
Dynamic and Systemic Perspective in Autism Spectrum Disorders: A Change of Gaze in Research Opens to A New Landscape of Needs and Solutions


{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Biological Basis of Autism Spectrum Disorders
2.1. Genetics in ASD
More Relevant ASD Susceptibility Genes
2.2. Non-Linear Epigenetics and Paradigm Shift for Understanding Neurodevelopmental Disorders
2.3. The Multifaceted Immune System along the Way of Neurodevelopment
3. The Web of Metabolic, Immunologic, and Microbiologic Imbalance in ASD People
4. Much More Than Behavioral Abnormalities in People with ASD
5. Lessons from Biological Research and Suggestions for Clinical Practice
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5), 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Bölte, S.; Mahdi, S.; de Vries, P.J.; Granlund, M.; Robison, J.E.; Shulman, C.; Swedo, S.; Tonge, B.; Wong, V.; Zwaigenbaum, L.; et al. The Gestalt of functioning in autism spectrum disorder: Results of the international conference to develop final consensus International Classification of Functioning, Disability and Health core sets. Autism 2019, 23, 449–467. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, U.; Alaie, I.; Löfgren Wilteus, A.; Zander, E.; Marschik, P.B.; Coghill, D.; Bölte, S. Annual Research Review: Quality of life and childhood mental and behavioural disorders a critical review of the research. J. Child Psychol. Psychiatry 2017, 58, 439–469. [Google Scholar] [CrossRef] [PubMed]
- Happé, F.; Frith, U. Annual Research Review: Looking back to look forward—Changes in the concept of autism and implications for future research. J. Child Psychol. Psychiatry Allied Discip. 2020, 61, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Charman, T.; Havdahl, A.; Carbone, P.; Anagnostou, E.; Boyd, B.; Carr, T.; de Vries, P.J.; Dissanayake, C.; Divan, G.; et al. The Lancet Commission on the future of care and clinical research in autism. Lancet 2021. [Google Scholar] [CrossRef]
- Van Naarden Braun, K.; Christensen, D.; Doernberg, N.; Schieve, L.; Rice, C.; Wiggins, L.; Schendel, D.; Yeargin-Allsopp, M. Trends in the prevalence of autism spectrum disorder, cerebral palsy, hearing loss, intellectual disability, and vision impairment, metropolitan Atlanta, 1991–2010. PLoS ONE 2015, 10, e0124120. [Google Scholar] [CrossRef]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States. MMWR Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef]
- Narzisi, A.; Posada, M.; Barbieri, F.; Chericoni, N.; Ciuffolini, D.; Pinzino, M.; Romano, R.; Scattoni, M.L.; Tancredi, R.; Calderoni, S.; et al. Prevalence of Autism Spectrum Disorder in a large Italian catchment area: A school-based population study within the ASDEU project. Epidemiol. Psychiatr. Sci. 2020, 29, e5. [Google Scholar] [CrossRef]
- Hertz-Picciotto, I.; Delwiche, L. The rise in autism and the role of age at diagnosis. Epidemiology 2009, 20, 84–90. [Google Scholar] [CrossRef]
- Rice, C.E.; Rosanoff, M.; Dawson, G.; Durkin, M.S.; Croen, L.A.; Singer, A.; Yeargin-Allsopp, M. Evaluating changes in the prevalence of the autism spectrum disorders (ASDs). Public Health Rev. 2012, 34, 17. [Google Scholar] [CrossRef]
- Grandjean, P.; Landrigan, P.J. Neurobehavioural effects of developmental toxicity. Lancet Neurol. 2014, 13, 330–338. [Google Scholar] [CrossRef]
- Francis, K.; Karantanos, G.; Al-Ozairi, A.; AlKhadhari, S. Prevention in Autism Spectrum Disorder: A Lifelong Focused Approach. Brain Sci. 2021, 11, 151. [Google Scholar] [CrossRef] [PubMed]
- Aldinger, K.A.; Lane, C.J.; Veenstra-VanderWeele, J.; Levitt, P. Patterns of Risk for Multiple Co-Occurring Medical Conditions Replicate Across Distinct Cohorts of Children with Autism Spectrum Disorder. Autism Res. 2015, 8, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Panisi, C.; Guerini, F.R.; Abruzzo, P.M.; Balzola, F.; Biava, P.M.; Bolotta, A.; Brunero, M.; Burgio, E.; Chiara, A.; Clerici, M.; et al. Autism Spectrum Disorder from the Womb to Adulthood: Suggestions for a Paradigm Shift. J. Pers. Med. 2021, 11, 70. [Google Scholar] [CrossRef]
- Engel, G.L. The need for a new medical model: A challenge for biomedicine. Science 1977, 196, 129–136. [Google Scholar] [CrossRef]
- Engel, G.L. The biopsychosocial model and the education of health professionals. Ann. N. Y. Acad. Sci. 1978, 310, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Cusick, S.E.; Georgieff, M.K. The Role of Nutrition in Brain Development: The Golden Opportunity of the “First 1000 Days”. J. Pediatr. 2016, 175, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.M.; Takumi, T. Genomic and genetic aspects of autism spectrum disorder. Biochem. Biophys. Res. Commun. 2014, 452, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhu, Y.; Wang, T.; Zhang, X.; Zhang, K.; Zhang, Z. Genetic risk factors for autism-spectrum disorders: A systematic review based on systematic reviews and meta-analysis. J. Neural Transm. 2021, 128, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Hu, V.W. From genes to environment: Using integrative genomics to build a “systems-level” understanding of autism spectrum disorders. Child Dev. 2013, 84, 89–103. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Muotri, A.R.; Sebat, J. Getting to the Cores of Autism. Cell 2019, 5, 1287–1298. [Google Scholar] [CrossRef]
- Ronald, A.; Hoekstra, R.A. Autism spectrum disorders and autistic traits: A decade of new twin studies. Am. J. Med. Genet. 2011, 156, 255–274. [Google Scholar] [CrossRef] [PubMed]
- Hu, V.W.; Devlin, C.A.; Debski, J.J. ASD Phenotype-Genotype Associations in Concordant and Discordant Monozygotic and Dizygotic Twins Stratified by Severity of Autistic Traits. Int. J. Mol. Sci. 2019, 20, 3804. [Google Scholar] [CrossRef] [PubMed]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatr. 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Castelbaum, L.; Sylvester, C.M.; Zhang, Y.; Yu, Q.; Constantino, J.N. On the Nature of Monozygotic Twin Concordance and Discordance for Autistic Trait Severity: A Quantitative Analysis. Behav. Genet. 2020, 50, 263–272. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Sahin, M. Autism and the synapse: Emerging mechanisms and mechanism-based therapies. Curr. Opin. Neurol. 2015, 28, 91–102. [Google Scholar] [CrossRef]
- Parikshak, N.N.; Gandal, M.J.; Geschwind, D.H. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet. 2015, 16, 441–458. [Google Scholar] [CrossRef]
- Keller, R.; Basta, R.; Salerno, L.; Elia, M. Autism, epilepsy, and synaptopathies: A not rare association. Neurol. Sci. 2017, 38, 1353–1361. [Google Scholar] [CrossRef]
- Fernandez, M.; Mollinedo-Gajate, I.; Penagarikano, O. Neural circuits for social cognition: Implications for autism. Neuroscience 2018, 370, 148–162. [Google Scholar] [CrossRef]
- Waxman, S.G. Channel, neuronal, and clinical function in sodium channelopathies: From genotype to phenotype. Nat. Neurosci. 2007, 10, 405–409. [Google Scholar] [CrossRef]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef]
- Kiefer, J.C. Epigenetics in development. Dev. Dyn. 2007, 236, 1144–1156. [Google Scholar] [CrossRef]
- Reichetzeder, C. Overweight and obesity in pregnancy: Its impact on epigenetics. Eur. J. Clin. Nutr. 2021, 75, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.P.; Velazquez, M.A.; Eckert, J.J. Embryos, DOHaD and David Barker. J. Dev. Orig. Health Dis. 2015, 6, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Burgio, E. Environment and Fetal Programming: The origins of some current “pandemics”. J. Pediatr. Neonatal Individ. Med. 2015, 4, 2. [Google Scholar]
- Strohman, R.C. Linear genetics, non-linear epigenetics: Complementary approaches to understanding complex diseases. Integr. Physiol. Behav. Sci. 1995, 30, 273–282. [Google Scholar] [CrossRef]
- Barker, D.J.; Eriksson, J.G.; Forsén, T.; Osmond, C. Fetal origins of adult disease: Strength of effects and biological basis. Int. J. Epidemiol. 2002, 31, 1235–1239. [Google Scholar] [CrossRef]
