
Advanced Analytical Tools for the Estimation of Gut Permeability of Compounds of Pharmaceutical Interest





Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Molecular Descriptors
2.2. Chemometric Modelling
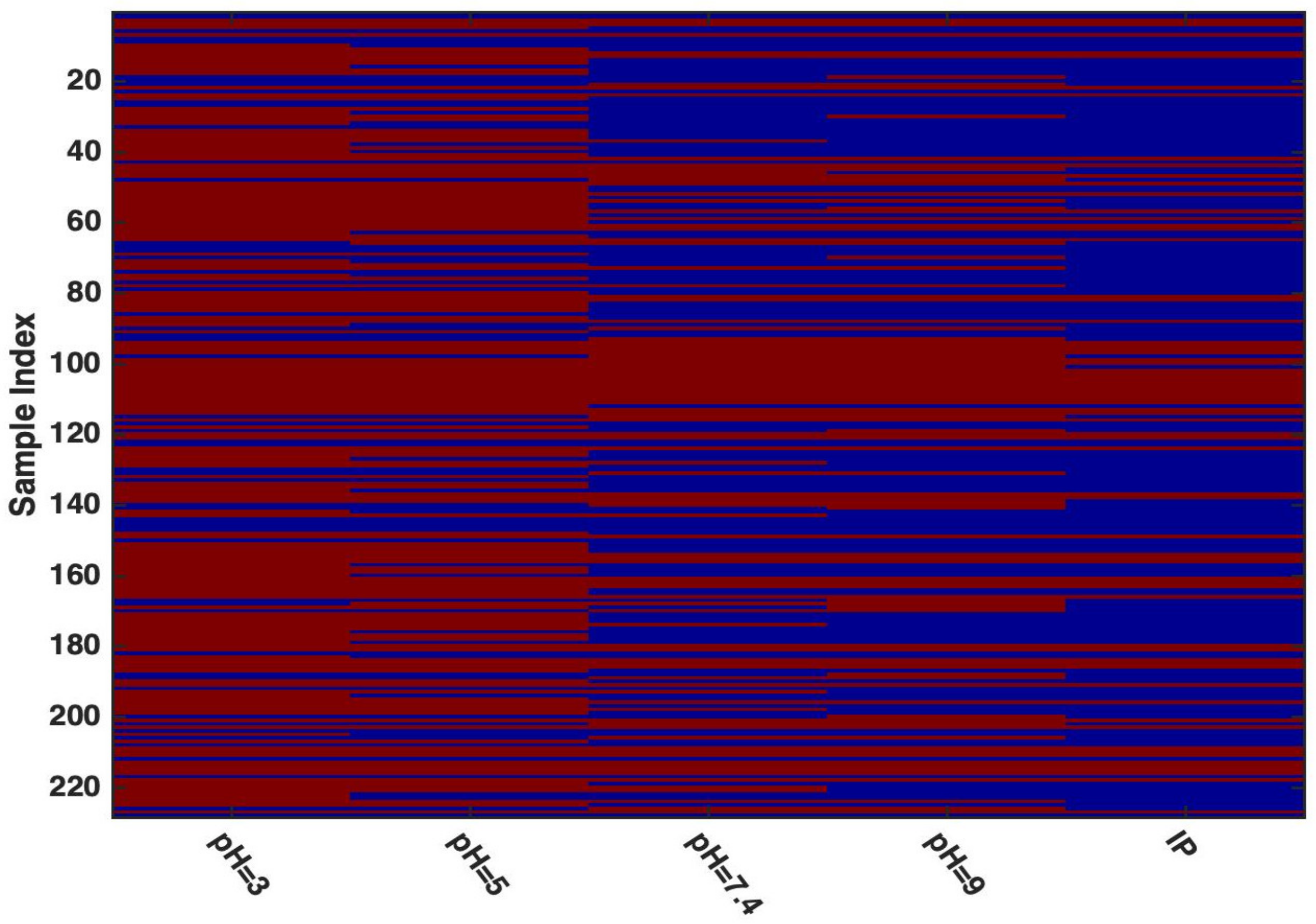
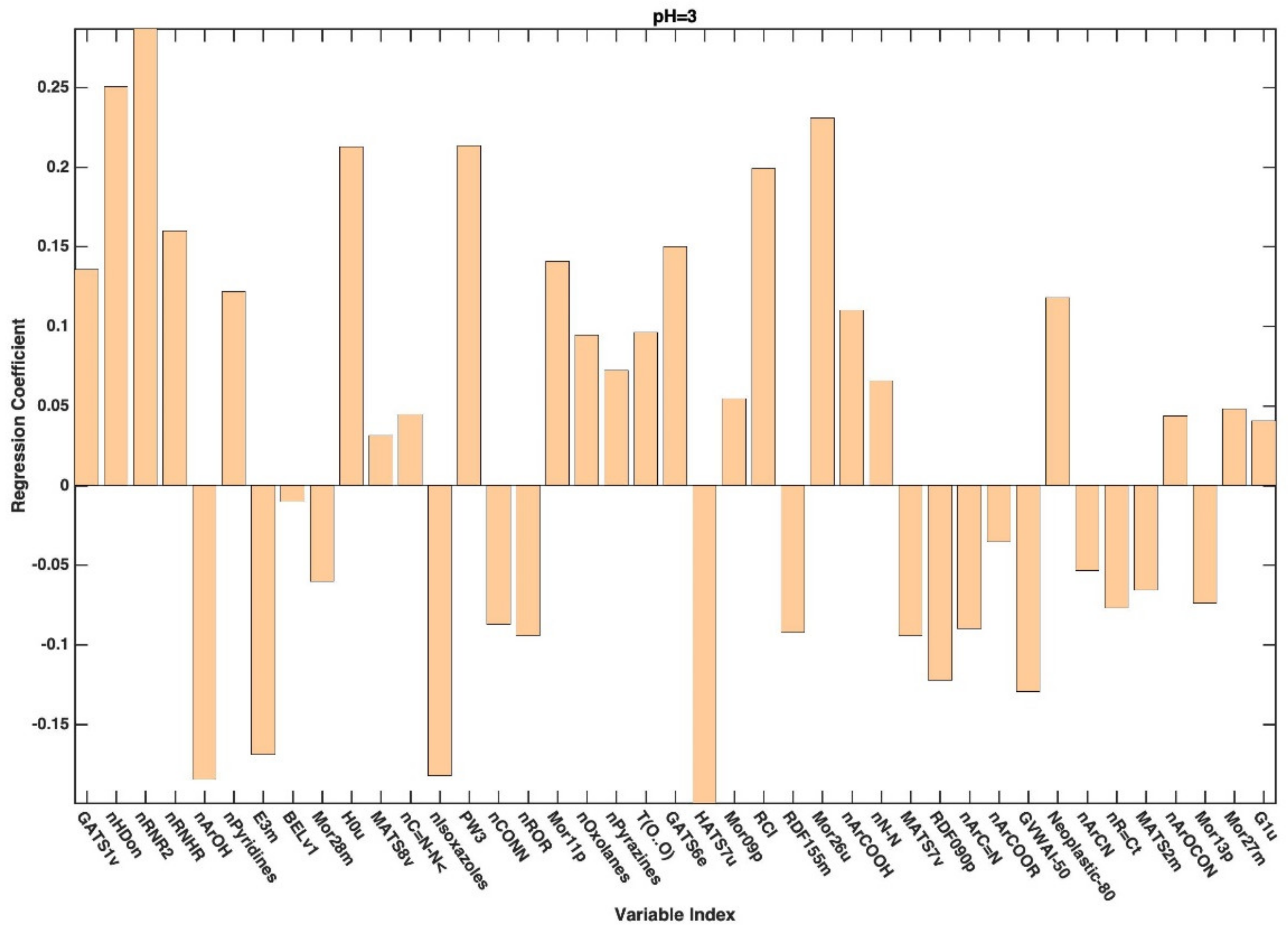
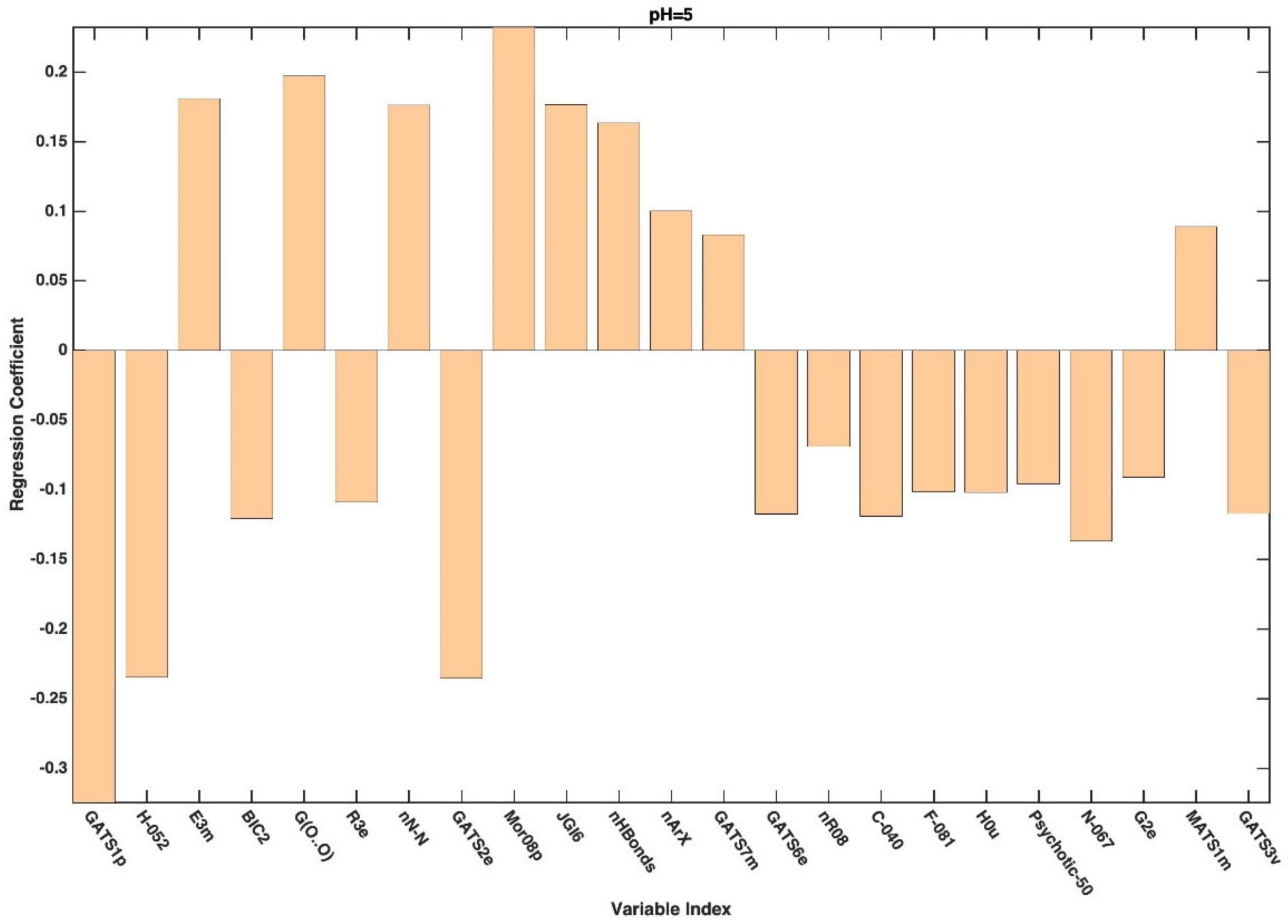
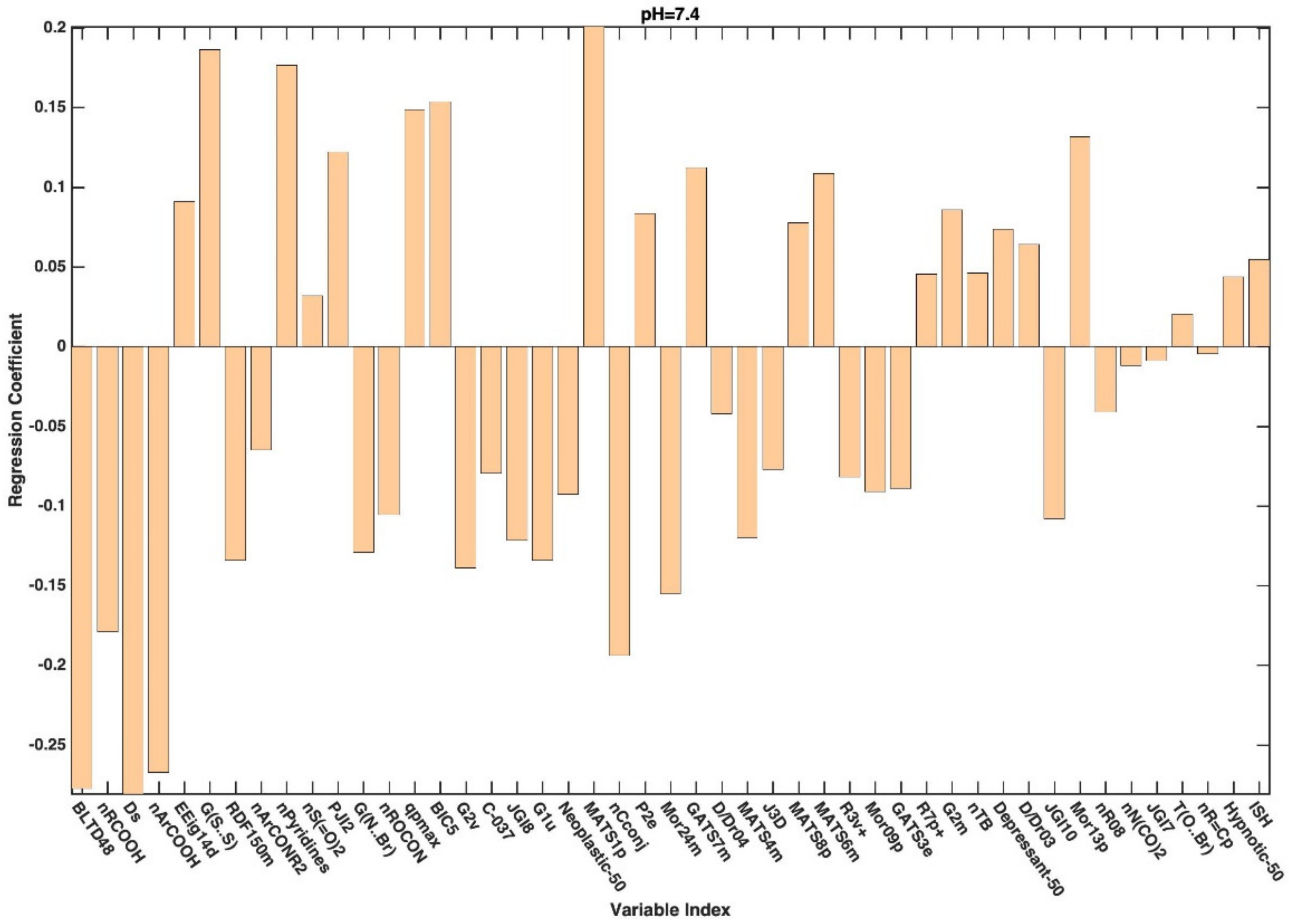
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
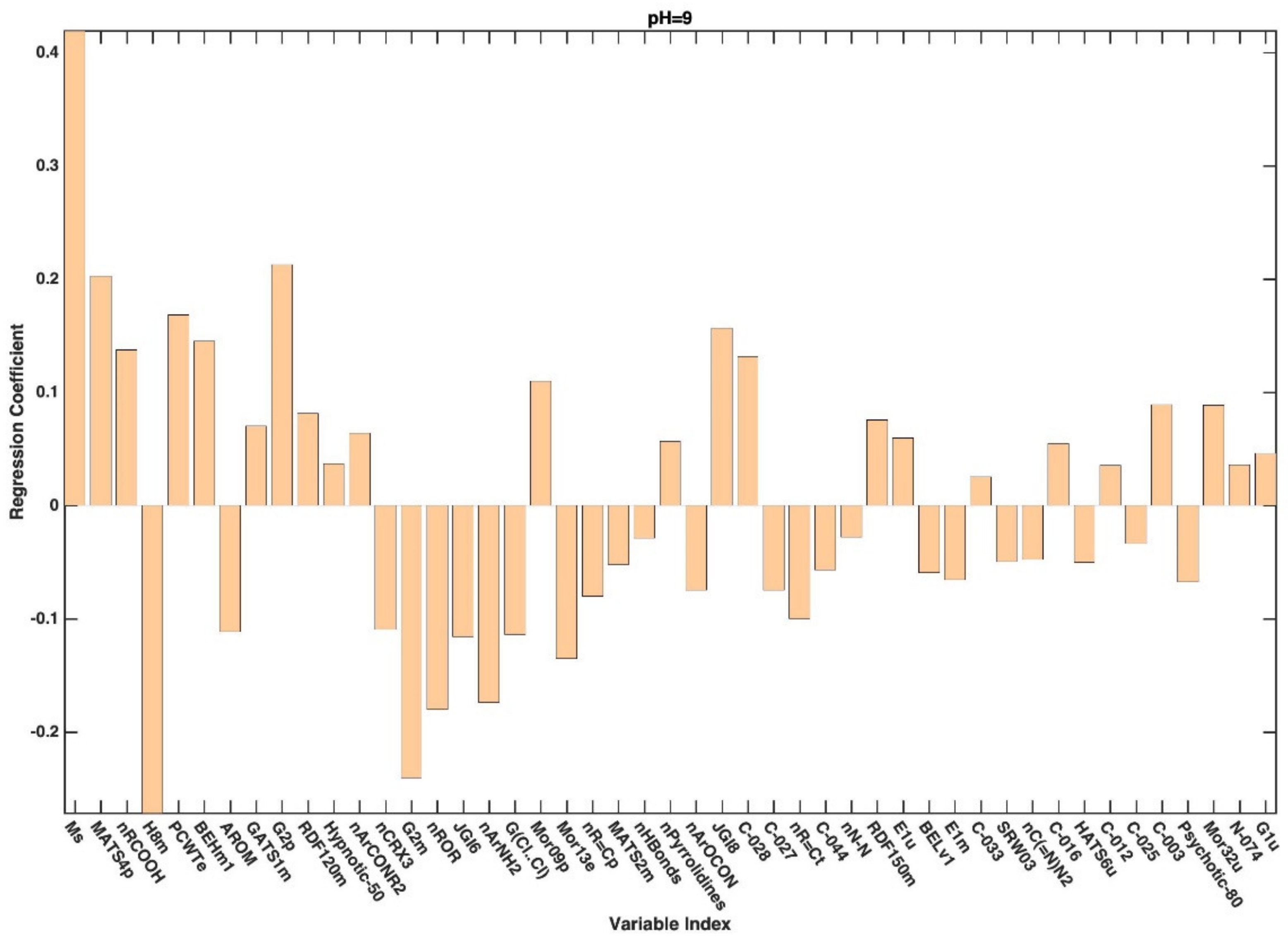{kind=link}
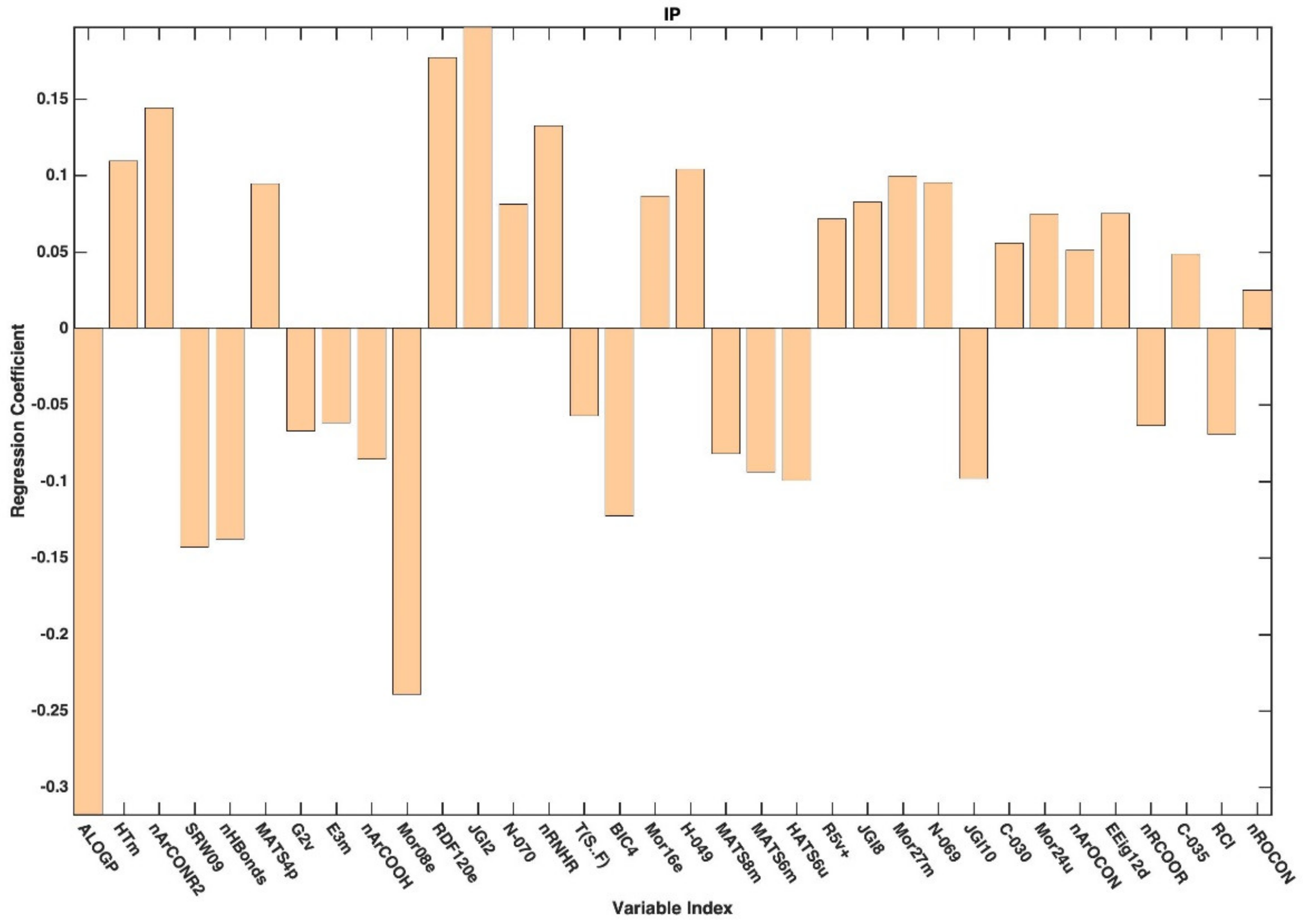{kind=link}
| Descriptor | Block | |
|---|---|---|
| ALOGP | Molecular properties | Other |
| AROM | Geometrical descriptors | 3D |
| BEHm1 | Burden eigenvalues | 2D |
| BELv1 | Burden eigenvalues | 2D |
| BIC2 | Information indices | 2D |
| BIC4 | Information indices | 2D |
| BIC5 | Information indices | 2D |
| BLTD48 | Molecular properties | Other |
| C-003 | Atom-centered fragments | 1D |
| C-012 | Atom-centered fragments | 1D |
| C-016 | Atom-centered fragments | 1D |
| C-025 | Atom-centered fragments | 1D |
| C-027 | Atom-centered fragments | 1D |
| C-028 | Atom-centered fragments | 1D |
| C-030 | Atom-centered fragments | 1D |
| C-033 | Atom-centered fragments | 1D |
| C-035 | Atom-centered fragments | 1D |
| C-037 | Atom-centered fragments | 1D |
| C-040 | Atom-centered fragments | 1D |
| C-044 | Atom-centered fragments | 1D |
| D/Dr03 | Ring descriptors | 2D |
| D/Dr04 | Ring descriptors | 2D |
| Depressant-50 | Drug-like indices | Other |
| Ds | WHIM descriptors | 3D |
| E1m | WHIM descriptors | 3D |
| E1u | WHIM descriptors | 3D |
| E3m | WHIM descriptors | 3D |
| EEig12d | Edge adjacency indices | 2D |
| EEig14d | Edge adjacency indices | 2D |
| F-081 | Atom-centered fragments | 1D |
| G(Cl…Cl) | 3D atom pairs | 3D |
| G(N—Br) | 3D atom pairs | 3D |
| G(O…O) | 3D atom pairs | 3D |
| G(S…S) | 3D atom pairs | 3D |
| G1u | WHIM descriptors | 3D |
| G2e | WHIM descriptors | 3D |
| G2m | WHIM descriptors | 3D |
| G2p | WHIM descriptors | 3D |
| G2v | WHIM descriptors | 3D |
| GATS1m | 2D autocorrelations | 2D |
| GATS1v | 2D autocorrelations | 2D |
| GATS3e | 2D autocorrelations | 2D |
