
Investigating the Effect of Imputed Structural Variants from Whole-Genome Sequence on Genome-Wide Association and Genomic Prediction in Dairy Cattle

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequencing Data
2.2. Structural Variation Detection and Genotyping
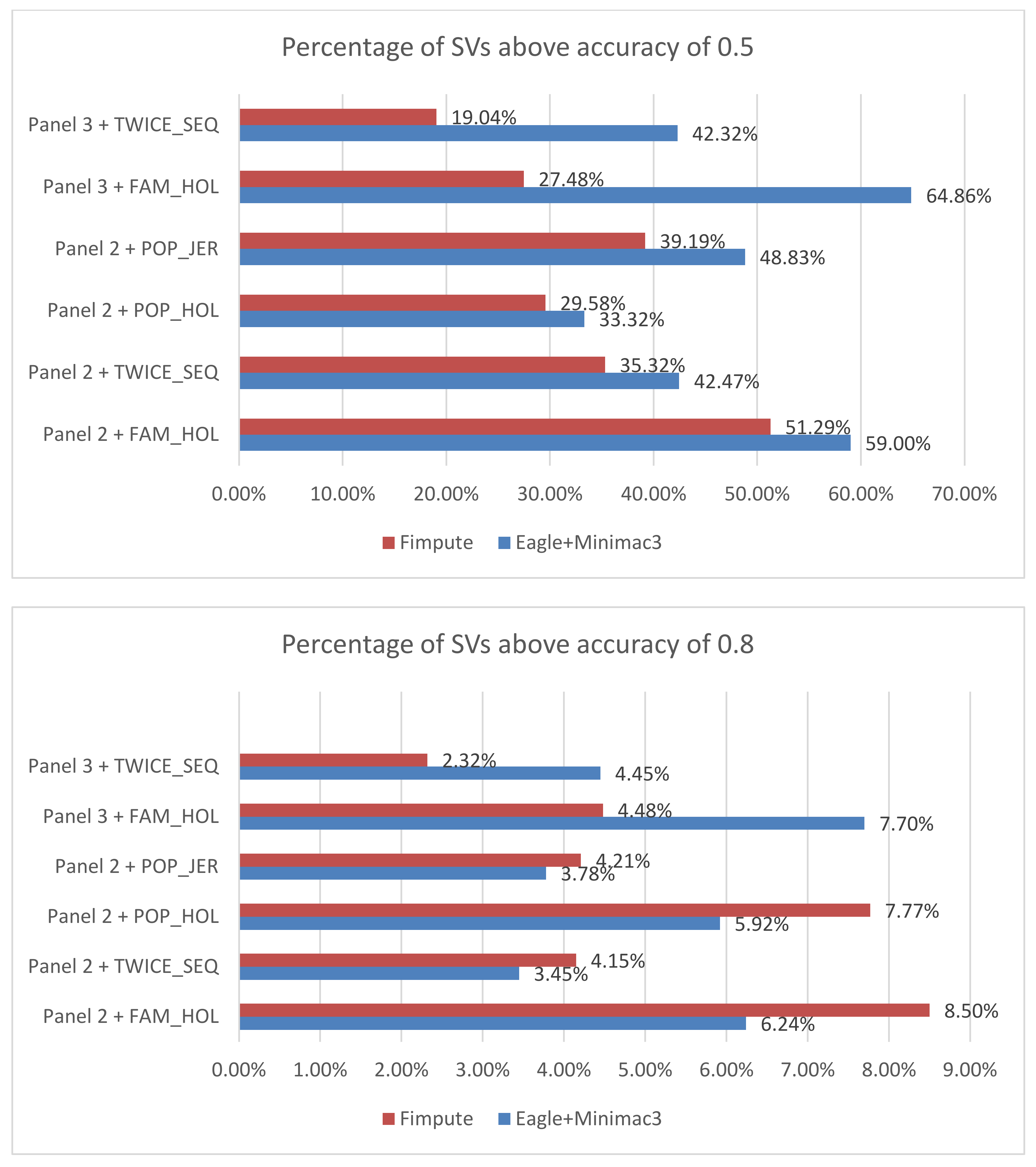
2.3. Structural Variation Imputation Accuracy Assessment
2.4. Genome-Wide Association Studies
2.5. Statistical Model for Variance Estimation and Genomic Prediction
3. Results
3.1. Structural Variant Sets
3.2. Imputation Accuracy
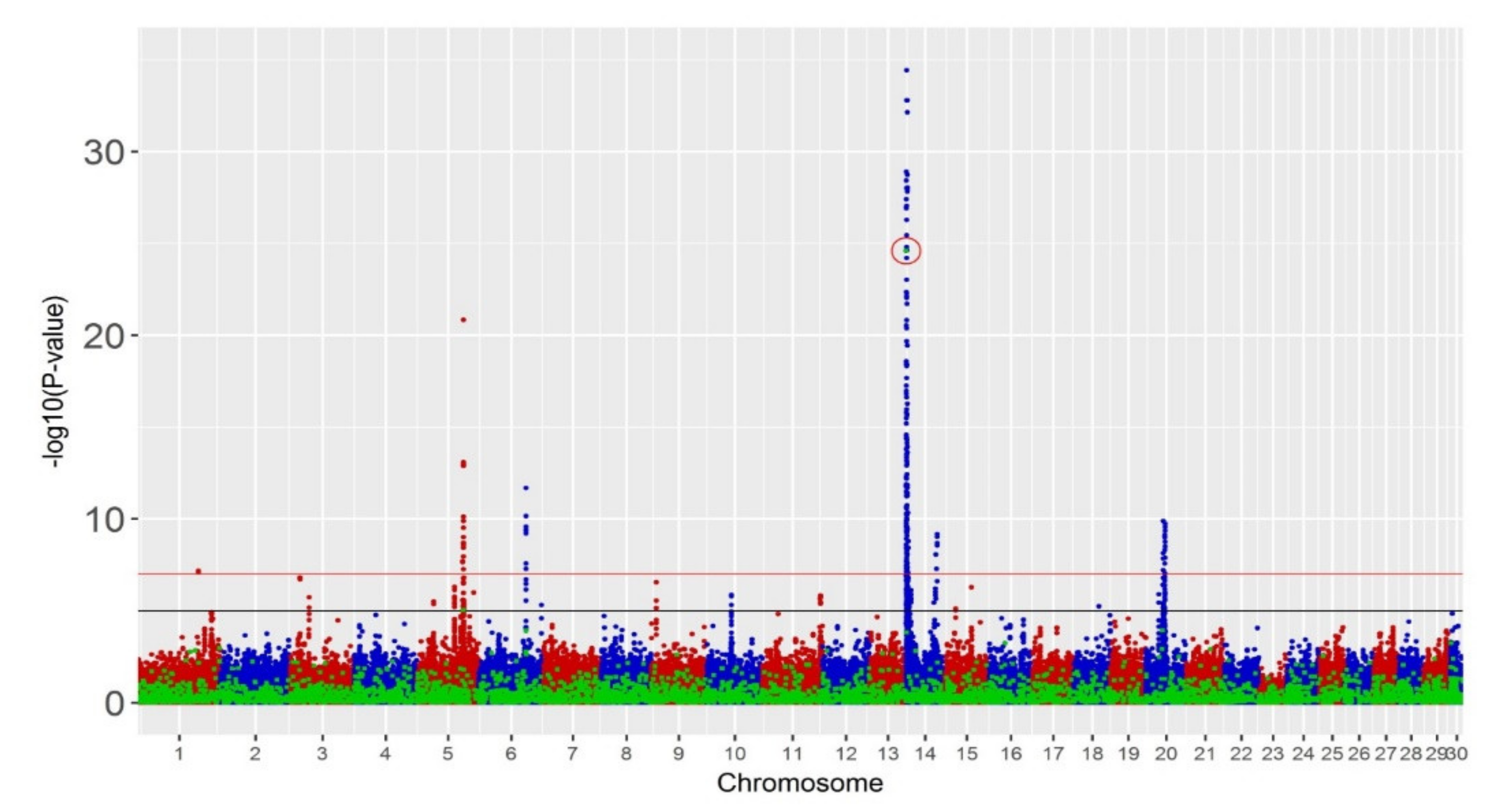
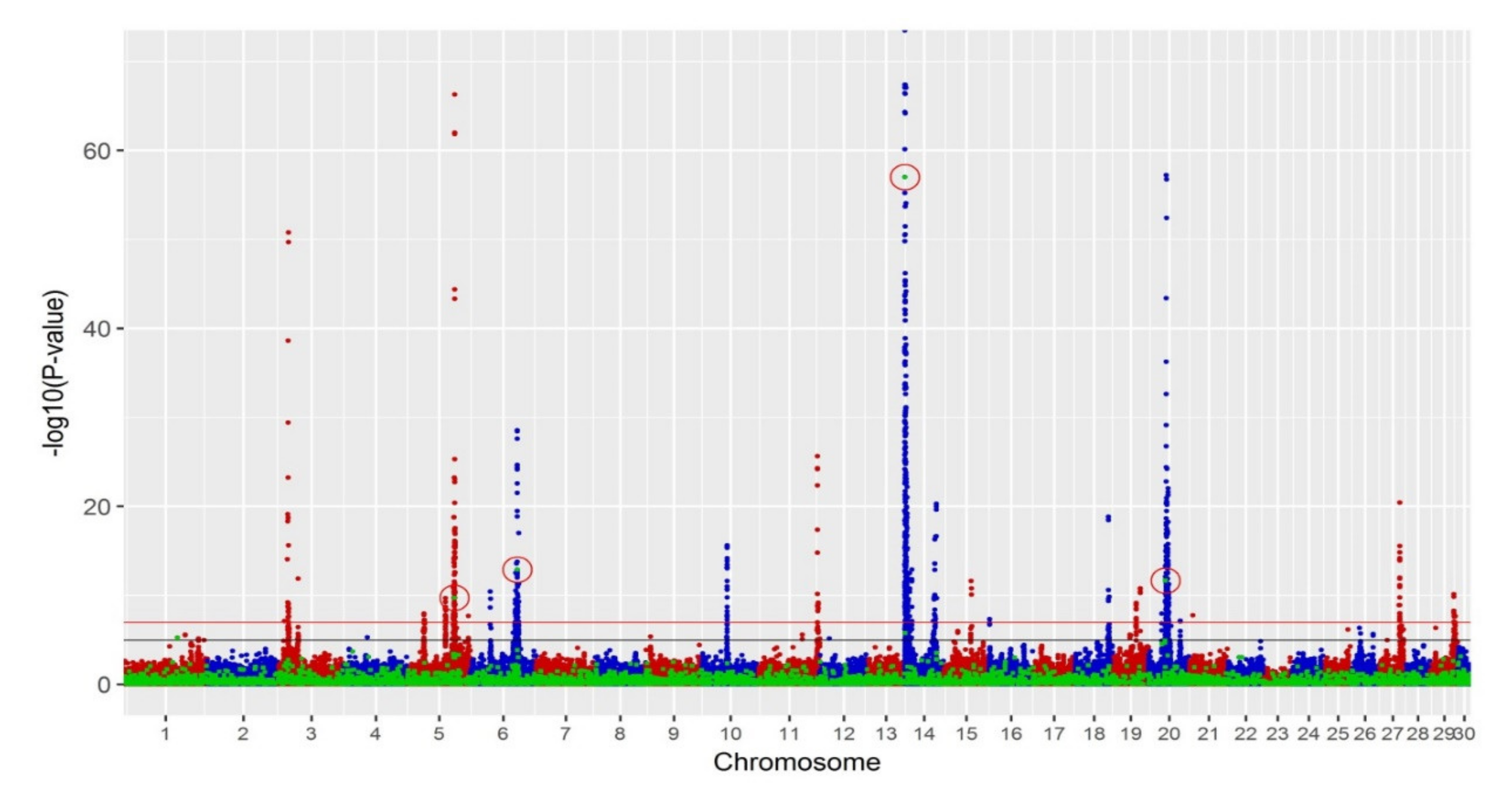
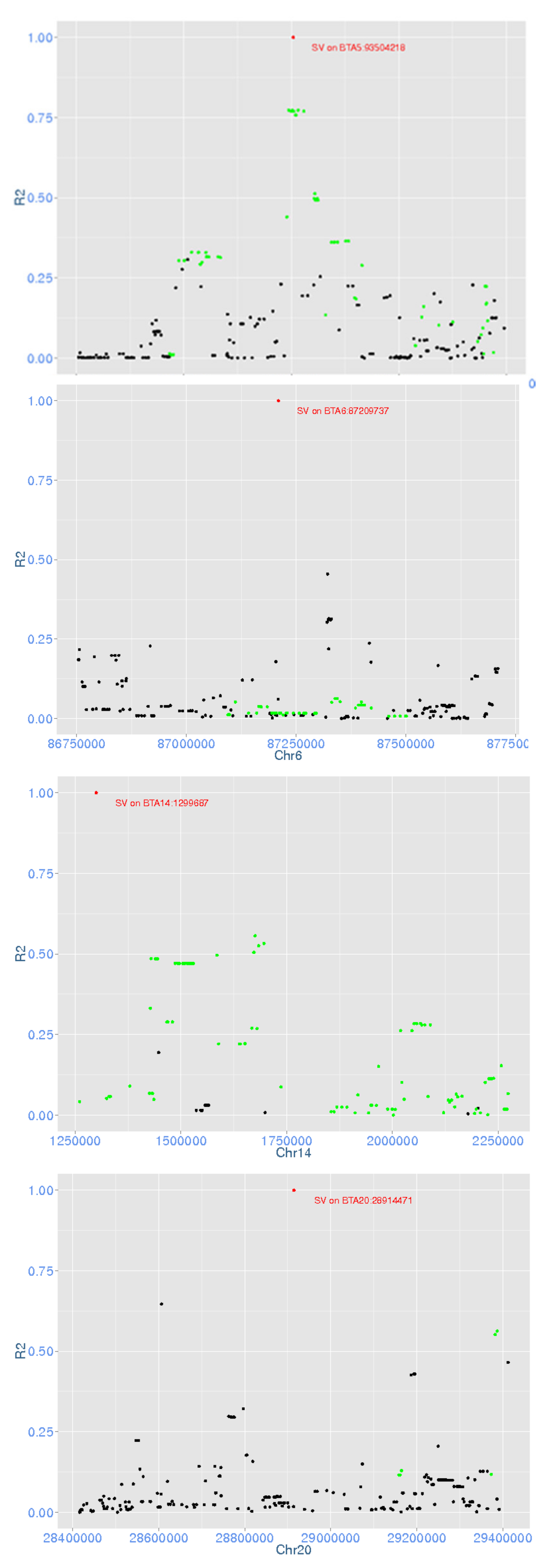
3.3. GWAS
3.4. Variance Components
3.5. Genomic Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Need, A.C.; Attix, D.K.; McEvoy, J.M.; Cirulli, E.T.; Linney, K.L.; Hunt, P.; Ge, D.; Heinzen, E.L.; Maia, J.M.; Shianna, K.V.; et al. A genome-wide study of common SNPs and CNVs in cognitive performance in the CANTAB. Hum. Mol. Genet. 2009, 18, 4650–4661. [Google Scholar] [CrossRef] [PubMed]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-Wide Detection of Single-Nucleotide and Copy-Number Variations of a Single Human Cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef]
- Olsson, L.M.; Nerstedt, A.; Lindqvist, A.K.; Johansson, S.C.; Medstrand, P.; Olofsson, P.; Holmdahl, R. Copy number variation of the gene NCF1 is associated with rheumatoid arthritis. Antioxid. Redox Sign. 2012, 16, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.E.; Hou, Y.; Zhu, B.; Cardone, M.F.; Jiang, L.; Cellamare, A.; Mitra, A.; Alexander, L.J.; Coutinho, L.L.; Dell’Aquila, M.E.; et al. Analysis of copy number variations among diverse cattle breeds. Genome Res. 2010, 20, 693–703. [Google Scholar] [CrossRef]
- Hou, Y.; Liu, G.E.; Bickhart, D.M.; Matukumalli, L.K.; Li, C.; Song, J.; Gasbarre, L.C.; Van Tassell, C.P.; Sonstegard, T.S. Genomic regions showing copy number variations associate with resistance or susceptibility to gastrointestinal nematodes in Angus cattle. Funct. Integr. Genom. 2012, 12, 81–92. [Google Scholar] [CrossRef]