- Heindel, J.J.; Vandenberg, L.N. Developmental origins of health and disease: A paradigm for understanding disease cause and prevention. Curr. Opin. Pediatr. 2015, 27, 248–253. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A.; Beedle, A.S. Early life events and their consequences for later disease: A life history and evolutionary perspective. Am. J. Hum. Biol. 2007, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Barouki, R.; Gluckman, P.D.; Grandjean, P.; Hanson, M.; Heindel, J.J. Developmental origins of noncommunicable disease: Implications for research and public health. Environ. Health 2012, 11, 42. [Google Scholar] [CrossRef]
- Burgio, E. The Raise of Neurodevelopmental Disorders (NDS): From Genetics to Epigenetics. Psychiatr. Danub. 2021, 33 (Suppl. 11), 40–41. [Google Scholar]
- Baxter, F.A.; Drake, A.J. Non-genetic inheritance via the male germline in mammals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180118. [Google Scholar] [CrossRef] [PubMed]
- Burgio, E.; Piscitelli, P.; Colao, A. Environmental Carcinogenesis and Transgenerational Transmission of Carcinogenic Risk: From Genetics to Epigenetics. Int. J. Environ. Res. Public Health 2018, 15, 1791. [Google Scholar] [CrossRef] [PubMed]
- Spiers, H.; Hannon, E.; Schalkwyk, L.C.; Smith, R.; Wong, C.C.; O’Donovan, M.C.; Bray, N.J.; Mill, J. Methylomic trajectories across human fetal brain development. Genome Res. 2015, 25, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Linnér, A.; Almgren, M. Epigenetic programming-The important first 1000 days. Acta Paediatr. 2020, 109, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Nugent, B.M.; Bale, T.L. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front. Neuroendocrinol. 2015, 39, 28–37. [Google Scholar] [CrossRef] [PubMed]
- La Salle, J.M. A genomic point-of-view on environmental factors influencing the human brain methylome. Epigenetics 2011, 6, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Dhulkifle, H.; Agouni, A.; Zeidan, A.; Saif Al-Kuwari, M.; Parray, A.; Tolefat, M.; Korashy, H.M. Influence of the Aryl Hydrocarbon Receptor Activating Environmental Pollutants on Autism Spectrum Disorder. Int. J. Mol. Sci. 2021, 22, 9258. [Google Scholar] [CrossRef]
- Abruzzo, P.M.; Matté, A.; Bolotta, A.; Federti, E.; Ghezzo, A.; Guarnieri, T.; Marini, M.; Posar, A.; Siciliano, A.; De Franceschi, L.; et al. Plasma peroxiredoxin changes and inflammatory cytokines support the involvement of neuro-inflammation and oxidative stress in Autism Spectrum Disorder. J. Transl. Med. 2019, 17, 332. [Google Scholar] [CrossRef]
- Emberti Gialloreti, L.; Mazzone, L.; Benvenuto, A.; Fasano, A.; Alcon, A.G.; Kraneveld, A.; Moavero, R.; Raz, R.; Riccio, M.P.; Siracusano, M.; et al. Risk and Protective Environmental Factors Associated with Autism Spectrum Disorder: Evidence-Based Principles and Recommendations. J. Clin. Med. 2019, 8, 217. [Google Scholar] [CrossRef]
- Dall’Aglio, L.; Muka, T.; Cecil, C.A.M.; Bramer, W.M.; Verbiest, M.M.P.J.; Nano, J.; Hidalgo, A.C.; Franco, O.H.; Tiemeier, H. The role of epigenetic modifications in neurodevelopmental disorders: A systematic review. Neurosci. Biobehav. Rev. 2018, 94, 17–30. [Google Scholar] [CrossRef]
- Nardone, S.; Elliott, E. The Interaction between the Immune System and Epigenetics in the Etiology of Autism Spectrum Disorders. Front. Neurosci. 2016, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Labouesse, M.A.; Dong, E.; Grayson, D.R.; Guidotti, A.; Meyer, U. Maternal immune activation induces GAD1and GAD2 promoter remodeling in the offspring pre-frontal cortex. Epigenetics 2015, 10, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Parker-Athill, E.C.; Tan, J. Maternal immune activation and autism spectrum disorder: Interleukin-6 signaling as a key mechanistic pathway. Neurosignals 2010, 18, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Gesundheit, B.; Rosenzweig, J.P.; Naor, D.; Lerer, B.; Zachor, D.A.; Procházka, V.; Melamed, M.; Kristt, D.A.; Steinberg, A.; Shulman, C.; et al. Immunological and autoimmune considerations of Autism Spectrum Disorders. J. Autoimmun. 2013, 44, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Filiano, A.J.; Gadani, S.P.; Kipnis, J. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 2015, 18, 18–27. [Google Scholar] [CrossRef]
- Bilbo, S.D.; Schwarz, J.M. The immune system and developmental programming of brain and behavior. Front. Neuroendocrinol. 2012, 33, 267–286. [Google Scholar] [CrossRef]
- Kipnis, J. Immune system: The “seventh sense”. J. Exp. Med. 2018, 215, 397–398. [Google Scholar] [CrossRef]
- Boulanger, L.M. Immune proteins in brain development and synaptic plasticity. Neuron 2009, 64, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.L.; McAllister, A.K. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 469–486. [Google Scholar] [CrossRef]
- Estes, M.L.; McAllister, A.K. Maternal immune activation: Implications for neuropsychiatric disorders. Science 2016, 19, 772–777. [Google Scholar] [CrossRef]
- Tang, B.; Jia, H.; Kast, R.J.; Thomas, E.A. Epigenetic changes at gene promoters in response to immune activation in utero. Brain Behav. Immun. 2013, 30, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Reisinger, S.N.; Kong, E.; Khan, D.; Schulz, S.; Ronovsky, M.; Berger, S.; Horvath, O.; Cabatic, M.; Berger, A.; Pollak, D.D. Maternal immune activation epigenetically regulates hippocampal serotonin transporter levels. Neurobiol. Stress 2016, 4, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, S.; Manca, S.; Gagliano, A.; Minutolo, A.; Melis, M.C.; Pisuttu, G.; Scoppola, C.; Bolognesi, E.; Clerici, M.; Guerini, F.R. Immune regulation of neurodevelopment at the mother-foetus interface: The case of autism. Clin. Transl. Immunol. 2020, 9, e1211. [Google Scholar] [CrossRef]
- Knuesel, I.; Chicha, L.; Britschgi, M.; Schobel, S.A.; Bodmer, M.; Hellings, J.A.; Toovey, S.; Prinssen, E.P. Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol. 2014, 10, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, V.; Sequeira, J.M.; DiDuca, M.; Vrancken, G.; Thomas, A.; Philippe, C.; Peters, M.; Jadot, A.; Quadros, E.V. Improving Outcome in Infantile Autism with Folate Receptor Autoimmunity and Nutritional Derangements: A Self-Controlled Trial. Autism Res. Treat. 2019, 18, 7486431. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, J.M.; Desai, A.; Berrocal-Zaragoza, M.I.; Murphy, M.M.; Fernandez-Ballart, J.D.; Quadros, E.V. Exposure to Folate Receptor Alpha Antibodies during Gestation and Weaning Leads to Severe Behavioral Deficits in Rats: A Pilot Study. PLoS ONE 2016, 11, e0152249. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Rossignol, D.; Scahill, L.; McDougle, C.J.; Huberman, H.; Quadros, E.V. Treatment of Folate Metabolism Abnormalities in Autism Spectrum Disorder. Semin. Pediatr. Neurol. 2020, 35, 100835. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Sequeira, J.M.; Quadros, E.V.; James, S.J.; Rossignol, D.A. Cerebral folate receptor autoantibodies in autism spectrum disorder. Mol. Psychiatry 2013, 18, 369–381. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Cerebral Folate Deficiency, Folate Receptor Alpha Autoantibodies and Leucovorin (Folinic Acid) Treatment in Autism Spectrum Disorders: A Systematic Review and Meta-Analysis. J. Pers. Med. 2021, 11, 1141. [Google Scholar] [CrossRef] [PubMed]