| GATS3v | 2D autocorrelations | 2D |
| GATS6e | 2D autocorrelations | 2D |
| GATS7m | 2D autocorrelations | 2D |
| GETS2e | 2D autocorrelations | 2D |
| GVWAI-50 | Drug-like indices | Other |
| H-049 | Atom-centered fragments | 1D |
| H-052 | Atom-centered fragments | 1D |
| H0u | GETAWAY descriptors | 3D |
| H8m | GETAWAY descriptors | 3D |
| HATS6u | GETAWAY descriptors | 3D |
| HATS7u | GETAWAY descriptors | 3D |
| HTm | GETAWAY descriptors | 3D |
| Hypnotic-50 | Drug-like indices | Other |
| ISH | GETAWAY descriptors | 3D |
| J3D | 2D autocorrelations | 2D |
| JGI10 | 2D autocorrelations | 2D |
| JGI2 | 2D autocorrelations | 2D |
| JGI6 | 2D autocorrelations | 2D |
| JGI7 | 2D autocorrelations | 2D |
| JGI8 | 2D autocorrelations | 2D |
| MAT1p | 2D autocorrelations | 2D |
| MATS1m | 2D autocorrelations | 2D |
| MATS2m | 2D autocorrelations | 2D |
| MATS4m | 2D autocorrelations | 2D |
| MATS4p | 2D autocorrelations | 2D |
| MATS6m | 2D autocorrelations | 2D |
| MATS7u | 2D autocorrelations | 2D |
| MATS8m | 2D autocorrelations | 2D |
| MATS8p | 2D autocorrelations | 2D |
| MATS8v | 2D autocorrelations | 2D |
| Mor08e | 3D-MoRSE descriptors | 3D |
| Mor08p | 3D-MoRSE descriptors | 3D |
| Mor09p | 3D-MoRSE descriptors | 3D |
| Mor09p | 2D autocorrelations | 2D |
| Mor11p | 3D-MoRSE descriptors | 3D |
| Mor13e | 3D-MoRSE descriptors | 1D |
| Mor13p | 3D-MoRSE descriptors | 3D |
| Mor16e | 3D-MoRSE descriptors | 3D |
| Mor24m | 2D autocorrelations | 2D |
| Mor24u | 2D autocorrelations | 2D |
| Mor26u | 3D-MoRSE descriptors | 3D |
| Mor27m | 3D-MoRSE descriptors | 3D |
| Mor28m | 3D-MoRSE descriptors | 3D |
| Mor32u | 3D-MoRSE descriptors | 3D |
| Ms | Topological descriptors | 2D |
| N-067 | Atom-centered fragments | 1D |
| N-069 | Atom-centered fragments | 1D |
| N-070 | Atom-centered fragments | 1D |
| N-074 | Atom-centered fragments | 1D |
| nArC=N | Functional group counts | 1D |
| nArCN | Functional group counts | 1D |
| nArCONR2 | Functional group counts | 1D |
| nArCOOH | Functional group counts | 1D |
| nArCOOR | Functional group counts | 1D |
| nArNH2 | Functional group counts | 1D |
| nArOCON | Functional group counts | 1D |