- Kadri, N.K.; Sahana, G.; Charlier, C.; Iso-Touru, T.; Guldbrandtsen, B.; Karim, L.; Nielsen, U.S.; Panitz, F.; Aamand, G.P.; Schulman, N.; et al. A 660-Kb Deletion with Antagonistic Effects on Fertility and Milk Production Segregates at High Frequency in Nordic Red Cattle: Additional Evidence for the Common Occurrence of Balancing Selection in Livestock. PLoS Genet. 2014, 10, e1004049. [Google Scholar] [CrossRef]
- Medugorac, I.; Seichter, D.; Graf, A.; Russ, I.; Blum, H.; Göpel, K.H.; Rothammer, S.; Förster, M.; Krebs, S. Bovine Polledness—An Autosomal Dominant Trait with Allelic Heterogeneity. PLoS ONE 2012, 7, e39477. [Google Scholar] [CrossRef]
- Rothammer, S.; Capitan, A.; Mullaart, E.; Seichter, D.; Russ, I.; Medugorac, I. The 80-kb DNA duplication on BTA1 is the only remaining candidate mutation for the polled phenotype of Friesian origin. Genet. Sel. Evol. 2014, 46, 44. [Google Scholar] [CrossRef]
- Zhang, H.; Du, Z.-Q.; Dong, J.-Q.; Wang, H.-X.; Shi, H.-Y.; Wang, N.; Wang, S.-Z.; Li, H. Detection of genome-wide copy number variations in two chicken lines divergently selected for abdominal fat content. BMC Genom. 2014, 15, 517. [Google Scholar] [CrossRef]
- Fadista, J.; Nygaard, M.; Holm, L.-E.; Thomsen, B.; Bendixen, C. A Snapshot of CNVs in the Pig Genome. PLoS ONE 2008, 3, e3916. [Google Scholar] [CrossRef] [PubMed]
- Kijas, J.W.; Barendse, W.; Barris, W.; Harrison, B.; McCulloch, R.; McWilliam, S.; Whan, V. Analysis of copy number variants in the cattle genome. Gene 2011, 482, 73–77. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Dekkers, J.C. Genomic Selection Using Low-density Marker Panels. Genetics 2009, 182, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Aliloo, H.; Pryce, J.E.; González-Recio, O.; Cocks, B.G.; Hayes, B.J. Validation of markers with non-additive effects on milk yield and fertility in Holstein and Jersey cows. BMC Genet. 2015, 16, 89. [Google Scholar] [CrossRef]
- Marchini, J.; Howie, B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010, 11, 499–511. [Google Scholar] [CrossRef]