- Shapira, I.; Sequeira, J.M.; Quadros, E.V. Folate receptor autoantibodies in pregnancy related complications. Birth Defects Research Part A. Clin. Mol. Teratol. 2015, 103, 1028–1030. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Sequeira, J.M.; Blau, N.; Quadros, E.V. A milk-free diet downregulates folate receptor autoimmunity in cerebral folate deficiency syndrome. Dev. Med. Child Neurol. 2008, 50, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M. Leaky gut: Mechanisms, measurement and clinical implications in humans. Gut 2019, 68, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A. Leaky gut and autoimmune diseases. Clin. Rev. Allergy Immunol. 2012, 42, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Patterson, P.H. Immune involvement in schizophrenia and autism: Etiology, pathology and animal models. Behav. Brain Res. 2009, 204, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Verhaeghe, J.; Van Herck, E.; Billen, J.; Moerman, P.; Van Assche, F.A.; Giudice, L.C. Regulation of insulin-like growth factor-I and insulin-like growth factor binding protein-1 concentrations in preterm fetuses. Am. J. Obstet. Gynecol. 2003, 188, 485–491. [Google Scholar] [CrossRef]
- Steinman, G. The putative etiology and prevention of autism. Prog. Mol. Biol. Transl. Sci. 2020, 173, 1–34. [Google Scholar] [CrossRef]
- Steinman, G.; Mankuta, D. Insulin-like growth factor and the etiology of autism. Med. Hypotheses 2013, 80, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Hellström, A.; Ley, D.; Hansen-Pupp, I.; Hallberg, B.; Ramenghi, L.A.; Löfqvist, C.; Smith, L.E.; Hård, A.L. Role of Insulinlike Growth Factor 1 in Fetal Development and in the Early Postnatal Life of Premature Infants. Am. J. Perinatol. 2016, 33, 1067–1071. [Google Scholar] [CrossRef]
- Riikonen, R.; Makkonen, I.; Vanhala, R.; Turpeinen, U.; Kuikka, J.; Kokki, H. Cerebrospinal fluid insulin-like growth factors IGF- 1 and IGF-2 in infantile autism. Dev. Med. Child Neurol. 2006, 48, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Audette, M.C.; Kingdom, J.C. Screening for fetal growth restriction and placental insufficiency. Semin. Fetal Neonatal Med. 2018, 23, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Bokobza, C.; Van Steenwinckel, J.; Mani, S.; Mezger, V.; Fleiss, B.; Gressens, P. Neuroinflammation in preterm babies and autism spectrum disorders. Pediatr. Res. 2019, 85, 155–165. [Google Scholar] [CrossRef]
- Grossi, E.; Migliore, L.; Muratori, F. Pregnancy risk factors related to autism: An Italian case-control study in mothers of children with autism spectrum disorders (ASD), their siblings and of typically developing children. J. Dev. Orig. Health Dis. 2018, 9, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Garbett, K.A.; Hsiao, E.Y.; Kálmán, S.; Patterson, P.H.; Mirnics, K. Effects of maternal immune activation on gene expression patterns in the fetal brain. Transl. Psychiatry 2012, 2, e98. [Google Scholar] [CrossRef] [PubMed]
- Valenza, M.; Steardo, L., Jr.; Steardo, L.; Verkhratsky, A.; Scuderi, C. Systemic Inflammation and Astrocyte Reactivity in the Neuropsychiatric Sequelae of COVID-19: Focus on Autism Spectrum Disorders. Front. Cell. Neurosci. 2021, 15, 748136. [Google Scholar] [CrossRef] [PubMed]
- Leal, C.R.V.; Maciel, R.A.M.; Corrêa Júnior, M.D. SARS-CoV-2 Infection and Placental Pathology. Rev. Bras. Ginecol. Obstet. 2021, 43, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Forestieri, S.; Marcialis, M.A.; Migliore, L.; Panisi, C.; Fanos, V. Relationship between pregnancy and coronavirus: What we know. J. Matern. Fetal Neonatal Med. 2020, 1–12. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, B.; Qu, Y.; Chen, Y.; Xiong, J.; Feng, Y.; Men, D.; Huang, Q.; Liu, Y.; Yan, B. Detectable serum severe acute respiratory syndrome coronavirus 2 viral load (RNAemia) is closely correlated with drastically elevated interleukin 6 level in critically ill patients with coronavirus disease 2019. Clin. Infect. Dis. 2020, 71, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Cai, T.; Fan, L.; Lou, K.; Hua, X.; Huang, Z.; Guao, G. Clinical value of immune-inflammatory parameters to assess the severity of coronavirus disease 2019. Int. J. Infect. Dis. 2020, 95, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Vismara, L. The Psychological Impact of COVID-19 Pandemic on Women’s Mental Health during Pregnancy: A Rapid Evidence Review. Int. J. Environ. Res. Public Health 2021, 18, 7112. [Google Scholar] [CrossRef] [PubMed]
- Yirmiya, K.; Yakirevich-Amir, N.; Preis, H.; Lotan, A.; Atzil, S.; Reuveni, I. Women’s Depressive Symptoms during the COVID-19 Pandemic: The Role of Pregnancy. Int. J. Environ. Res. Public Health 2021, 18, 4298. [Google Scholar] [CrossRef] [PubMed]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Bäckhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Aagaard, K.; Riehle, K.; Ma, J.; Segata, N.; Mistretta, T.A.; Coarfa, C.; Raza, S.; Rosenbaum, S.; Van den Veyver, I.; Milosavljevic, A.; et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS ONE 2012, 7, e36466. [Google Scholar] [CrossRef]
- Gomez de Agüero, M.; Ganal-Vonarburg, S.C.; Fuhrer, T.; Rupp, S.; Uchimura, Y.; Li, H.; Steinert, A.; Heikenwalder, M.; Hapfelmeier, S.; Sauer, U.; et al. The maternal microbiota drives early postnatal innate immune development. Science 2016, 18, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Romano-Keeler, J.; Weitkamp, J.H. Maternal influences on fetal microbial colonization and immune development. Pediatr. Res. 2015, 77, 189–195. [Google Scholar] [CrossRef]
- Hoffman, D.J.; Reynolds, R.M.; Hardy, D.B. Developmental origins of health and disease: Current knowledge and potential mechanisms. Nutr. Rev. 2017, 75, 951–970. [Google Scholar] [CrossRef]
- Weaver, I.C.; Korgan, A.C.; Lee, K.; Wheeler, R.V.; Hundert, A.S.; Goguen, D. Stress and the Emerging Roles of Chromatin Remodeling in Signal Integration and Stable Transmission of Reversible Phenotypes. Front. Behav. Neurosci. 2017, 15, 41. [Google Scholar] [CrossRef]
- Blaser, M.J.; Dominguez-Bello, M.G. The Human Microbiome before Birth. Cell Host Microbe 2016, 20, 558–560. [Google Scholar] [CrossRef]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Matta, S.M.; Hill-Yardin, E.L.; Crack, P.J. The influence of neuroinflammation in Autism Spectrum Disorder. Brain Behav. Immun. 2019, 79, 75–90. [Google Scholar] [CrossRef]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.; Van de Water, J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 2011, 25, 40–45. [Google Scholar] [CrossRef]
- Saresella, M.; Piancone, F.; Marventano, I.; Zoppis, M.; Hernis, A.; Zanette, M.; Trabattoni, D.; Chiappedi, M.; Ghezzo, A.; Canevini, M.P.; et al. Multiple inflammasome complexes are activated in autistic spectrum disorders. Brain Behav. Immun. 2016, 57, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Saghazadeh, A.; Ataeinia, B.; Keynejad, K.; Abdolalizadeh, A.; Hirbod-Mobarakeh, A.; Rezaei, N. Anti-inflammatory cytokines in autism spectrum disorders: A systematic review and meta-analysis. Cytokine 2019, 123, 154740. [Google Scholar] [CrossRef]
- Onore, C.; Careaga, M.; Ashwood, P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav. Immun. 2012, 26, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Marventano, I.; Guerini, F.R.; Mancuso, R.; Ceresa, L.; Zanzottera, M.; Rusconi, B.; Maggioni, E.; Tinelli, C.; Clerici, M. An autistic endophenotype results in complex immune dysfunction in healthy siblings of autistic children. Biol. Psychiatry 2009, 66, 978–984. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, J.M. The microbiota-gut-brain axis and its potential therapeutic role in autism spectrum disorder. Neuroscience 2016, 2, 131–139. [Google Scholar] [CrossRef]
- Kulkarni, S.; Ganz, J.; Bayrer, J.; Becker, L.; Bogunovic, M.; Rao, M. Advances in enteric neurobiology: The “brain” in the gut in health and disease. J. Neurosci. 2018, 38, 9346–9354. [Google Scholar] [CrossRef]
- Naveed, M.; Zhou, Q.G.; Xu, C.; Taleb, A.; Meng, F.; Ahmed, B.; Zhang, Y.; Fukunaga, K.; Han, F. Gut-brain axis: A matter of concern in neuropsychiatric disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 104, 110051. [Google Scholar] [CrossRef]
- Gars, A.; Ronczkowski, N.M.; Chassaing, B.; Castillo-Ruiz, A.; Forger, N.G. First Encounters: Effects of the Microbiota on Neonatal Brain Development. Front. Cell. Neurosci. 2021, 15, 682505. [Google Scholar] [CrossRef]