| nArOH | Functional group counts | 1D |
| nArX | Functional group counts | 1D |
| nC(=N)N2 | Functional group counts | 1D |
| nC=N-N< | Functional group counts | 1D |
| nCconj | Functional group counts | 1D |
| nCONN | Functional group counts | 1D |
| nCRX3 | Functional group counts | 1D |
| Neoplastic-50 | Drug-like indices | Other |
| Neoplastic-80 | Drug-like indices | Other |
| nHBonds | Functional group counts | 1D |
| nHDON | Functional group counts | 1D |
| nlsoxazoles | Functional group counts | 1D |
| nN(CO)2 | Functional group counts | 1D |
| nN-N | Functional group counts | 1D |
| nOxolanes | Functional group counts | 1D |
| nPyrazines | Functional group counts | 1D |
| nPyridines | Functional group counts | 1D |
| nR=Cp | Functional group counts | 1D |
| nR=Ct | Functional group counts | 1D |
| nR08 | Ring descriptors | 2D |
| nR08 | Ring descriptors | 2D |
| nRNHR | Functional group counts | 1D |
| nRNR2 | Functional group counts | 1D |
| nROCON | Functional group counts | 1D |
| nROR | Functional group counts | 1D |
| nS(=O)2 | Functional group counts | 1D |
| nTB | Constitutional indices | 0D |
| P2e | WHIM descriptors | 3D |
| PCWTe | Charge descriptors | Other |
| PJI2 | Topological indices | 2D |
| Psicotic-80 | Drug-like indices | Other |
| Psychotic-50 | Drug-like indices | Other |
| PW3 | Topological indices | 2D |
| qpmax | Charge descriptors | Other |
| R3e | GETAWAY descriptors | 3D |
| R3v+ | GETAWAY descriptors | 3D |
| R5v+ | GETAWAY descriptors | 3D |
| R7p+ | GETAWAY descriptors | 3D |
| RCI | Ring descriptors | 2D |
| RDF090p | RDF descriptors | 3D |
| RDF120e | RDF descriptors | 3D |
| RDF120m | RDF descriptors | 3D |
| RDF150m | RDF descriptors | 3D |
| RDF155m | RDF descriptors | 3D |
| SRW03 | Walk and path counts | 2D |
| SRW09 | Walk and path counts | 2D |
| T(O…Br) | 2D atomic pairs | 2D |
| T(O…O) | 2D atom pairs | 2D |
| T(S…F) | 2D atom pairs | 2D |
References
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Oja, M.; Sild, S.; Maran, U. Logistic Classification Models for pH-Permeability Profile: Predicting Permeability Classes for the Biopharmaceutical Classification System. J. Chem. Inf. Model. 2019, 59, 2442–2455. [Google Scholar] [CrossRef] [PubMed]