- Hayes, B.J.; Bowman, P.J.; Daetwyler, H.D.; Kijas, J.W.; van der Werf, J.H.J. Accuracy of genotype imputation in sheep breeds. Anim. Genet. 2011, 43, 72–80. [Google Scholar] [CrossRef]
- Hehir-Kwa, J.Y.; Marschall, T.; Kloosterman, W.P.; Francioli, L.C.; Baaijens, J.A.; Dijkstra, L.J.; Abdellaoui, A.; Koval, V.; Thung, D.T.; Wardenaar, R.; et al. A high-quality human reference panel reveals the complexity and distribution of genomic structural variants. Nat. Commun. 2016, 7, 12989. [Google Scholar] [CrossRef]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef]
- Sargolzaei, M.; Chesnais, J.; Schenkel, F. A new approach for efficient genotype imputation using information from relatives. BMC Genom. 2014, 15, 478. [Google Scholar] [CrossRef]
- Loh, P.-R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; Reshef, A.Y.; Finucane, H.K.; Schoenherr, S.; Forer, S.S.L.; McCarthy, S.; Abecasis, C.F.G.R.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schonherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Daetwyler, H.D.; Capitan, A.; Pausch, H.; Stothard, P.; Binsbergen, R.; Brondum, R.F. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 2014, 46. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Daetwyler, H.D. 1000 Bull Genomes Project to Map Simple and Complex Genetic Traits in Cattle: Applications and Outcomes. Ann. Rev. Anim. Biosci. 2019, 7. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Erbe, M.; Hayes, B.J.; Matukumalli, L.K.; Goswami, S.; Bowman, P.J.; Reich, C.M. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels. J. Dairy Sci. 2012, 95. [Google Scholar] [CrossRef]
- Chen, L.; Chamberlain, A.J.; Reich, C.M.; Daetwyler, H.D.; Hayes, B.J. Detection and validation of structural variations in bovine whole-genome sequence data. Genet. Sel. Evol. 2017, 49, 13. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.; Lunter, G.; Marth, G.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Bolormaa, S.; Pryce, J.E.; Reverter, A.; Zhang, Y.; Barendse, W.; Kemper, K.; Tier, B.; Savin, K.; Hayes, B.J.; Goddard, M.E. A multi-trait, meta-analysis for detecting pleiotropic polymorphisms for stature, fatness and reproduction in beef cattle. PLoS Genet. 2014, 10, e1004198. [Google Scholar] [CrossRef] [PubMed]