- Deidda, G.; Biazzo, M. Gut and Brain: Investigating Physiological and Pathological Interactions Between Microbiota and Brain to Gain New Therapeutic Avenues for Brain Diseases. Front. Neurosci. 2021, 15, 753915. [Google Scholar] [CrossRef]
- Bonaz, B.; Sinniger, V.; Pellissier, S. Therapeutic Potential of Vagus Nerve Stimulation for Inflammatory Bowel Diseases. Front. Neurosci. 2021, 15, 300. [Google Scholar] [CrossRef]
- Frasch, M.G.; Herry, C.L.; Niu, Y.; Giussani, D.A. First evidence that intrinsic fetal heart rate variability exists and is affected by hypoxic pregnancy. J. Physiol. 2020, 598, 249–263. [Google Scholar] [CrossRef]
- Bystrova, K. Novel mechanism of human fetal growth regulation: A potential role of lanugo, vernix caseosa and a second tactile system of unmyelinated low-threshold C-afferents. Med. Hypotheses 2009, 72, 143–146. [Google Scholar] [CrossRef]
- Cerritelli, F.; Frasch, M.G.; Antonelli, M.C.; Viglione, C.; Vecchi, S.; Chiera, M.; Manzotti, A. A Review on the Vagus Nerve and Autonomic Nervous System During Fetal Development: Searching for Critical Windows. Front. Neurosci. 2021, 15, 721605. [Google Scholar] [CrossRef]
- Mulkey, S.B.; du Plessis, A.J. Autonomic nervous system development and its impact on neuropsychiatric outcome. Pediatr. Res. 2019, 85, 120–126. [Google Scholar] [CrossRef]
- Kong, X.; Liu, J.; Liu, K.; Koh, M.; Tian, R.; Hobbie, C.; Fong, M.; Chen, Q.; Zhao, M.; Budjan, C.; et al. Altered Autonomic Functions and Gut Microbiome in Individuals with Autism Spectrum Disorder (ASD): Implications for Assisting ASD Screening and Diagnosis. J. Autism Dev. Disord. 2021, 51, 144–157. [Google Scholar] [CrossRef]
- Kushki, A.; Brian, J.; Dupuis, A.; Anagnostou, E. Functional autonomic nervous system profile in children with autism spectrum disorder. Mol. Autism 2014, 5, 39. [Google Scholar] [CrossRef]
- Mouridsen, S.E.; Brønnum-Hansen, H.; Rich, B.; Isager, T. Mortality and causes of death in autism spectrum disorders. Autism 2008, 12, 403–414. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, M.; Kong, X. Gut microbiome and autism: Recent advances and future perspectives. Am. J. Med. Sci. 2016, 9, 3. [Google Scholar] [CrossRef]
- El Aidy, S.; Dinan, T.G.; Cryan, J.F. Gut Microbiota: The Conductor in the Orchestra of Immune-Neuroendocrine Communication. Clin. Ther. 2015, 37, 954–967. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Ratsika, A.; Codagnone, M.C.; O’Mahony, S.; Stanton, C.; Cryan, J.F. Priming for Life: Early Life Nutrition and the Microbiota-Gut-Brain Axis. Nutrients 2021, 13, 423. [Google Scholar] [CrossRef]
- Stilling, R.M.; Dinan, T.G.; Cryan, J.F. Microbial genes, brain & behavior—Epigenetic regulation of the gut-brain axis. Genes Brain Behav. 2014, 13, 69–86. [Google Scholar] [CrossRef]
- Ho, P.; Ross, D.A. More Than a Gut Feeling: The Implications of the Gut Microbiota in Psychiatry. Biol. Psychiatry. 2017, 81, e35–e37. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 19, 1451–1463. [Google Scholar] [CrossRef]
- Finegold, S.M.; Molitoris, D.; Song, Y.; Liu, C.; Vaisanen, M.-L.; Bolte, E.; McTeague, M.; Sandler, R.; Wexler, H.; Marlowe, E.M.; et al. Gastrointestinal microflora studies in late-onset autism. Clin. Infect. Dis. 2002, 35 (Suppl. 1), S6–S16. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Macfabe, D.F. Short-chain fatty acid fermentation products of the gut microbiome: Implications in autism spectrum disorders. Microb. Ecol. Health Dis. 2012, 23. [Google Scholar] [CrossRef]
- De Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS ONE 2013, 8, e76993. [Google Scholar] [CrossRef]
- Littman, D.R.; Pamer, E.G. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe 2011, 20, 311–323. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef]
- Vuong, H.E.; Hsiao, E.Y. Emerging Roles for the Gut Microbiome in Autism Spectrum Disorder. Biol. Psychiatry 2017, 1, 411–423. [Google Scholar] [CrossRef]
- Borre, Y.E.; O’Keeffe, G.W.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota and neurodevelopmental windows: Implications for brain disorders. Trends Mol. Med. 2014, 20, 509–518. [Google Scholar] [CrossRef]
- Jiang, C.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef]
- Nishiwaki, H.; Ito, M.; Ishida, T.; Hamaguchi, T.; Maeda, T.; Kashihara, K.; Tsuboi, Y.; Ueyama, J.; Shimamura, T.; Mori, H.; et al. Meta-Analysis of Gut Dysbiosis in Parkinson’s Disease. Mov. Disord. 2020, 35, 1626–1635. [Google Scholar] [CrossRef]
- Liu, G.; Gao, J.; Zhu, M.; Liu, K.; Zhang, H.L. Gut Microbiota and Dysbiosis in Alzheimer’s Disease: Implications for Pathogenesis and Treatment. Mol. Neurobiol. 2020, 57, 5026–5043. [Google Scholar] [CrossRef]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal flora and gastrointestinal status in children with autism—Comparisons to typical children and correlation with autism severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef]
- Chaidez, V.; Hansen, R.L.; Hertz-Picciotto, I. Gastrointestinal Problems in Children with Autism, Developmental Delays or Typical Development. J. Autism Dev. Disord. 2014, 44, 1117–1127. [Google Scholar] [CrossRef]
- Marler, S.; Ferguson, B.J.; Lee, E.B.; Peters, B.; Williams, K.C.; McDonnell, E.; Macklin, E.A.; Levitt, P.; Margolis, K.G.; Beversdorf, D.Q.; et al. Association of Rigid-Compulsive Behavior with Functional Constipation in Autism Spectrum Disorder. J. Autism Dev. Disord. 2017, 47, 1673–1681. [Google Scholar] [CrossRef]
- Bundgaard-Nielsen, C.; Knudsen, J.; Leutscher, P.D.C.; Lauritsen, M.B.; Nyegaard, M.; Hagstrom, S.; Sørensen, S. Gut Microbiota Profiles of Autism Spectrum Disorder and Attention Deficit/Hyperactivity Disorder: A Systematic Literature Review. Gut Microbes 2020, 11, 1172–1187. [Google Scholar] [CrossRef]
- Ding, H.; Yi, X.; Zhang, X.; Wang, H.; Liu, H.; Mou, W.W. Imbalance in the Gut Microbiota of Children with Autism Spectrum Disorders. Front. Cell. Infect. Microbiol. 2021, 11, 572752. [Google Scholar] [CrossRef] [PubMed]
- Rudzki, L.; Stone, T.W.; Maes, M.; Misiak, B.; Samochowiec, J.; Szulc, A. Gut microbiota-derived vitamins—Underrated powers of a multipotent ally in psychiatric health and disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 107, 110240. [Google Scholar] [CrossRef] [PubMed]
- Firth, J.; Carney, R.; Stubbs, B.; Teasdale, S.B.; Vancampfort, D.; Ward, P.B.; Berk, M.; Sarris, J. Nutritional Deficiencies and Clinical Correlates in First-Episode Psychosis: A Systematic Review and Meta-analysis. Schizophr. Bull. 2018, 44, 1275–1292. [Google Scholar] [CrossRef]
- Douaud, G.; Refsum, H.; de Jager, C.A.; Jacoby, R.; Nichols, T.E.; Smith, S.M.; Smith, A.D. Preventing Alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. Proc. Natl. Acad. Sci. USA 2013, 110, 9523–9528. [Google Scholar] [CrossRef] [PubMed]
- Magnúsdottir, S.; Ravcheev, D.; de Cŕecy-Lagard, V.; Thiele, I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front. Genet. 2015, 6, 148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hodgson, N.W.; Trivedi, M.S.; Abdolmaleky, H.M.; Fournier, M.; Cuenod, M.; Do, K.Q.; Deth, R.C. Decreased Brain Levels of Vitamin B12 in Aging, Autism and Schizophrenia. PLoS ONE 2016, 11, e0146797. [Google Scholar] [CrossRef]
- Belardo, A.; Gevi, F.; Zolla, L. The concomitant lower concentrations of vitamins B6, B9 and B12 may cause methylation deficiency in autistic children. J. Nutr. Biochem. 2019, 70, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Nardone, S.; Sams, D.S.; Reuveni, E.; Getselter, D.; Oron, O.; Karpuj, M.; Karpuj, M.; Elliott, E. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 2014, 4, e433. [Google Scholar] [CrossRef] [PubMed]
- Hamza, M.; Halayem, S.; Mrad, R.; Bourgou, S.; Charfi, F.; Belhadj, A. Epigenetics’ implication in autism spectrum disorders: A review. Encephale 2017, 43, 374–381. [Google Scholar] [CrossRef]
- Chauhan, A.; Chauhan, V. Oxidative stress in autism. Pathophysiology 2006, 13, 171–181. [Google Scholar] [CrossRef]