- Kansy, M.; Senner, F.; Gubernator, K. Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeation Assay in the Description of Passive Absorption Processes. J. Med. Chem. 1998, 41, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Siramshetty, V.; Williams, J.; Nguyễn, Ð.T.; Neyra, J.; Southall, N.; Mathé, E.; Xu, X.; Shah, P. Validating ADME QSAR Models Using Marketed Drugs. SLAS Discov. 2021, 26, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Diukendjieva, A.; Tsakovska, I.; Alov, P.; Pencheva, T.; Pajeva, I.; Worth, A.P.; Madden, J.C.; Cronin, M.T.D. Advances in the prediction of gastrointestinal absorption: Quantitative Structure-Activity Relationship (QSAR) modelling of PAMPA permeability. Comput. Toxicol. 2019, 10, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Chi, C.T.; Lee, M.H.; Weng, C.F.; Leong, M.K. In silico prediction of PAMPA effective permeability using a two-QSAR approach. Int. J. Mol. Sci. 2019, 20, 3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diukendjieva, A.; Alov, P.; Tsakovska, I.; Pencheva, T.; Richarz, A.; Kren, V.; Cronin, M.T.D.; Pajeva, I. In vitro and in silico studies of the membrane permeability of natural flavonoids from Silybum marianum (L.) Gaertn. and their derivatives. Phytomedicine 2019, 53, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Savić, J.; Dobričić, V.; Nikolic, K.; Vladimirov, S.; Dilber, S.; Brborić, J. In vitro prediction of gastrointestinal absorption of novel β-hydroxy-β-arylalkanoic acids using PAMPA technique. Eur. J. Pharm. Sci. 2017, 100, 36–41. [Google Scholar] [CrossRef] [PubMed]
- DeSesso, J.M.; Jacobson, C.F. Anatomical and physiological parameters affecting gastrointestinal absorption in humans and rats. Food Chem. Toxicol. 2001, 39, 209–228. [Google Scholar] [CrossRef]