- Bolormaa, S.; Hayes, B.J.; Savin, K.; Hawken, R.; Barendse, W.; Arthur, P.F.; Herd, R.M.; Goddard, M.E. Genome-wide association studies for feedlot and growth traits in cattle. J. Anim. Sci. 2011, 89, 1684–1697. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Nejati-Javaremi, A.; Smith, C.; Gibson, J.P. Effect of total allelic relationship on accuracy of evaluation and response to selection. J. Anim. Sci. 1997, 75, 1738–1745. [Google Scholar] [CrossRef]
- Gilmour, A.R.; Gogel, B.J.; Cullis, B.R.; Welham, S.J.; Thompson, R. ASReml User Guide Release 4.1 Structural Specification; VSN International Ltd.: Hemel Hempstead, UK, 2015. [Google Scholar]
- Butty, A.M.; Chud, T.C.S.; Miglior, F.; Schenkel, F.S.; Kommadath, A.; Krivushin, K.; Grant, J.R.; Häfliger, I.M.; Drögemüller, C.; Cánovas, A.; et al. High confidence copy number variants identified in Holstein dairy cattle from whole genome sequence and genotype array data. Sci. Rep. 2020, 10, 8044. [Google Scholar] [CrossRef] [PubMed]
- Rowan, T.N.; Hoff, J.L.; Crum, T.E.; Taylor, J.F.; Schnabel, R.D.; Decker, J.E. A multi-breed reference panel and additional rare variants maximize imputation accuracy in cattle. Genet. Sel. Evol. 2019, 51, 77. [Google Scholar] [CrossRef]
- Littlejohn, M.D.; Tiplady, K.; Fink, T.A.; Lehnert, K.; Lopdell, T.; Johnson, T.; Couldrey, C.; Keehan, M.; Sherlock, R.G.; Harland, C.; et al. Sequence-based Association Analysis Reveals an MGST1 eQTL with Pleiotropic Effects on Bovine Milk Composition. Sci. Rep. 2016, 6, 25376. [Google Scholar] [CrossRef] [PubMed]
- Do, D.N.; Bissonnette, N.; Lacasse, P.; Miglior, F.; Sargolzaei, M.; Zhao, X.; Ibeagha-Awemu, E.M. Genome-wide association analysis and pathways enrichment for lactation persistency in Canadian Holstein cattle. J. Dairy Sci. 2017, 100, 1955–1970. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.E.; Reich, C.M.; Bowman, P.J.; Vander Jagt, C.J.; Chamberlain, A.J.; Mason, B.A. Improved precision of QTL mapping using a nonlinear Bayesian method in a multi-breed population leads to greater accuracy of across-breed genomic predictions. Genet. Sel. Evol. 2015, 47. [Google Scholar] [CrossRef]