- Jurnak, F. The Pivotal Role of Aldehyde Toxicity in Autism Spectrum Disorder: The Therapeutic Potential of Micronutrient Supplementation. Nutr. Metab. Insights 2016, 8 (Suppl. 1), 57–77. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Kałużna-Czaplińska, J.; Gątarek, P.; Chirumbolo, S.; Chartrand, M.S.; Bjørklund, G. How important is tryptophan in human health? Crit. Rev. Food Sci. Nutr. 2019, 59, 72–88. [Google Scholar] [CrossRef]
- Anwar, A.; Marini, M.; Abruzzo, P.M.; Bolotta, A.; Ghezzo, A.; Visconti, P.; Thornalley, P.J.; Rabbani, N. Quantitation of plasma thiamine, related metabolites and plasma protein oxidative damage markers in children with autism spectrum disorder and healthy controls. Free Radic. Res. 2016, 50 (Suppl. 1), S85–S90. [Google Scholar] [CrossRef] [PubMed]
- Gevi, F.; Zolla, L.; Gabriele, S.; Persico, A.M. Urinary metabolomics of young Italian autistic children supports abnormal tryptophan and purine metabolism. Mol. Autism 2016, 7, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.J.; Yin, C.H.; Guo, Y.Q.; Li, S.O.; Chen, H.; Long, B.H. Serum homocysteine concentrations in Chinese children with autism. Clin. Chem. Lab. Med. 2013, 51, e19–e22. [Google Scholar] [CrossRef] [PubMed]
- Józefczuk, J.; Kasprzycka, W.; Czarnecki, R.; Graczyk, A.; Józefczuk, P.; Magda, K.; Lampart, U. Homocysteine as a diagnostic and etiopathogenic factor in children with autism spectrum disorder. J. Med. Food 2017, 20, 744–749. [Google Scholar] [CrossRef]
- Yektaş, Ç.; Alpay, M.; Tufan, A.E. Comparison of serum B12, folate and homocysteine concentrations in children with autism spectrum disorder or attention deficit hyperactivity disorder and healthy controls. Neuropsychiatr. Dis. Treat. 2019, 15, 2213. [Google Scholar] [CrossRef]
- Zieminska, E.; Matyja, E.; Kozlowska, H.; Stafiej, A.; Lazarewicz, J.W. Excitotoxic neuronal injury in acute homocysteine neurotoxicity: Role of calcium and mitochondrial alterations. Neurochem. Int. 2006, 48, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Herrmann, W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006, 580, 2994–3005. [Google Scholar] [CrossRef]
- Tetreault, N.A.; Hakeem, A.Y.; Jiang, S.; Williams, B.A.; Allman, E.; Wold, B.J.; Allman, J.M. Microglia in the cerebral cortex in autism. J. Autism Dev. Disord. 2012, 4, 2569–2584. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sugihara, G.; Ouchi, Y.; Nakamura, K.; Futatsubashi, M.; Takebayashi, K.; Yoshihara, Y.; Omata, K.; Matsumoto, K.; Tsuchiya, K.J.; et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 2013, 70, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Carey, N.; La Thangue, N.B. Histone deacetylase inhibitors: Gathering pace. Curr. Opin. Pharmacol. 2006, 6, 369–375. [Google Scholar] [CrossRef]
- Lanza, M.; Campolo, M.; Casili, G.; Filippone, A.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Sodium Butyrate Exerts Neuroprotective Effects in Spinal Cord Injury. Mol. Neurobiol. 2019, 56, 3937–3947. [Google Scholar] [CrossRef] [PubMed]
- Kratsman, N.; Getselter, D.; Elliott, E. Sodium butyrate attenuates social behavior deficits and modifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model. Neuropharmacology 2016, 102, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Jia, Y.; Pan, S.; Jia, L.; Li, H.; Han, Z.; Cai, D.; Zhao, R. Butyrate alleviates high fat diet-induced obesity through activation of adiponectin-mediated pathway and stimulation of mitochondrial function in the skeletal muscle of mice. Oncotarget 2016, 7, 56071–56082. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, E.; Sun, Z.; Fu, D.; Duan, G.; Jiang, M.; Yu, Y.; Mei, L.; Yang, P.; Tang, Y.; et al. Altered gut microbiota and short chain fatty acids in Chinese children with autism spectrum disorder. Sci. Rep. 2019, 9, 287. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Bennuri, S.C.; Davis, J.E.; Wynne, R.; Slattery, J.C.; Tippett, M.; Delhey, L.; Melnyk, S.; Kahler, S.G.; MacFabe, D.F.; et al. Butyrate enhances mitochondrial function during oxidative stress in cell lines from boys with autism. Transl. Psychiatry 2018, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. A review of research trends in physiological abnormalities in autism spectrum disorders: Immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol. Psychiatry 2012, 17, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E. Biomarkers of abnormal energy metabolism in children with autism spectrum disorder. N. Am. J. Med. Sci. 2012, 5, 141–147. [Google Scholar] [CrossRef]
- Wright, R.H.; Fernandez-Fuentes, N.; Oliva, B.; Beato, M. Insight into the machinery that oils chromatin dynamics. Nucleus 2016, 7, 532–539. [Google Scholar] [CrossRef][Green Version]
- Khacho, M.; Harris, R.; Slack, R.S. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 2019, 20, 34–48. [Google Scholar] [CrossRef]
- Wang, S.; Qian, J.; Sun, F.; Li, M.; Ye, J.; Li, M.; Du, M.; Li, D. Bidirectional regulation between 1st trimester HTR8/SVneo trophoblast cells and in vitro differentiated Th17/Treg cells suggest a fetal-maternal regulatory loop in human pregnancy. Am. J. Reprod. Immunol. 2019, 81, e13106. [Google Scholar] [CrossRef] [PubMed]
- Mandò, C.; Anelli, G.M.; Novielli, C.; Panina-Bordignon, P.; Massari, M.; Mazzocco, M.I.; Cetin, I. Impact of Obesity and Hyperglycemia on Placental Mitochondria. Oxidative Med. Cell. Longev. 2018, 2378189. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.N.; Chan, S.S.L. Sources, mechanisms, and consequences of chemical-induced mitochondrial toxicity. Toxicology 2017, 391, 2–4. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Genuis, S.J.; Frye, R.E. Environmental toxicants and autism spectrum disorders: A systematic review. Transl. Psychiatry 2014, 4, e360. [Google Scholar] [CrossRef]
- Frye, R.E.; Cakir, J.; Rose, S.; Palmer, R.F.; Austin, C.; Curtin, P.; Arora, M. Mitochondria May Mediate Prenatal Environmental Influences in Autism Spectrum Disorder. J. Pers. Med. 2021, 11, 218. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Giulivi, C.; Zhang, Y.F.; Omanska-Klusek, A.; Ross-Inta, C.; Wong, S.; Hertz-Picciotto, I.; Tassone, F.; Pessah, I.N. Mitochondrial dysfunction in autism. JAMA 2010, 304, 2389–2396. [Google Scholar] [CrossRef]
- Bargiela, D.; Chinnery, P.F. Mitochondria in neuroinflammation—Multiple sclerosis (MS), leber hereditary optic neuropathy (LHON) and LHON-MS. Neurosci. Lett. 2019, 710, 132932. [Google Scholar] [CrossRef]
- Rose, S.; Niyazov, D.M.; Rossignol, D.A.; Goldenthal, M.; Kahler, S.G.; Frye, R.E. Clinical and Molecular Characteristics of Mitochondrial Dysfunction in Autism Spectrum Disorder. Mol. Diagn. Ther. 2018, 22, 571–593. [Google Scholar] [CrossRef] [PubMed]
- Ghezzo, A.; Visconti, P.; Abruzzo, P.M.; Bolotta, A.; Ferreri, C.; Gobbi, G.; Malisardi, G.; Manfredini, S.; Marini, M.; Nanetti, L.; et al. Oxidative Stress and Erythrocyte Membrane Alterations in Children with Autism: Correlation with Clinical Features. PLoS ONE 2013, 8, e66418. [Google Scholar] [CrossRef] [PubMed]
- Ferreri, C.; Masi, A.; Sansone, A.; Giacometti, G.; Larocca, A.V.; Menounou, G.; Scanferlato, R.; Tortorella, S.; Rota, D.; Conti, M.; et al. Fatty Acids in Membranes as Homeostatic, Metabolic and Nutritional Biomarkers: Recent Advancements in Analytics and Diagnostics. Diagnostics 2016, 22, 1. [Google Scholar] [CrossRef]
- Giacometti, G.; Ferreri, C.; Sansone, A.; Chatgilialoglu, C.; Marzetti, C.; Spyratou, E.; Georgakilas, A.G.; Marini, M.; Abruzzo, P.M.; Bolotta, A.; et al. High predictive values of RBC membrane-based diagnostics by biophotonics in an integrated approach for Autism Spectrum Disorders. Sci. Rep. 2017, 29, 9854. [Google Scholar] [CrossRef] [PubMed]
- Bolotta, A.; Battistelli, M.; Falcieri, E.; Ghezzo, A.; Manara, M.C.; Manfredini, S.; Marini, M.; Posar, A.; Visconti, P.; Abruzzo, P.M. Oxidative Stress in Autistic Children Alters Erythrocyte Shape in the Absence of Quantitative Protein Alterations and of Loss of Membrane Phospholipid Asymmetry. Oxidative Med. Cell. Longev. 2018, 2018, 6430601. [Google Scholar] [CrossRef] [PubMed]