- Charman, W.N.; Porter, C.J.H.; Mithani, S.; Dressman, J.B. Physicochemical and Physiological Mechanisms for the Effects of Food on Drug Absorption: The Role of Lipids and pH. J. Pharm. Sci. 1997, 86, 269–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oja, M.; Maran, U. pH-permeability profiles for drug substances: Experimental detection, comparison with human intestinal absorption and modelling. Eur. J. Pharm. Sci. 2018, 123, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Ruusmann, V.; Sild, S.; Maran, U. QSAR DataBank repository: Open and linked qualitative and quantitative structure–activity relationship models. J. Cheminform. 2015, 7, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oja, M.; Sild, S.; Maran, U. Data for: Logistic Classification Models for pH-Permeability Profile: Predicting Permeability Classes for the Biopharmaceutical Classification System. QsarDB Repos. 2019. [Google Scholar] [CrossRef]
- Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W.C. Macromodel—An integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar] [CrossRef]
- Talete, S.r.l. DRAGON 6.0 for Windows (Software for Molecular Descriptor Calculations); Talete S.r.l.: Milan, Italy, 2015. [Google Scholar]
- Sjöström, M.; Wold, S.; Söderström, B. PLS Discriminant Plots. In Pattern Recognition in Practice; Elsevier: Amsterdam, The Netherlands, 1986. [Google Scholar]
- Ståhle, L.; Wold, S. Partial least squares analysis with cross-validation for the two-class problem: A Monte Carlo study. J. Chemom. 1987, 1, 185–196. [Google Scholar] [CrossRef]
- Wold, S.; Martens, H.; Wold, H. The Multivariate Calibration Problem in Chemistry Solved by the PLS Method. In Matrix Pencils; Lecture Notes in Mathematics; Kågström, B., Ruhe, A., Eds.; Springer: Berlin/Heidelberg, Germany, 1983; Volume 973. [Google Scholar]
- Fisher, R.A. The use of multiple measurements in taxonomic problems. Ann. Eugen. 1936, 7, 179–188. [Google Scholar] [CrossRef]
- Pérez, N.F.; Ferré, J.; Boqué, R. Calculation of the reliability of classification in discriminant partial least-squares binary classification. Chemom. Intell. Lab. Syst. 2009, 95, 122–128. [Google Scholar] [CrossRef]
- Snee, R.D. Validation of Regression Models: Methods and Examples. Technometrics 1977, 19, 415–428. [Google Scholar] [CrossRef]