- Elsik, C.G.; Tellam, R.L.; Worley, K.C. The Genome Sequence of Taurine Cattle: A Window to Ruminant Biology and Evolution. Science 2009, 324, 522–528. [Google Scholar] [CrossRef]
- Yang, S.; Qi, C.; Xie, Y.; Cui, X.; Gao, Y.; Jiang, J.; Jiang, L.; Zhang, S.; Zhang, Q.; Sun, D. A post-GWAS replication study confirming the association of C14H8orf33 gene with milk production traits in dairy cattle. Front. Agric. Sci. Eng. 2014, 1, 321–330. [Google Scholar] [CrossRef]
- Macciotta, N.P.P.; Biffani, S.; Bernabucci, U.; Lacetera, N.; Vitali, A.; Ajmone-Marsan, P.; Nardone, A. Derivation and genome-wide association study of a principal component-based measure of heat tolerance in dairy cattle. J. Dairy Sci. 2017, 100, 4683–4697. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, X.; Yang, J.; Wang, H.; Jiang, J.; Liu, L.; He, S.; Ding, X.; Liu, J.; Zhang, Q. Targeted resequencing of GWAS loci reveals novel genetic variants for milk production traits. BMC Genom. 2014, 15, 1105. [Google Scholar] [CrossRef] [PubMed]
- Grisart, B.; Coppieters, W.; Farnir, F.; Karim, L.; Ford, C.; Berzi, P.; Cambisano, N.; Mni, M.; Reid, S.; Simon, P.; et al. Positional Candidate Cloning of a QTL in Dairy Cattle: Identification of a Missense Mutation in the Bovine DGAT1 Gene with Major Effect on Milk Yield and Composition. Genome Res. 2002, 12, 222–231. [Google Scholar] [CrossRef]
- Park, T.; Casella, G. The Bayesian Lasso. J. Am. Stat. Assoc. 2008, 103, 681–686. [Google Scholar] [CrossRef]
- Ni, G.; Cavero, D.; Fangmann, A.; Erbe, M.; Simianer, H. Whole-genome sequence-based genomic prediction in laying chickens with different genomic relationship matrices to account for genetic architecture. Genet. Sel. Evol. 2017, 49, 8. [Google Scholar] [CrossRef]