- Abruzzo, P.M.; Panisi, C.; Marini, M. The Alteration of Chloride Homeostasis/GABAergic Signaling in Brain Disorders: Could Oxidative Stress Play a Role? Antioxidants 2021, 10, 1316. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.; Patisaul, H.; Liedtke, W. Decoding the language of epigenetics during neural development is key for understanding development as well as developmental neurotoxicity. Epigenetics 2013, 8, 1128–1132. [Google Scholar] [CrossRef]
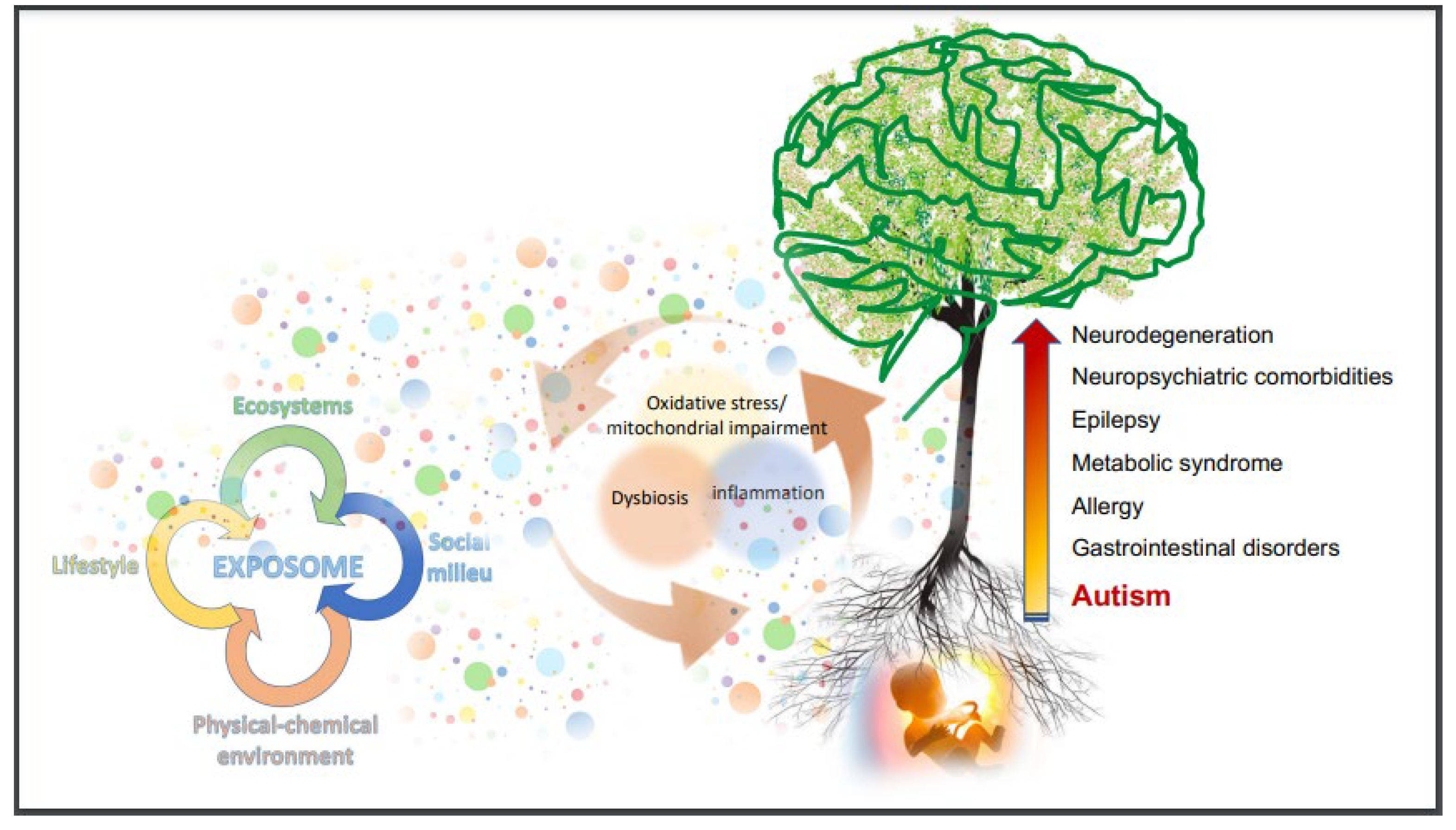
- Fanos, V.; Noto, A.; Mussap, M. The juniper bush of autism spectrum disorder (ASD): Metabolomics, microbiomics, acetaminophen. What else? J. Pediatr. Neonatal Individ. Med. (JPNIM) 2018, 7, e070205. [Google Scholar] [CrossRef]
- Zamboni, N.; Saghatelian, A.; Patti, G.J. Defining the metabolome: Size, flux, and regulation. Mol. Cell. 2015, 21, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Rajula, H.S.R.; Mauri, M.; Fanos, V. Scale-free networks in metabolomics. Bioinformation 2018, 14, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Faa, G.; Manchia, M.; Pintus, R.; Gerosa, C.; Marcialis, M.A.; Fanos, V. Fetal programming of neuropsychiatric disorders. Birth Defects Res. C Embryo Today 2016, 108, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Fanni, D.; Gerosa, C.; Rais, M.; Ravarino, A.; Van Eyken, P.; Fanos, V.; Faa, G. The role of neuropathological markers in the interpretation of neuropsychiatric disorders: Focus on fetal and perinatal programming. Neurosci. Lett. 2018, 669, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Hagenbeek, F.A.; Kluft, C.; Hankemeier, T.; Bartels, M.; Draisma, H.H.; Middeldorp, C.M.; Berger, R.; Noto, A.; Lussu, M.; Pool, R.; et al. Discovery of biochemical biomarkers for aggression: A role for metabolomics in psychiatry. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.L.; McAllister, A.K. Maternal TH17 cells take a toll on baby’s brain. Science 2016, 351, 919–920. [Google Scholar] [CrossRef]
- Manchia, M.; Fanos, V. Targeting aggression in severe mental illness: The predictive role of genetic, epigenetic, and metabolomic markers. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 77, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Elsabbagh, M.; Johnson, M.H. Getting answers from babies about autism. Trends. Cogn. Sci. 2010, 14, 81–87. [Google Scholar] [CrossRef]
- Mussap, M.; Siracusano, M.; Noto, A.; Fanos, V. The Urine Metabolome of Young Autistic Children Correlates with Their Clinical Profile Severity. Metabolites 2020, 10, 476. [Google Scholar] [CrossRef]
- Mussap, M.; Noto, A.; Fanos, V. Metabolomics of autism spectrum disorders: Early insights regarding mammalian-microbial cometabolites. Expert Rev. Mol. Diagn. 2016, 16, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Bitar, T.; Mavel, S.; Emond, P.; Nadal-Desbarats, L.; Lefèvre, A.; Mattar, H.; Soufia, M.; Blasco, H.; Vourc’h, P.; Hleihel, W.; et al. Identification of metabolic pathway disturbances using multimodal metabolomics in autistic disorders in a Middle Eastern population. J. Pharm. Biomed. Anal. 2018, 152, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Lussu, M.; Noto, A.; Masili, A.; Rinaldi, A.C.; Dessì, A.; De Angelis, M.; De Giacomo, A.; Fanos, V.; Atzori, L.; Francavilla, R. The urinary 1 H-NMR metabolomics profile of an italian autistic children population and their unaffected siblings. Autism Res. 2017, 10, 1058–1066. [Google Scholar] [CrossRef]
- Shedlock, K.; Susi, A.; Gorman, G.H.; Hisle-Gorman, E.; Erdie-Lalena, C.R.; Nylund, C.M. Autism Spectrum Disorders and Metabolic Complications of Obesity. J. Pediatr. 2016, 178, 183–187.e1. [Google Scholar] [CrossRef] [PubMed]
- Lago, S.G.; Tomasik, J.; van Rees, G.F.; Rubey, M.; Gonzalez-Vioque, E.; Ramsey, J.M.; Haenisch, F.; Broek, J.A.; Vázquez-Bourgon, J.; Papiol, S.; et al. Exploring cellular markers of metabolic syndrome in peripheral blood mononuclear cells across the neuropsychiatric spectrum. Brain Behav. Immun. 2021, 91, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Madra, M.; Ringel, R.; Margolis, K.G. Gastrointestinal Issues and Autism Spectrum Disorder. Child Adolesc. Psychiatr. Clin. N. Am. 2020, 29, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.T.; Taur, Y.; Walkup, J.T. Gut Microbiota and Autism: Key Concepts and Findings. J. Autism Dev. Disord. 2017, 47, 480–489. [Google Scholar] [CrossRef] [PubMed]
- de Magistris, L.; Picardi, A.; Sapone, A.; Cariello, R.; Siniscalco, D.; Bravaccio, C.; Pascotto, A. Intestinal barrier in autism. In Comprehensive Guide to Autism; Patel, V.B., Preedy, V.R., Martin, C.R., Eds.; Springer: New York, NY, USA, 2014; pp. 2047–2060. [Google Scholar] [CrossRef]
- Keller, R.; Chieregato, S.; Bari, S.; Castaldo, R.; Rutto, F.; Chiocchetti, A.; Dianzani, U. Autism in Adulthood: Clinical and Demographic Characteristics of a Cohort of Five Hundred Persons with Autism Analyzed by a Novel Multistep Network Model. Brain Sci. 2020, 10, 416. [Google Scholar] [CrossRef] [PubMed]
- Nikolov, R.N.; Bearss, K.E.; Lettinga, J.; Erickson, C.; Rodowski, M.; Aman, M.G.; McCracken, J.T.; McDougle, C.J.; Tierney, E.; Vitiello, B.; et al. Gastrointestinal symptoms in a sample of children with pervasive developmental disorders. J. Autism Dev. Disord. 2009, 39, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Buie, T.; Campbell, D.B.; Fuchs, G.J., 3rd; Furuta, G.T.; Levy, J.; Vandewater, J.; Whitaker, A.H.; Atkins, D.; Bauman, M.L.; Beaudet, A.L.; et al. Evaluation, diagnosis, and treatment of gastrointestinal disorders in individuals with ASDs: A consensus report. Pediatrics 2010, 125 (Suppl. 1), S1–S18. [Google Scholar] [CrossRef]
- McElhanon, B.O.; McCracken, C.; Karpen, S.; Sharp, W.G. Gastrointestinal symptoms in autism spectrum disorder: A meta-analysis. Pediatrics 2014, 133, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, A.; Vatta, F.; Votto, M.; Licari, A.; Ruffoli, M.; Brunero, M.; Marseglia, G.; Riccipetitoni, G. Eosinophilic colitis in children: A new and elusive enemy? Pediatr. Surg. Int. 2021, 37, 485–490. [Google Scholar] [CrossRef]
- Whitney, D.G.; Shapiro, D.N. National Prevalence of Pain Among Children and Adolescents with Autism Spectrum Disorders. JAMA Pediatr. 2019, 173, 1203–1205. [Google Scholar] [CrossRef]
- Iwata, B.A.; Deleon, I.G.; Roscoe, E.M. Reliability and validity of the functional analysis screening tool. J. Appl. Behav. Anal. 2013, 46, 271–284. [Google Scholar] [CrossRef]
- Lukmanji, S.; Manji, S.A.; Kadhim, S. The co-occurrence of epilepsy and autism: A systematic review. Epilepsy Behav. 2019, 98, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Mouridsen, S.E.; Rich, B.; Isager, T. A longitudinal study of epilepsy and other central nervous system diseases in individuals with and without a history of infantile autism. Brain Dev. 2011, 33, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.T.; Plioplys, S. Epilepsy and autism: Is there a special relationship? Epilepsy Behav. 2012, 23, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Zhang, B. Neuro-inflammation, blood-brain barrier, seizures and autism. J. Neuroinflamm. 2011, 30, 168. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.J. Childhood epilepsy and autism spectrum disorders: Psychiatric problems, phenotypic expression, and anticonvulsants. Neuropsychol. Rev. 2012, 22, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.M.; Cowan, M.; Moonah, S.N.; Petri, W.A., Jr. The Impact of Systemic Inflammation on Neurodevelopment. Trends Mol. Med. 2018, 24, 794–804. [Google Scholar] [CrossRef]
- Hawks, Z.W.; Constantino, J.N. Neuropsychiatric “Comorbidity” as Causal Influence in Autism. J. Am. Acad. Child Adolesc. Psychiatry 2020, 59, 229–235. [Google Scholar] [CrossRef]
- Dale, R.C. Autistic regression and central nervous system autoimmunity. Dev. Med. Child Neurol. 2016, 58, 1002–1003. [Google Scholar] [CrossRef]