- Roger, J.M.; Palagos, B.; Bertrand, D.; Fernandez-Ahumada, E. CovSel: Variable selection for highly multivariate and multi-response calibration: Application to IR spectroscopy. Chemom. Intell. Lab. Syst. 2011, 106, 216–223. [Google Scholar] [CrossRef] [Green Version]


| Compound Name | Compound Name |
|---|---|
| 8-hydroxyquinoline | Fluvoxamine |
| Acebutolol | Furosemide |
| Acetanilide | Gemfibrozil |
| Acrivastine | Guanabenz |
| Alfuzosin | Haloperidol |
| Alosetron | Hydralazine |
| Aminosalicylic acid | Hydrochlorothiazide |
| Amodiaquine | Hydroxychloroquine |
| Bendroflumethiazide | Imipramine |
| Benzocaine | Indomethacin |
| Brimonidine | Iopromide |
| Capecitabine | Irbesartan |
| Chloropyramine | Isoniazid |
| Chlorzoxazone | Isradipine |
| Chlorthalidone | Ketoconazole |
| Ciclopirox | Ketoprofen |
| Coumarin | Ketotifen |
| Cyclobenzaprine | Labetalol |
| Cyproheptadine | Lamivudine |
| Deferiprone | Lamotrigine |
| Dipyridamole | Lidocaine |
| Donepezil | Linezolid |
| Flupirtine | Lomefloxacin |
| Galantamine | Marbofloxacin |
| Granisetron | Mazipredone |
| Hesperetin | Medroxyprogesterone acetate |
| Hydrocortisone | Methylprednisolone |
| Isoxepac | Metoclopramide |
| Levofloxacin | Metoprolol |
| Levosimendan | Metyrapone |
| Loxapine | Midazolam |
| Melatonin | Nafcillin |
| Menadione | Naloxone |
| Mepivacaine | Naproxen |
| Mycophenolate mofetil | Naratriptan |
| Oxybuprocaine | Nevirapine |
| Phenazopyridine | Nicotinamide |
| Phenol | Nicotine |
| Physostigmine | Norfloxacin |
| Prilocaine | Ofloxacin |
| Primaquine | Omeprazole |
| Procainamide | Ondansetron |
| Procarbazine | Orbifloxacin |
| Proparacaine | Oxacillin |
| Protionamide | Oxcarbazepine |
| Pyridoxal | Oxprenolol |
| Salicylamide | Pantoprazole |
| Succinylsulfathiazole | Papaverine |
| Sulpiride | Paroxetine |
| Zolmitriptan | Pentamidine |
| Tamsulosin | Pentoxifylline |
| Tolmetin | Perphenazine |
| Trichlormethiazide | Pheniramine |
| Tripelennamine | Phenylbutazone |
| Vardenafil | Pindolol |
| Yohimbine | Pitofenone |
| Aciclovir | Prednisolone |
| Acitretin | Prednisone |
| Allopurinol | Procaine |
| Ambroxol | Promethazine |
| Aminophenazone | Propafenone |
| Amitriptyline | Propofol |
| Amoxapine | Propranolol |
| Antipyrine | Pyrazinamide |