- Druet, T.; Macleod, I.M.; Hayes, B.J. Toward genomic prediction from whole-genome sequence data: Impact of sequencing design on genotype imputation and accuracy of predictions. Heredity 2013, 112, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Van Binsbergen, R.; Bink, M.C.; Calus, M.P.; van Eeuwijk, F.A.; Hayes, B.J.; Hulsegge, I.; Veerkamp, R.F. Accuracy of imputation to whole-genome sequence data in Holstein Friesian cattle. Genet. Sel. Evol. 2014, 46, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bickhart, D.M.; Liu, G.E. The challenges and importance of structural variation detection in livestock. Front. Genet. 2014, 5, 37. [Google Scholar] [CrossRef]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. GigaScience 2020, 9, giaa021. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- English, A.C.; Salerno, W.J.; Hampton, O.A.; Gonzaga-Jauregui, C.; Ambreth, S.; Ritter, D.I.; Beck, C.R.; Davis, C.F.; Dahdouli, M.; Ma, S.; et al. Assessing structural variation in a personal genome—Towards a human reference diploid genome. BMC Genom. 2015, 16, 286. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Chaisson, M.; Nakhleh, L.; Chen, K. HySA: A Hybrid Structural variant Assembly approach using next-generation and single-molecule sequencing technologies. Genome Res. 2017, 27, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteome Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Ritz, A.; Bashir, A.; Sindi, S.; Hsu, D.; Hajirasouliha, I.; Raphael, B.J. Characterization of structural variants with single molecule and hybrid sequencing approaches. Bioinformatics 2014, 30, 3458–3466. [Google Scholar] [CrossRef] [PubMed]
- Couldrey, C.; Keehan, M.; Johnson, T.; Tiplady, K.; Winkelman, A.; Littlejohn, M.D.; Scott, A.; Kemper, K.E.; Hayes, B.; Davis, S.R.; et al. Detection and assessment of copy number variation using PacBio long-read and Illumina sequencing in New Zealand dairy cattle. J. Dairy Sci. 2017, 100, 5472–5478. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | p-Value Threshold 10−7 | p-Value Threshold 10−5 | p-Value Threshold 10−4 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP Number | SV Number | FDR for SV | SNP Number | SV Number | FDR for SV | SNP Number | SV Number | FDR for SV | |
