A, Kwiecinska P, Zabel BA, Osyczka A, Kwitniewski M, Cichy J. Redox Active Antimicrobial Peptides in Controlling Growth of Microorganisms at Body Barriers. Antioxidants. 2021; 10(3):446. https://doi.org/10.3390/antiox10030446
Chicago/Turabian StyleBrzoza, Piotr, Urszula Godlewska, Arkadiusz Borek, Agnieszka Morytko, Aneta Zegar, Patrycja Kwiecinska, Brian A. Zabel, Artur Osyczka, Mateusz Kwitniewski, and Joanna Cichy. 2021. "Redox Active Antimicrobial Peptides in Controlling Growth of Microorganisms at Body Barriers" Antioxidants 10, no. 3: 446. https://doi.org/10.3390/antiox10030446
APA StyleBrzoza, P., Godlewska, U., Borek, A., Morytko, A., Zegar, A., Kwiecinska, P., Zabel, B. A., Osyczka, A., Kwitniewski, M., & Cichy, J. (2021). Redox Active Antimicrobial Peptides in Controlling Growth of Microorganisms at Body Barriers. Antioxidants, 10(3), 446. https://doi.org/10.3390/antiox10030446