| Astemizole | Quetiapine |
| Azelastine | Quinine |
| Beclomethasone dipropionate | Rabeprazole |
| Benzbromarone | Salicylic acid |
| Benzoic acid | Sarafloxacin |
| Betahistine | Sulfacetamide |
| Betaxolol | Sulfachloropyridazine |
| Bicalutamide | Sulfadiazine |
| Bisacodyl | Sulfadimethoxine |
| Bisoprolol | Sulfadoxine |
| Budesonide | Sulfaguanidine |
| Caffeine | Sulfamerazine |
| Carbamazepine | Sulfamethazine |
| Carvedilol | Sulfamethizole |
| Cefadroxil | Sulfamethoxazole |
| Chlormadinone acetate | Sulfamethoxypyridazine |
| Chloroquine | Sulfamonomethoxine |
| Chlorphenamine | Sulfamoxole |
| Chlorpromazine | Sulfanilamide |
| Cinoxacin | Sulfapyridine |
| Ciprofloxacin | Sulfaquinoxaline |
| Citalopram | Sulfathiazole |
| Clomipramine | Sulfisoxazole |
| Clonidine | Sulindac |
| Clozapine | Zalcitabine |
| Cromoglicic acid | Zaleplon |
| Danofloxacin | Tacrine |
| Dapsone | Tenidap |
| Desipramine | Theobromine |
| Dexamethasone | Theophylline |
| Diclofenac | Thiabendazole |
| Dicloxacillin | Thiamphenicol |
| Difloxacin | Thiethylperazine |
| Diflunisal | Thioridazine |
| Dihydroergotamine | Tiagabine |
| Diltiazem | Timolol |
| Dimetindene | Tizanidine |
| Disopyramide | Tofisopam |
| Dolasetron | Tolbutamide |
| Domperidone | Tolperisone |
| Doxazosin | Tolterodine |
| Doxepin | Tramadol |
| Enoxacin | Trazodone |
| Enrofloxacin | Triamcinolone |
| Ethinyl estradiol | Trimethoprim |
| Etoricoxib | Tropisetron |
| Famotidine | Valsartan |
| Fenoterol | Warfarin |
| Fluconazole | Verapamil |
| Flumequine | |
| Fluorescein |
| pH | Training Set | Test Set | |||||
|---|---|---|---|---|---|---|---|
| Class High-Permeable | Class Low-Permeable | Total | Class High-Permeable | Class Low-Permeable | Total | ||
| Dataset 1 | 3 | 35 | 118 | 153 | 23 | 52 | 75 |
| Dataset 2 | 5 | 54 | 99 | 153 | 27 | 48 | 75 |
| Dataset 3 | 7.4 | 82 | 71 | 153 | 51 | 24 | 75 |
| Dataset 4 | 9 | 87 | 65 | 152 | 40 | 36 | 76 |
| Dataset 5 | IP | 106 | 47 | 153 | 57 | 18 | 75 |
| pH | LVs | Accuracy | CCR (%) | ||
|---|---|---|---|---|---|
| Class High | Class Low | ||||
| 3 | Calibration | 16 | 100.00 | 100.00 | 100.00 |