| FY | 237 | 1 | 0.04% | 365 | 1 | 4.49% | 1159 | 3 | 14.95% |
| MY | 289 | 1 | 0.04% | 505 | 2 | 2.24% | 1644 | 2 | 22.43% |
| PY | 178 | 1 | 0.04% | 268 | 1 | 4.49% | 1152 | 2 | 14.95% |
| Fert | 34 | 0 | 99 | 0 | 888 | 0 | |||
| Otype | 0 | 0 | 11 | 0 | 763 | 2 | 22.43% | ||
| Chr | Start (bp) | End (bp) | SV Type | p-Value | Single-Trait (p-Value < 10−7) | Single-Trait (p-Value < 10−5) |
|---|---|---|---|---|---|---|
| Chr5 | 93,504,218 | 93,505,234 | Deletion | 1.9 × 10−10 | MY | |
| Chr6 | 87,209,737 | 87,211,122 | Deletion | 1.3 × 10−13 | ||
| Chr14 | 1,299,687 | 1,299,831 | Deletion | 1.0 × 10−57 | FY, MY, PY | FY, MY, PY |
| Chr20 | 28,914,471 | 28,915,027 | Deletion | 2.2 × 10−12 |
| Breed | Model | Variance Item | Traits | ||||
|---|---|---|---|---|---|---|---|
| FY | MY | PY | Fertility | Otype | |||
| Holstein | SNP | 0.708(0.022) | 0.79(0.019) | 0.763(0.035) | 0.495(0.028) | 0.476(0.019) | |
| SNP + SV | 0.692(0.027) | 0.766(0.023) | 0.738(0.041) | 0.471(0.033) | 0.486(0.024) | ||
| 0.022(0.021) | 0.038(0.019) | 0.038(0.03) | 0.031(0.024) | 0(0.02) | |||
| 0.031 | 0.047 | 0.049 | 0.062 | 0.000 | |||
| 0.715(0.023) | 0.804(0.019) | 0.777(0.036) | 0.502(0.029) | 0.486(0.02) | |||
| Jersey | SNP | 0.855(0.037) | 0.812(0.043) | 0.835(0.08) | 0.296(0.065) | 0.623(0.038) | |
| SNP + SV | 0.841(0.058) | 0.759(0.068) | 0.842(0.11) | 0.32(0.089) | 0.609(0.06) | ||
| 0.019(0.063) | 0.073(0.072) | 0(0.109) | 0(0.094) | 0.02(0.068) | |||
| 0.022 | 0.088 | 0.000 | 0.000 | 0.032 | |||
| 0.86(0.041) | 0.833(0.047) | 0.842(0.087) | 0.32(0.075) | 0.629(0.043) |
| Bulls | SNP Variance | SV Variance | SNP+SV Variance | SV Variance |
|---|---|---|---|---|
| Traits | ||||
| FY | 0.728(0.021) | 0.014(0.016) | 0.743(0.019) | 1.94% |
| Fert | 0.433(0.030) | 0.038(0.021) | 0.471(0.026) | 8.11% |
| MY | 0.758(0.02) | 0.036(0.016) | 0.794(0.017) | 4.57% |
| Otype | 0.526(0.036) | 0.000(0.026) | 0.526(0.033) | 0.00% |
| PY | 0.747(0.020) | 0.028(0.016) | 0.775(0.017) | 3.58% |
| Cows | ||||
| Traits | ||||