- Gagliano, A.; Galati, C.; Ingrassia, M.; Ciuffo, M.; Alquino, M.A.; Tanca, M.G.; Carucci, S.; Zuddas, A.; Grossi, E. Pediatric Acute-Onset Neuropsychiatric Syndrome: A Data Mining Approach to a Very Specific Constellation of Clinical Variables. J. Child Adolesc. Psychopharmacol. 2020, 30, 495–511. [Google Scholar] [CrossRef]
- Godoy-Vitorino, F. Human microbial ecology and the rising new medicine. Ann. Transl. Med. 2019, 7, 342. [Google Scholar] [CrossRef] [PubMed]
- Perlman, S.J.; Hodson, C.N.; Hamilton, P.T.; Opit, G.P.; Gowen, B.E. Maternal transmission, sex ratio distortion, and mitochondria. Proc. Natl. Acad. Sci. USA 2015, 112, 10162–10168. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Tessing, J.; Lee, B.K.; Lyall, K. Maternal Dietary Factors and the Risk of Autism Spectrum Disorders: A Systematic Review of Existing Evidence. Autism Res. 2020, 13, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.E.; Barry, C.; Sabhlok, A. Maternal pre-pregnancy obesity and child neurodevelopmental outcomes: A meta-analysis. Obes. Rev. 2018, 19, 464–484. [Google Scholar] [CrossRef]
- Goodrich, A.J.; Volk, H.E.; Tancredi, D.J. Joint effects of prenatal air pollutant exposure and maternal folic acid supplementation on risk of autism spectrum disorder. Autism Res. 2018, 11, 69–80. [Google Scholar] [CrossRef]
- May, P.A.; Gossage, J.P. Maternal risk factors for fetal alcohol spectrum disorders: Not as simple as it might seem. Alcohol Res. Health 2011, 34, 15–26. [Google Scholar]
- Tenenbaum, A.; Mandel, A.; Dor, T.; Sapir, A.; Sapir-Bodnaro, O.; Hertz, P.; Wexler, I.D. Fetal alcohol spectrum disorder among pre-adopted and foster children. BMC Pediatr. 2020, 20, 275. [Google Scholar] [CrossRef]
- Lange, S.; Rehm, J.; Anagnostou, E.; Popova, S. Prevalence of externalizing disorders and Autism Spectrum Disorders among children with Fetal Alcohol Spectrum Disorder: Systematic review and meta-analysis. Biochem. Cell. Biol. 2018, 96, 241–251. [Google Scholar] [CrossRef]
- Anderson, B.L.; Dang, E.P.; Floyd, R.L.; Sokol, R.; Mahoney, J.; Schulkin, J. Knowledge, opinions, and practice patterns of obstetrician-gynecologists regarding their patients’ use of alcohol. J. Addict. Med. 2010, 4, 114–121. [Google Scholar] [CrossRef]
- Chiodo, L.M.; Cosmian, C.; Pereira, K.; Kent, N.; Sokol, R.J.; Hannigan, J.H. Prenatal Alcohol Screening During Pregnancy by Midwives and Nurses. Alcohol. Clin. Exp. Res. 2019, 43, 1747–1758. [Google Scholar] [CrossRef]
- Payne, J.M.; France, K.E.; Henley, N. Paediatricians’ knowledge, attitudes and practice following provision of educational resources about prevention of prenatal alcohol exposure and Fetal Alcohol Spectrum Disorder. J. Paediatr. Child Health 2011, 47, 704–710. [Google Scholar] [CrossRef]
- Ciafrè, S.; Ferraguti, G.; Greco, A.; Polimeni, A.; Ralli, M.; Ceci, F.M.; Ceccanti, M.; Fiore, M. Alcohol as an early life stressor: Epigenetics, metabolic, neuroendocrine and neurobehavioral implications. Neurosci. Biobehav. Rev. 2020, 118, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, B.R.H.; van den Heuvel, M.I.; Lahti, M.; Braeken, M.; de Rooij, S.R.; Entringer, S.; Hoyer, D.; Roseboom, T.; Räikkönen, K.; King, S.; et al. Prenatal developmental origins of behavior and mental health: The influence of maternal stress in pregnancy. Neurosci. Biobehav. Rev. 2020, 117, 26–64. [Google Scholar] [CrossRef] [PubMed]
- Pohl, A.; Crockford, S.K.; Blakemore, M.; Allison, C.; Baron-Cohen, S. A comparative study of autistic and non-autistic women’s experience of motherhood. Mol. Autism 2020, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Hirvikoski, T.; Mittendorfer-Rutz, E.; Boman, M.; Larsson, H.; Lichtenstein, P.; Bölte, S. Premature mortality in autism spectrum disorder. Br. J. Psychiatry 2016, 208, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Gillberg, C.; Billstedt, E.; Sundh, V.; Gillberg, I.C. Mortality in autism: A prospective longitudinal community-based study. J. Autism Dev. Disord. 2010, 40, 352–357. [Google Scholar] [CrossRef]
- Baxter, A.J.; Brugha, T.S.; Erskine, H.E.; Scheurer, R.W.; Vos, T.; Scott, J.G. The epidemiology and global burden of autism spectrum disorders. Psychol. Med. 2014, 11, 1–13. [Google Scholar] [CrossRef]
- Ritter, C.; Hewitt, K.; McMorris, C.A. Psychotropic Polypharmacy Among Children and Youth with Autism: A Systematic Review. J. Child Adolesc. Psychopharmacol. 2021, 31, 244–258. [Google Scholar] [CrossRef]
- Guinchat, V.; Cravero, C.; Diaz, L.; Périsse, D.; Xavier, J.; Amiet, C.; Gourfinkel-An, I.; Bodeau, N.; Wachtel, L.; Cohen, D.; et al. Acute behavioral crises in psychiatric inpatients with autism spectrum disorder (ASD): Recognition of concomitant medical or non-ASD psychiatric conditions predicts enhanced improvement. Res. Dev. Disabil. 2015, 38, 242–255. [Google Scholar] [CrossRef]
- Benninga, M.A.; Nurko, S.; Faure, C.; Hyman, P.E.; St James Roberts, I.; Schechter, N.L. Childhood functional gastrointestinal disorders: Neonate/toddler. Gastroenterology 2016, 150, 1443–1455. [Google Scholar] [CrossRef]
- Bağ, Ö.; Alşen Güney, S.; Cevher Binici, N.; Tuncel, T.; Şahin, A.; Berksoy, E.; Ecevit, Ç. Infant colic or early symptom of autism spectrum disorder? Pediatr. Int. 2018, 60, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Zwaigenbaum, L.; Bauman, M.L.; Choueiri, R.; Kasari, C.; Carter, A.; Granpeesheh, D.; Mailloux, Z.; Smith Roley, S.; Wagner, S.; Fein, D.; et al. Early intervention for children with autism spectrum disorder under 3 years of age: Recommendations for practice and research. Pediatrics 2015, 136 (Suppl. 1), S60–S81. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, A.; Kantsjö, J.B.; Ronchi, F. The gut-brain axis: How microbiota and host inflammasome influence brain physiology and pathology. Front. Immunol. 2020, 11, 3237. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Zhu, H.; Feng, Y.; Guo, R.; Wan, D. The impact of gut microbiota disorders on the blood-brain barrier. Infect. Drug Resist. 2020, 13, 3351–3363. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Bertocchi, A.; Mancinelli, S.; Bellini, M.; Erreni, M.; Borreca, A.; Braga, D.; Giugliano, S.; Mozzarelli, A.M.; Manganaro, D.; et al. Identification of a choroid plexus vascular barrier closing during intestinal inflammation. Science 2021, 374, 439–448. [Google Scholar] [CrossRef]
- Dawson, G.; Rogers, S.; Munson, J.; Smith, M.; Winter, J.; Greenson, J.; Donaldson, A.; Varley, J. Randomized, controlled trial of an intervention for toddlers with autism: The Early Start Denver Model. Pediatrics 2010, 125, e17–e23. [Google Scholar] [CrossRef]
- Fountain, C.; Winter, A.S.; Bearman, P.S. Six developmental trajectories characterize children with autism. Pediatrics 2012, 129, e1112–e1120. [Google Scholar] [CrossRef]
- Kushak, R.; Buie, T.M.; Murray, K.F.; Newburg, D.S.; Chen, C.; Nestoridi, E.; Winter, H.S. Evaluation of Intestinal Function in Children with Autism and Gastrointestinal Symptoms. J. Pediatr. Gastroenterol. Nutr. 2016, 62, 687–691. [Google Scholar] [CrossRef]
- Babinská, K.; Tomova, A.; Celušáková, H.; Babková, J.; Repiská, G.; Kubranská, A.; Filčíková, D.; Siklenková, L.; Ostatníková, D. Fecal calprotectin levels correlate with main domains of the autism diagnostic interview-revised (ADI-R) in a sample of individuals with autism spectrum disorders from Slovakia. Physiol. Res. 2017, 66 (Suppl. 4), S517–S522. [Google Scholar] [CrossRef]
- Fasano, A. Zonulin and its regulation of intestinal barrier function: The biological door to inflammation, autoimmunity, and cancer. Physiol. Rev. 2011, 91, 151–175. [Google Scholar] [CrossRef]
- Fasano, A.; Hill, I. Serum Zonulin, Gut Permeability, and the Pathogenesis of Autism Spectrum Disorders: Cause, Effect, or an Epiphenomenon? J. Pediatr. 2017, 188, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Huke, V.; Turk, J.; Saeidi, S.; Kent, A.; Morgan, J.F. Autism spectrum disorders in eating disorder populations: A systematic review. Eur. Eat. Disord. Rev. 2013, 21, 345–351. [Google Scholar] [CrossRef]