| CV | 92.16 | 77.14 | 96.61 | ||
| Prediction | 82.67 | 65.22 | 90.38 | ||
| 5 | Calibration | 10 | 100.00 | 100.00 | 100.00 |
| CV | 89.54 | 85.19 | 91.92 | ||
| Prediction | 89.33 | 81.48 | 93.75 | ||
| 7.4 | Calibration | 7 | 100.00 | 100.00 | 100.00 |
| CV | 91.50 | 93.90 | 88.73 | ||
| Prediction | 89.33 | 84.31 | 100.00 | ||
| 9 | Calibration | 7 | 100.00 | 100.00 | 100.00 |
| CV | 90.13 | 93.10 | 86.15 | ||
| Prediction | 88.16 | 77.50 | 100.00 | ||
| IP | Calibration | 11 | 100.00 | 100.00 | 100.00 |
| CV | 96.08 | 96.23 | 95.74 | ||
| Prediction | 93.33 | 91.23 | 100.00 | ||
| pH | nVar | LVs | Accuracy | CCR (%) | ||
|---|---|---|---|---|---|---|
| Class High | Class Low | |||||
| 3 | CV | 41 | 12 | 96.73 | 91.43 | 98.30 |
| Prediction | 80.00 | 65.22 | 86.54 | |||
| 5 | CV | 23 | 6 | 96.73 | 94.44 | 97.98 |
| Prediction | 84.49 | 81.48 | 87.50 | |||
| 7.4 | CV | 47 | 6 | 97.38 | 97.56 | 97.18 |
| Prediction | 96.08 | 92.16 | 100.00 | |||
| 9 | CV | 47 | 7 | 98.68 | 98.85 | 98.46 |
| Prediction | 92.36 | 87.50 | 97.22 | |||
| IP | CV | 34 | 5 | 99.35 | 99.06 | 100.00 |
| Prediction | 93.86 | 100.00 | 87.72 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biancolillo, A.; Mennitti, L.; Foschi, M.; Marini, F. Advanced Analytical Tools for the Estimation of Gut Permeability of Compounds of Pharmaceutical Interest. Appl. Sci. 2022, 12, 1326. https://doi.org/10.3390/app12031326
Biancolillo A, Mennitti L, Foschi M, Marini F. Advanced Analytical Tools for the Estimation of Gut Permeability of Compounds of Pharmaceutical Interest. Applied Sciences. 2022; 12(3):1326. https://doi.org/10.3390/app12031326
Chicago/Turabian StyleBiancolillo, Alessandra, Luca Mennitti, Martina Foschi, and Federico Marini. 2022. "Advanced Analytical Tools for the Estimation of Gut Permeability of Compounds of Pharmaceutical Interest" Applied Sciences 12, no. 3: 1326. https://doi.org/10.3390/app12031326
APA StyleBiancolillo, A., Mennitti, L., Foschi, M., & Marini, F. (2022). Advanced Analytical Tools for the Estimation of Gut Permeability of Compounds of Pharmaceutical Interest. Applied Sciences, 12(3), 1326. https://doi.org/10.3390/app12031326