| FY | 0.394(0.009) | 0.008(0.004) | 0.402(0.009) | 1.94% |
| Fert | 0.105(0.008) | 0.000(0.004) | 0.105(0.007) | 0.00% |
| MY | 0.466(0.009) | 0.007(0.004) | 0.474(0.009) | 1.54% |
| Otype | 0.139(0.015) | 0.000(0.010) | 0.139(0.014) | 0.00% |
| PY | 0.366(0.009) | 0.013(0.004) | 0.379(0.009) | 3.53% |
| Sample Set | Model | Trait | ||||
|---|---|---|---|---|---|---|
| FY | MY | PY | Fertility | OType | ||
| Holstein | SNP | 0.645(0.008) | 0.751(0.007) | 0.808(0.005) | 0.525(0.011) | 0.588(0.014) |
| SNP+SV | 0.645(0.008) | 0.751(0.007) | 0.808(0.005) | 0.525(0.011) | 0.588(0.014) | |
| Jersey | SNP | 0.76(0.013) | 0.697(0.018) | 0.794(0.011) | 0.262(0.026) | 0.549(0.023) |
| SNP+SV | 0.76(0.013) | 0.698(0.018) | 0.795(0.011) | 0.262(0.026) | 0.55(0.023) | |
| Holstein+Jersey | SNP | 0.657(0.013) | 0.835(0.005) | 0.788(0.007) | 0.623(0.009) | 0.553(0.013) |
| SNP+SV | 0.656(0.013) | 0.838(0.005) | 0.789(0.007) | 0.623(0.009) | 0.553(0.013) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Pryce, J.E.; Hayes, B.J.; Daetwyler, H.D. Investigating the Effect of Imputed Structural Variants from Whole-Genome Sequence on Genome-Wide Association and Genomic Prediction in Dairy Cattle. Animals 2021, 11, 541. https://doi.org/10.3390/ani11020541
Chen L, Pryce JE, Hayes BJ, Daetwyler HD. Investigating the Effect of Imputed Structural Variants from Whole-Genome Sequence on Genome-Wide Association and Genomic Prediction in Dairy Cattle. Animals. 2021; 11(2):541. https://doi.org/10.3390/ani11020541
Chicago/Turabian StyleChen, Long, Jennie E. Pryce, Ben J. Hayes, and Hans D. Daetwyler. 2021. "Investigating the Effect of Imputed Structural Variants from Whole-Genome Sequence on Genome-Wide Association and Genomic Prediction in Dairy Cattle" Animals 11, no. 2: 541. https://doi.org/10.3390/ani11020541
APA StyleChen, L., Pryce, J. E., Hayes, B. J., & Daetwyler, H. D. (2021). Investigating the Effect of Imputed Structural Variants from Whole-Genome Sequence on Genome-Wide Association and Genomic Prediction in Dairy Cattle. Animals, 11(2), 541. https://doi.org/10.3390/ani11020541