- Fahmie, T.A.; Iwata, B.A.; Jann, K.E. Comparison of edible and leisure reinforcers. J. Appl. Behav. Anal. 2015, 48, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Engin, A. Endothelial Dysfunction in Obesity. In Obesity and Lipotoxicity; Springer: Cham, Switzerland, 2017; Volume 960. [Google Scholar] [CrossRef]
- Newsholme, P.; Fernandes Cruzat, V.; Noel Keane, K.; Carlessi, R.; Ivo Homem de Bittencourt, P. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, J.R.; Sartor, R.B. The role of diet on intestinal microbiota metabolism: Downstream impacts on host immune function and health, and therapeutic implications. J. Gastroenterol. 2014, 49, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Conlon, M.A.; Bird, A.R. The impact of diet and lifestyle on gut microbiota and human health. Nutrients 2014, 24, 17–44. [Google Scholar] [CrossRef]
- Berding, K.; Donovan, S.M. Diet Can Impact Microbiota Composition in Children with Autism Spectrum Disorder. Front. Neurosci. 2018, 31, 515. [Google Scholar] [CrossRef]
- Murphy, K.; Meyer, B.; Mori, T.; Burke, V.; Mansour, J.; Patch, C.; Tapsell, L.C.; Noakes, M.; Clifton, P.A.; Barden, A.; et al. Impact of foods enriched with n-3 long-chain polyunsaturated fatty acids on erythrocyte n-3 levels and cardiovascular risk factors. Br. J. Nutr. 2007, 97, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Rossignol, D.A. Identification and Treatment of Pathophysiological Comorbidities of Autism Spectrum Disorder to Achieve Optimal Outcomes. Clin. Med. Insights Pediatr. 2016, 15, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Guinchat, V.; Cravero, C.; Lefèvre-Utile, J.; Cohen, D. Multidisciplinary treatment plan for challenging behaviors in neurodevelopmental disorders. Handb. Clin. Neurol. 2020, 174, 301–321. [Google Scholar] [CrossRef] [PubMed]
- van’t Hof, M.; Tisseur, C.; van Berckelear-Onnes, I.; van Nieuwenhuyzen, A.; Daniels, A.M.; Deen, M. Age at autism spectrum disorder diagnosis: A systematic review and meta-analysis from 2012 to 2019. Autism 2021, 25, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Fraguas, D.; Díaz-Caneja, C.M.; Pina-Camacho, L.; Moreno, C.; Durán-Cutilla, M.; Ayora, M.; González-Vioque, E.; de Matteis, M.; Hendren, R.L.; Arango, C.; et al. Dietary Interventions for Autism Spectrum Disorder: A Meta-analysis. Pediatrics 2019, 144, e20183218. [Google Scholar] [CrossRef] [PubMed]
- Hollin, G. Autistic heterogeneity: Linking uncertainties and indeterminacies. Sci. Cult. 2017, 26, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxidative Med. Cell. Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, V.C.; De Meirleir, K.L.; Subramanian, K.; Nourani, S.M.; Dagda, R.K.; Delaney, S.L.; Palotás, A. Nutritional modulation of the intestinal microbiota; future opportunities for the prevention and treatment of neuroimmune and neuroinflammatory disease. J. Nutr. Biochem. 2018, 61, 1–16. [Google Scholar] [CrossRef]
- Haigh, S.M.; Walford, T.P.; Brosseau, P. Heart Rate Variability in Schizophrenia and Autism. Front. Psychiatry 2021, 12, 760396. [Google Scholar] [CrossRef]
- Torres, E.B. Reframing Psychiatry for Precision Medicine. J. Pers. Med. 2020, 10, 144. [Google Scholar] [CrossRef]
- Borell-Carrió, F.; Suchman, A.L.; Epstein, R.M. The biopsychosocial model 25 years later: Principles, practice, and scientific inquiry. Ann. Fam. Med. 2004, 2, 576–582. [Google Scholar] [CrossRef]
- Adler, R.H. Engel’s biopsychosocial model is still relevant today. J. Psychosom. Res. 2009, 67, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Grossi, E. Artificial Adaptive Systems and predictive medicine: A revolutionary paradigm shift. Immun. Ageing 2010, 7 (Suppl. 1). [Google Scholar] [CrossRef]
- Ristori, M.V.; Mortera, S.L.; Marzano, V.; Guerrera, S.; Vernocchi, P.; Ianiro, G.; Gardini, S.; Torre, G.; Valeri, G.; Vicari, S.; et al. Proteomics and metabolomics approaches towards a functional insight onto autism spectrum disorders: Phenotype stratification and biomarker discovery. Int. J. Mol. Sci. 2020, 21, 6274. [Google Scholar] [CrossRef] [PubMed]
- Myatt, L. Placental adaptive responses and fetal programming. J. Physiol. 2006, 572 Pt 1, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Hughes, H.K.; Ko, E.; Rose, D.; Ashwood, P. Immune Dysfunction and Autoimmunity as Pathological Mechanisms in Autism Spectrum Disorders. Front. Cell. Neurosci. 2018, 12, 405. [Google Scholar] [CrossRef] [PubMed]
- Tersigni, C.; D’Ippolito, S.; Di Nicuolo, F.; Marana, R.; Valenza, V.; Masciullo, V.; Scaldaferri, F.; Malatacca, F.; de Waure, C.; Gasbarrini, A.; et al. Recurrent pregnancy loss is associated to leaky gut: A novel pathogenic model of endometrium inflammation? [published correction appears. J. Transl. Med. 2018, 16, 102. [Google Scholar] [CrossRef]
- Mokkala, K.; Röytiö, H.; Munukka, E.; Pietilä, S.; Ekblad, U.; Rönnemaa, T.; Eerola, E.; Laiho, A.; Laitinen, K. Gut Microbiota Richness and Composition and Dietary Intake of Overweight Pregnant Women Are Related to Serum Zonulin Concentration, a Marker for Intestinal Permeability. J. Nutr. 2016, 146, 1694–1700. [Google Scholar] [CrossRef]
- Cândido, F.G.; Valente, F.X.; Grześkowiak, Ł.M.; Moreira, A.P.B.; Rocha, D.M.; Alfenas, R.C.G. Impact of dietary fat on gut microbiota and low-grade systemic inflammation: Mechanisms and clinical implications on obesity. Int. J. Food Sci. Nutr. 2018, 69, 125–143. [Google Scholar] [CrossRef]
- Mulder, K.A.; Elango, R.; Innis, S.M. Fetal DHA inadequacy and the impact on child neurodevelopment: A follow-up of a randomised trial of maternal DHA supplementation in pregnancy. Br. J. Nutr. 2018, 119, 271–279. [Google Scholar] [CrossRef]
- Lee, E.; Cho, J.; Kim, K.Y. The Association between Autism Spectrum Disorder and Pre- and Postnatal Antibiotic Exposure in Childhood-A Systematic Review with Meta-Analysis. Int. J. Environ. Res. Public Health 2019, 16, 4042. [Google Scholar] [CrossRef] [PubMed]
- Imbriani, G.; Panico, A.; Grassi, T.; Idolo, A.; Serio, F.; Bagordo, F.; De Filippis, G.; De Giorgi, D.; Antonucci, G.; Piscitelli, P.; et al. Early-Life Exposure to Environmental Air Pollution and Autism Spectrum Disorder: A Review of Available Evidence. Int. J. Environ. Res. Public Health 2021, 18, 1204. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, B.Y.; Tanaka., E.; Yamamoto., N.; van Kruining, D.; Iinuma, K.; Nakamuta, Y.; Yamaguchi, H.; Yamasaki, R.; Matsumoto, K.; Kira, J.-I. Early postnatal allergic airway inflammation induces dystrophic microglia leading to excitatory postsynaptic surplus and autism-like behavior. Brain Behav. Immun. 2021, 95, 362–380. [Google Scholar] [CrossRef] [PubMed]
- Sabourin, K.R.; Reynolds, A.; Schendel, D.; Rosenberg, S.; Croen, L.A.; Pinto-Martin, J.A.; Schieve, L.A.; Newschaffer, C.; Lee, L.C.; DiGuiseppi, C. Infections in children with autism spectrum disorder: Study to Explore Early Development (SEED). Autism Res. 2019, 12, 136–146. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panisi, C.; Marini, M. Dynamic and Systemic Perspective in Autism Spectrum Disorders: A Change of Gaze in Research Opens to A New Landscape of Needs and Solutions. Brain Sci. 2022, 12, 250. https://doi.org/10.3390/brainsci12020250
Panisi C, Marini M. Dynamic and Systemic Perspective in Autism Spectrum Disorders: A Change of Gaze in Research Opens to A New Landscape of Needs and Solutions. Brain Sciences. 2022; 12(2):250. https://doi.org/10.3390/brainsci12020250
Chicago/Turabian StylePanisi, Cristina, and Marina Marini. 2022. "Dynamic and Systemic Perspective in Autism Spectrum Disorders: A Change of Gaze in Research Opens to A New Landscape of Needs and Solutions" Brain Sciences 12, no. 2: 250. https://doi.org/10.3390/brainsci12020250
APA StylePanisi, C., & Marini, M. (2022). Dynamic and Systemic Perspective in Autism Spectrum Disorders: A Change of Gaze in Research Opens to A New Landscape of Needs and Solutions. Brain Sciences, 12(2), 250. https://doi.org/10.3390/brainsci12020250